
Senescent T Cells: The Silent Culprit in Acute Myeloid Leukemia Progression?

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Characteristics of Senescent T Cells
3. T Cell Senescence in the AML Microenvironment
3.1. Evidence of T Cell Senescence in the AML Microenvironment
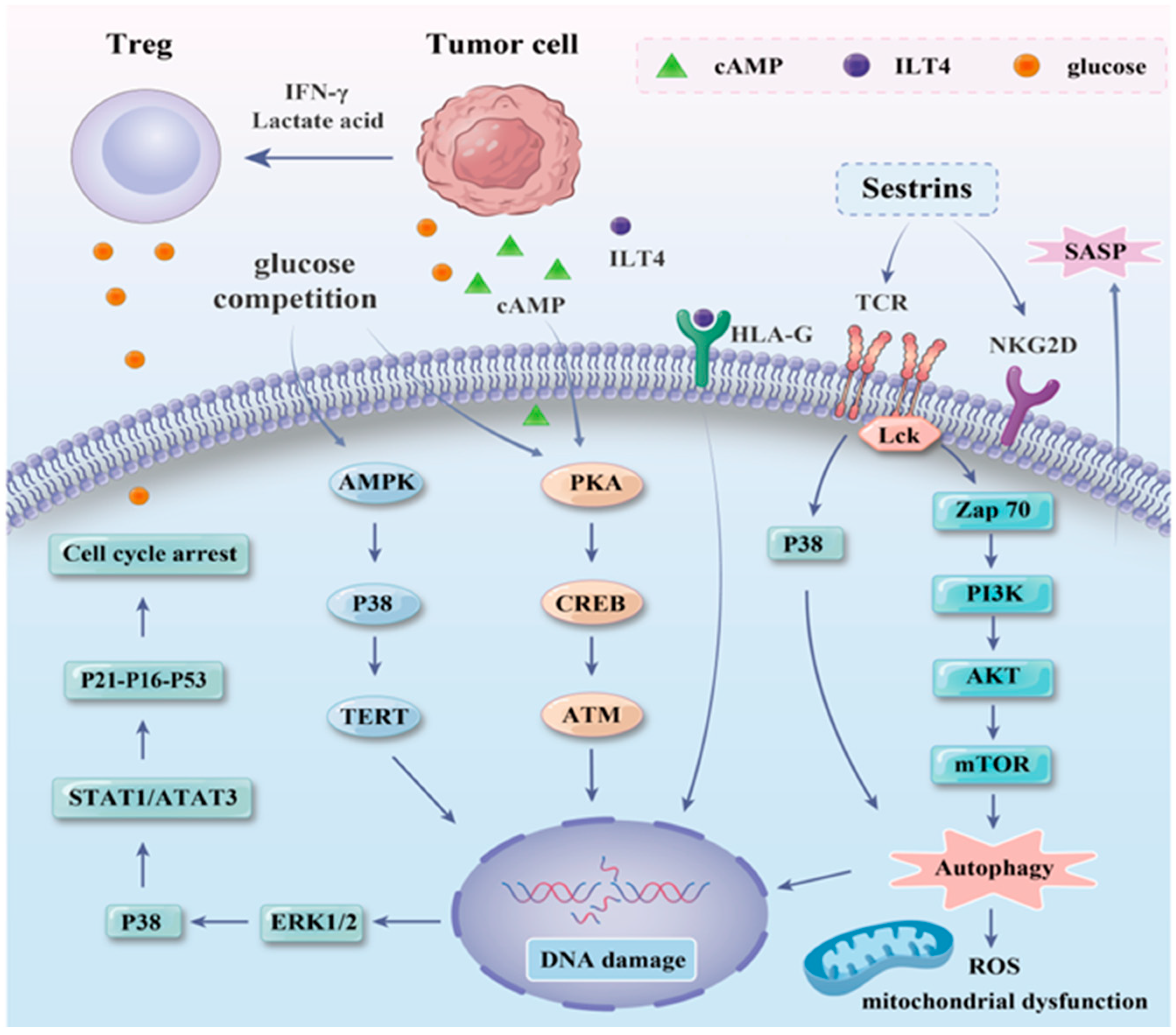
3.2. Inducing Mechanism of T Cell Senescence in the AML Microenvironment
3.3. Functional Alterations in Senescent T Cells in the AML Microenvironment
4. Intervention Strategies for T Cell Senescence in the AML Microenvironment
4.1. Blocking the Key Signaling Pathway of T Cell Senescence
4.2. Metabolic Regulation Targeting T Cell Senescence
4.3. Cellular Immunotherapy Targeting T Cell Senescence
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Zeng, A.G.X.; Bansal, S.; Jin, L.; Mitchell, A.; Chen, W.C.; Abbas, H.A.; Chan-Seng-Yue, M.; Voisin, V.; van Galen, P.; Tierens, A.; et al. A cellular hierarchy framework for understanding heterogeneity and predicting drug response in acute myeloid leukemia. Nat. Med. 2022, 28, 1212–1223. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Erba, H.P.; Freeman, S.D.; Wei, A.H. Acute myeloid leukaemia. Lancet 2023, 401, 2073–2086. [Google Scholar] [CrossRef] [PubMed]
- Vago, L.; Gojo, I. Immune escape and immunotherapy of acute myeloid leukemia. J. Clin. Investig. 2020, 130, 1552–1564. [Google Scholar] [CrossRef]
- Restelli, C.; Ruella, M.; Paruzzo, L.; Tarella, C.; Pelicci, P.G.; Colombo, E. Recent Advances in Immune-Based Therapies for Acute Myeloid Leukemia. Blood Cancer Discov. 2024, 5, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Mazziotta, F.; Biavati, L.; Rimando, J.; Rutella, S.; Borcherding, N.; Parbhoo, S.; Mukhopadhyay, R.; Chowdhury, S.; Knaus, H.A.; Valent, P.; et al. CD8+ T-cell differentiation and dysfunction inform treatment response in acute myeloid leukemia. Blood 2024, 144, 1168–1182. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liang, Q.; Ren, Y.; Guo, C.; Ge, X.; Wang, L.; Cheng, Q.; Luo, P.; Zhang, Y.; Han, X. Immunosenescence: Molecular mechanisms and diseases. Signal Transduct. Target. Ther. 2023, 8, 200. [Google Scholar] [CrossRef]
- Rutella, S.; Vadakekolathu, J.; Mazziotta, F.; Reeder, S.; Yau, T.-O.; Mukhopadhyay, R.; Dickins, B.; Altmann, H.; Kramer, M.; Knaus, H.A.; et al. Immune dysfunction signatures predict outcomes and define checkpoint blockade-unresponsive microenvironments in acute myeloid leukemia. J. Clin. Investig. 2022, 132, e159579. [Google Scholar] [CrossRef]
- Knaus, H.A.; Berglund, S.; Hackl, H.; Blackford, A.L.; Zeidner, J.F.; Montiel-Esparza, R.; Mukhopadhyay, R.; Vanura, K.; Blazar, B.R.; Karp, J.E.; et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight 2018, 3, e120974. [Google Scholar] [CrossRef]
- Zelle-Rieser, C.; Thangavadivel, S.; Biedermann, R.; Brunner, A.; Stoitzner, P.; Willenbacher, E.; Greil, R.; Jöhrer, K. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J. Hematol. Oncol. 2016, 9, 116. [Google Scholar] [CrossRef]
- Ye, J.; Ma, C.; Hsueh, E.C.; Dou, J.; Mo, W.; Liu, S.; Han, B.; Huang, Y.; Zhang, Y.; Varvares, M.A.; et al. TLR8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol. Med. 2014, 6, 1294–1311. [Google Scholar] [CrossRef]
- Vadakekolathu, J.; Rutella, S. Escape from T-cell-targeting immunotherapies in acute myeloid leukemia. Blood 2024, 143, 2689–2700. [Google Scholar] [CrossRef] [PubMed]
- ElTanbouly, M.A.; Noelle, R.J. Rethinking peripheral T cell tolerance: Checkpoints across a T cell’s journey. Nat. Rev. Immunol. 2021, 21, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Elyahu, Y.; Monsonego, A. Thymus involution sets the clock of the aging T-cell landscape: Implications for declined immunity and tissue repair. Ageing Res. Rev. 2021, 65, 101231. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hoft, D.F.; Peng, G. Senescent T cells within suppressive tumor microenvironments: Emerging target for tumor immunotherapy. J. Clin. Investig. 2020, 130, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Zamudio, R.I.; Dewald, H.K.; Vasilopoulos, T.; Gittens-Williams, L.; Fitzgerald-Bocarsly, P.; Herbig, U. Senescence-associated β-galactosidase reveals the abundance of senescent CD8+ T cells in aging humans. Aging Cell 2021, 20, e13344. [Google Scholar] [CrossRef]
- Ye, J.; Huang, X.; Hsueh, E.C.; Zhang, Q.; Ma, C.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Human regulatory T cells induce T-lymphocyte senescence. Blood 2012, 120, 2021–2031. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Liu, J.; Liu, J.; Qin, G.; Li, J.; Fu, Z.; Li, J.; Li, M.; Guo, C.; Zhao, M.; Zhang, Z.; et al. MDSCs-derived GPR84 induces CD8+ T-cell senescence via p53 activation to suppress the antitumor response. J. Immunother. Cancer 2023, 11, e007802. [Google Scholar] [CrossRef]
- Zhao, B.; Wu, B.; Feng, N.; Zhang, X.; Zhang, X.; Wei, Y.; Zhang, W. Aging microenvironment and antitumor immunity for geriatric oncology: The landscape and future implications. J. Hematol. Oncol. 2023, 16, 28. [Google Scholar] [CrossRef]
- Liu, X.; Mo, W.; Ye, J.; Li, L.; Zhang, Y.; Hsueh, E.C.; Hoft, D.F.; Peng, G. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat. Commun. 2018, 9, 249. [Google Scholar] [CrossRef]
- Mittelbrunn, M.; Kroemer, G. Hallmarks of T cell aging. Nat. Immunol. 2021, 22, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, S.; Tang, X.; Peng, Y.; Jiang, T.; Zhang, X.; Li, J.; Liu, Y.; Yang, Z. The role of KLRG1: A novel biomarker and new therapeutic target. Cell Commun. Signal 2024, 22, 337. [Google Scholar] [CrossRef] [PubMed]
- Henson, S.M.; Franzese, O.; Macaulay, R.; Libri, V.; Azevedo, R.I.; Kiani-Alikhan, S.; Plunkett, F.J.; Masters, J.E.; Jackson, S.E.; Griffiths, S.J.; et al. KLRG1 signaling induces defective Akt (ser473) phosphorylation and proliferative dysfunction of highly differentiated CD8+ T cells. Blood 2009, 113, 6619–6628. [Google Scholar] [CrossRef] [PubMed]
- Libri, V.; Azevedo, R.I.; Jackson, S.E.; Di Mitri, D.; Lachmann, R.; Fuhrmann, S.; Vukmanovic-Stejic, M.; Yong, K.; Battistini, L.; Kern, F.; et al. Cytomegalovirus infection induces the accumulation of short-lived, multifunctional CD4+ CD45RA+ CD27− T cells: The potential involvement of interleukin-7 in this process. Immunology 2011, 132, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Schmitt, A.; Rojewski, M.T.; Chen, J.; Giannopoulos, K.; Fei, F.; Yu, Y.; Götz, M.; Heyduk, M.; Ritter, G.; et al. RHAMM-R3 peptide vaccination in patients with acute myeloid leukemia, myelodysplastic syndrome, and multiple myeloma elicits immunologic and clinical responses. Blood 2008, 111, 1357–1365. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Karandikar, N.J.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Crotty, L.E.; Casazza, J.P.; Kuruppu, J.; Migueles, S.A.; Connors, M.; et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood 2003, 101, 2711–2720. [Google Scholar] [CrossRef]
- Pieren, D.K.J.; Smits, N.A.M.; Postel, R.J.; Kandiah, V.; de Wit, J.; van Beek, J.; van Baarle, D.; Guichelaar, T. Co-Expression of TIGIT and Helios Marks Immunosenescent CD8+ T Cells During Aging. Front. Immunol. 2022, 13, 833531. [Google Scholar] [CrossRef]
- Song, Y.; Wang, B.; Song, R.; Hao, Y.; Wang, D.; Li, Y.; Jiang, Y.; Xu, L.; Ma, Y.; Zheng, H.; et al. T-cell Immunoglobulin and ITIM Domain Contributes to CD8+ T-cell Immunosenescence. Aging Cell 2018, 17, e12716. [Google Scholar] [CrossRef]
- Vallejo, A.N. CD28 extinction in human T cells: Altered functions and the program of T-cell senescence. Immunol. Rev. 2005, 205, 158–169. [Google Scholar] [CrossRef]
- Ye, J.; Ma, C.; Hsueh, E.C.; Eickhoff, C.S.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Tumor-Derived γδ Regulatory T Cells Suppress Innate and Adaptive Immunity through the Induction of Immunosenescence. J. Immunol. 2013, 190, 2403–2414. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Weyand, C.M. Mechanisms underlying T cell ageing. Nat. Rev. Immunol. 2019, 19, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Soto-Heredero, G.; Gómez de Las Heras, M.M.; Escrig-Larena, J.I.; Mittelbrunn, M. Extremely Differentiated T Cell Subsets Contribute to Tissue Deterioration During Aging. Annu. Rev. Immunol. 2023, 41, 181–205. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, A.S. Tipping the balance in CD4+ T cells. Nat. Immunol. 2023, 24, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Larbi, A.; Pawelec, G.; Wong, S.C.; Goldeck, D.; Tai, J.J.Y.; Fulop, T. Impact of age on T cell signaling: A general defect or specific alterations? Ageing Res. Rev. 2011, 10, 370–378. [Google Scholar] [CrossRef]
- Stelmach, P.; Trumpp, A. Leukemic stem cells and therapy resistance in acute myeloid leukemia. Haematologica 2023, 108, 353–366. [Google Scholar] [CrossRef]
- Carter, J.L.; Hege, K.; Yang, J.; Kalpage, H.A.; Su, Y.; Edwards, H.; Hüttemann, M.; Taub, J.W.; Ge, Y. Targeting multiple signaling pathways: The new approach to acute myeloid leukemia therapy. Signal Transduct. Target. Ther. 2020, 5, 288. [Google Scholar] [CrossRef]
- Le Dieu, R.; Taussig, D.C.; Ramsay, A.G.; Mitter, R.; Miraki-Moud, F.; Fatah, R.; Lee, A.M.; Lister, T.A.; Gribben, J.G. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood 2009, 114, 3909–3916. [Google Scholar] [CrossRef]
- Beatty, G.L.; Smith, J.S.; Reshef, R.; Patel, K.P.; Colligon, T.A.; Vance, B.A.; Frey, N.V.; Johnson, F.B.; Porter, D.L.; Vonderheide, R.H. Functional unresponsiveness and replicative senescence of myeloid leukemia antigen-specific CD8+ T cells after allogeneic stem cell transplantation. Clin. Cancer Res. 2009, 15, 4944–4953. [Google Scholar] [CrossRef]
- Tang, L.; Wu, J.; Li, C.-G.; Jiang, H.-W.; Xu, M.; Du, M.; Yin, Z.; Mei, H.; Hu, Y. Characterization of Immune Dysfunction and Identification of Prognostic Immune-Related Risk Factors in Acute Myeloid Leukemia. Clin. Cancer Res. 2020, 26, 1763–1772. [Google Scholar] [CrossRef]
- Han, X.; Zhou, Z.; Fei, L.; Sun, H.; Wang, R.; Chen, Y.; Chen, H.; Wang, J.; Tang, H.; Ge, W.; et al. Construction of a human cell landscape at single-cell level. Nature 2020, 581, 303–309. [Google Scholar] [CrossRef]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.M.; Zhang, N.; Luo, T.; Yang, M.; Shen, W.K.; Tan, Z.L.; Xia, Y.; Zhang, L.; Zhou, X.; Lei, Q.; et al. TCellSI: A novel method for T cell state assessment and its applications in immune environment prediction. Imeta 2024, 3, e231. [Google Scholar] [CrossRef] [PubMed]
- Skelding, K.A.; Barry, D.L.; Theron, D.Z.; Lincz, L.F. Bone Marrow Microenvironment as a Source of New Drug Targets for the Treatment of Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2022, 24, 563. [Google Scholar] [CrossRef] [PubMed]
- Ciciarello, M.; Corradi, G.; Forte, D.; Cavo, M.; Curti, A. Emerging Bone Marrow Microenvironment-Driven Mechanisms of Drug Resistance in Acute Myeloid Leukemia: Tangle or Chance? Cancers 2021, 13, 5319. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wu, Z.; Deng, W.; Xu, R.; Ban, C.; Sun, X.; Zhao, Q. Spatiotemporal evolution of AML immune microenvironment remodeling and RNF149-driven drug resistance through single-cell multidimensional analysis. J. Transl. Med. 2023, 21, 760. [Google Scholar] [CrossRef]
- Kasakovski, D.; Xu, L.; Li, Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018, 11, 91. [Google Scholar] [CrossRef]
- Desdín-Micó, G.; Soto-Heredero, G.; Aranda, J.F.; Oller, J.; Carrasco, E.; Gabandé-Rodríguez, E.; Blanco, E.M.; Alfranca, A.; Cussó, L.; Desco, M.; et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 2020, 368, 1371–1376. [Google Scholar] [CrossRef]
- Reimann, M.; Lee, S.; Schmitt, C.A. Cellular senescence: Neither irreversible nor reversible. J. Exp. Med. 2024, 221, e20232136. [Google Scholar] [CrossRef]
- Harman, D. The free radical theory of aging. Antioxid. Redox Signal 2003, 5, 557–561. [Google Scholar] [CrossRef]
- Cao, J.; Liao, S.; Zeng, F.; Liao, Q.; Luo, G.; Zhou, Y. Effects of altered glycolysis levels on CD8+ T cell activation and function. Cell Death Dis. 2023, 14, 407. [Google Scholar] [CrossRef]
- Lanna, A.; Gomes, D.C.; Muller-Durovic, B.; McDonnell, T.; Escors, D.; Gilroy, D.W.; Lee, J.H.; Karin, M.; Akbar, A.N. A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging. Nat. Immunol. 2017, 18, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, T.; Unno, M.; Yoneda, N.; Motomura, Y.; Mochizuki, M.; Sasaki, T.; Pasparakis, M.; Saito, T. RIPK1 blocks T cell senescence mediated by RIPK3 and caspase-8. Sci. Adv. 2023, 9, eadd6097. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Tan, X.H.; Yang, H.H.; Jin, L.; Hong, J.R.; Zhou, Y.; Huang, X.T. COX-2/sEH Dual Inhibitor Alleviates Hepatocyte Senescence in NAFLD Mice by Restoring Autophagy through Sirt1/PI3K/AKT/mTOR. Int. J. Mol. Sci. 2022, 23, 8267. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood 2007, 109, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Shenghui, Z.; Yixiang, H.; Jianbo, W.; Kang, Y.; Laixi, B.; Yan, Z.; Xi, X. Elevated frequencies of CD4+ CD25+ CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int. J. Cancer 2011, 129, 1373–1381. [Google Scholar] [CrossRef]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481. [Google Scholar] [CrossRef]
- Mathiasen, S.L.; Gall-Mas, L.; Pateras, I.S.; Theodorou, S.D.; Namini, M.R.; Hansen, M.B.; Martin, O.C.; Vadivel, C.K.; Ntostoglou, K.; Butter, D.; et al. Bacterial genotoxins induce T cell senescence. Cell Rep. 2021, 35, 109220. [Google Scholar] [CrossRef]
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. AMPK-TAB1 activated p38 drives human T cell senescence. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, M.; Bai, D.; Qu, Y. Deciphering the impact of TERT/telomerase on immunosenescence and T cell revitalization. Front. Immunol. 2024, 15, 1465006. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, Y.; Hong, Y.; Lin, Z.; Zha, J.; Zhu, Y.; Li, Z.; Wang, C.; Fang, Z.; Zhou, Z.; et al. Lactate acid promotes PD-1+ Tregs accumulation in the bone marrow with high tumor burden of Acute myeloid leukemia. Int. Immunopharmacol. 2024, 130, 111765. [Google Scholar] [CrossRef]
- Xiang, H.; Tao, Y.; Jiang, Z.; Huang, X.; Wang, H.; Cao, W.; Li, J.; Ding, R.; Shen, M.; Feng, R.; et al. Vps33B controls Treg cell suppressive function through inhibiting lysosomal nutrient sensing complex-mediated mTORC1 activation. Cell Rep. 2022, 39, 110943. [Google Scholar] [CrossRef]
- Toffalori, C.; Zito, L.; Gambacorta, V.; Riba, M.; Oliveira, G.; Bucci, G.; Barcella, M.; Spinelli, O.; Greco, R.; Crucitti, L.; et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat. Med. 2019, 25, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.W.; Johmura, Y.; Suzuki, N.; Omori, S.; Migita, T.; Yamaguchi, K.; Hatakeyama, S.; Yamazaki, S.; Shimizu, E.; Imoto, S.; et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature 2022, 611, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch me if you can: How AML and its niche escape immunotherapy. Leukemia 2022, 36, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Corradi, G.; Bassani, B.; Simonetti, G.; Sangaletti, S.; Vadakekolathu, J.; Fontana, M.C.; Pazzaglia, M.; Gulino, A.; Tripodo, C.; Cristiano, G.; et al. Release of IFNγ by Acute Myeloid Leukemia Cells Remodels Bone Marrow Immune Microenvironment by Inducing Regulatory T Cells. Clin. Cancer Res. 2022, 28, 3141. [Google Scholar] [CrossRef] [PubMed]
- Bopp, T.; Becker, C.; Klein, M.; Klein-Heßling, S.; Palmetshofer, A.; Serfling, E.; Heib, V.; Becker, M.; Kubach, J.; Schmitt, S.; et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J. Exp. Med. 2007, 204, 1303–1310. [Google Scholar] [CrossRef]
- Gao, A.; Liu, X.; Lin, W.; Wang, J.; Wang, S.; Si, F.; Huang, L.; Zhao, Y.; Sun, Y.; Peng, G. Tumor-derived ILT4 induces T cell senescence and suppresses tumor immunity. J. Immunother. Cancer 2021, 9, e001536. [Google Scholar] [CrossRef]
- Chen, Y.; Wen, J.; Li, Q.; Peng, D.; Liao, C.; Ma, X.; Wang, M.; Niu, J.; Wang, D.; Li, Y.; et al. RAB27B-regulated exosomes mediate LSC maintenance via resistance to senescence and crosstalk with the microenvironment. Leukemia 2024, 38, 266–280. [Google Scholar] [CrossRef]
- Zhao, Y.; Shao, Q.; Peng, G. Exhaustion and senescence: Two crucial dysfunctional states of T cells in the tumor microenvironment. Cell Mol. Immunol. 2020, 17, 27–35. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Akbar, A.N.; Henson, S.M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 2011, 11, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qin, Y.; Li, B. CD8+ T cell exhaustion and cancer immunotherapy. Cancer Lett. 2023, 559, 216043. [Google Scholar] [CrossRef]
- Giles, J.R.; Manne, S.; Freilich, E.; Oldridge, D.A.; Baxter, A.E.; George, S.; Chen, Z.; Huang, H.; Chilukuri, L.; Carberry, M.; et al. Human epigenetic and transcriptional T cell differentiation atlas for identifying functional T cell-specific enhancers. Immunity 2022, 55, 557–574.e7. [Google Scholar] [CrossRef]
- Zhang, J.; He, T.; Xue, L.; Guo, H. Senescent T cells: A potential biomarker and target for cancer therapy. EBioMedicine 2021, 68, 103409. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Huang, M.C.; Kon, J.; Patel, K.; Schwartz, J.B.; Fast, K.; Ferrucci, L.; Madara, K.; Taub, D.D.; Longo, D.L. Gender specificity of altered human immune cytokine profiles in aging. FASEB J. 2010, 24, 3580–3589. [Google Scholar] [CrossRef]
- Pereira, B.I.; De Maeyer, R.P.H.; Covre, L.P.; Nehar-Belaid, D.; Lanna, A.; Ward, S.; Marches, R.; Chambers, E.S.; Gomes, D.C.O.; Riddell, N.E.; et al. Sestrins induce natural killer function in senescent-like CD8+ T cells. Nat. Immunol. 2020, 21, 684–694. [Google Scholar] [CrossRef]
- Kakuda, T.; Suzuki, J.; Matsuoka, Y.; Kikugawa, T.; Saika, T.; Yamashita, M. Senescent CD8+ T cells acquire NK cell-like innate functions to promote antitumor immunity. Cancer Sci. 2023, 114, 2810–2820. [Google Scholar] [CrossRef]
- Henson, S.M.; Lanna, A.; Riddell, N.E.; Franzese, O.; Macaulay, R.; Griffiths, S.J.; Puleston, D.J.; Watson, A.S.; Simon, A.K.; Tooze, S.A.; et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J. Clin. Investig. 2014, 124, 4004–4016. [Google Scholar] [CrossRef]
- Henson, S.M.; Macaulay, R.; Riddell, N.E.; Nunn, C.J.; Akbar, A.N. Blockade of PD-1 or p38 MAP kinase signaling enhances senescent human CD8(+) T-cell proliferation by distinct pathways. Eur. J. Immunol. 2015, 45, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Si, F.; Bagley, D.; Ma, F.; Zhang, Y.; Tao, Y.; Shaw, E.; Peng, G. Blockades of effector T cell senescence and exhaustion synergistically enhance antitumor immunity and immunotherapy. J. Immunother. Cancer 2022, 10, e005020. [Google Scholar] [CrossRef] [PubMed]
- Battram, A.M.; Bachiller, M.; Martín-Antonio, B. Senescence in the Development and Response to Cancer with Immunotherapy: A Double-Edged Sword. Int. J. Mol. Sci. 2020, 21, 4346. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Sosman, J.A. Update on the targeted therapy of melanoma. Curr. Treat. Options Oncol. 2013, 14, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Lenaers, G.; Bonneau, D.; Delneste, Y.; Papon, N. Dysfunctional T Cell Mitochondria Lead to Premature Aging. Trends Mol. Med. 2020, 26, 799–800. [Google Scholar] [CrossRef]
- Liu, X.; Li, L.; Peng, G. TLR8 reprograms human Treg metabolism and function. Aging 2019, 11, 6614–6615. [Google Scholar] [CrossRef]
- Li, L.; Liu, X.; Sanders, K.L.; Edwards, J.L.; Ye, J.; Si, F.; Gao, A.; Huang, L.; Hsueh, E.C.; Ford, D.A.; et al. TLR8-Mediated Metabolic Control of Human Treg Function: A Mechanistic Target for Cancer Immunotherapy. Cell Metab. 2019, 29, 103–123.e5. [Google Scholar] [CrossRef]
- Wu, M.; Fu, X.; Xu, R.; Liu, S.; Li, R.; Xu, J.; Shang, W.; Chen, X.; Wang, T.; Wang, F. Glucose metabolism and function of CD4+ Tregs are regulated by the TLR8/mTOR signal in an environment of SKOV3 cell growth. Cancer Med. 2023, 12, 16310–16322. [Google Scholar] [CrossRef]
- Yang, W.; Sun, X.; Liu, S.; Xu, Y.; Li, Y.; Huang, X.; Liu, K.; Mao, L.; Min, S.; Liu, L.; et al. TLR8 agonist Motolimod-induced inflammatory death for treatment of acute myeloid leukemia. Biomed. Pharmacother. 2023, 163, 114759. [Google Scholar] [CrossRef]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, H.; Li, H.; Zhao, S.; Zeng, Y.; Zhang, P.; Lin, X.; Sun, X.; Wang, L.; Fu, G.; et al. Development of a novel TLR8 agonist for cancer immunotherapy. Mol. Biomed. 2020, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.-F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Yue, Y.; Yu, W.; Zhang, Y. Immunosenescence: A key player in cancer development. J. Hematol. Oncol. 2020, 13, 151. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55.e6. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, X.; Liu, N.; Ma, S.; Zhang, H.; Zhang, Z.; Yang, K.; Jiang, M.; Zheng, Z.; Qiao, Y.; et al. Metformin decelerates aging clock in male monkeys. Cell 2024, 187, 6358–6378. [Google Scholar] [CrossRef]
- Liu, X.; Hartman, C.L.; Li, L.; Albert, C.J.; Si, F.; Gao, A.; Huang, L.; Zhao, Y.; Lin, W.; Hsueh, E.C.; et al. Reprogramming lipid metabolism prevents effector T cell senescence and enhances tumor immunotherapy. Sci. Transl. Med. 2021, 13, eaaz6314. [Google Scholar] [CrossRef]
- Ho, W.Y.; Blattman, J.N.; Dossett, M.L.; Yee, C.; Greenberg, P.D. Adoptive immunotherapy: Engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell 2003, 3, 431–437. [Google Scholar] [CrossRef]
- Huang, Q.; Zhang, X.H.; Liu, D. CAR T cell therapy for T cell leukemia and lymphoma: Latest updates from 2022 ASH Annual Meeting. J. Hematol. Oncol. 2023, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, S.; Yazdanpanah, N.; Rezaei, N. Natural killer cells and acute myeloid leukemia: Promises and challenges. Cancer Immunol. Immunother. 2022, 71, 2849–2867. [Google Scholar] [CrossRef] [PubMed]
- Krakow, E.F.; Brault, M.; Summers, C.; Cunningham, T.M.; A Biernacki, M.; Black, R.G.; Woodward, K.B.; Vartanian, N.; Kanaan, S.B.; Yeh, A.C.; et al. HA-1-targeted T-cell receptor T-cell therapy for recurrent leukemia after hematopoietic stem cell transplantation. Blood 2024, 144, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- Noll, J.H.; Levine, B.L.; June, C.H.; Fraietta, J.A. Beyond youth: Understanding CAR T cell fitness in the context of immunological aging. Semin. Immunol. 2023, 70, 101840. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.; Putavet, D.A.; Klein, J.D.; Derks, K.W.; Bourgeois, B.R.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef]
- Yoshida, S.; Nakagami, H.; Hayashi, H.; Ikeda, Y.; Sun, J.; Tenma, A.; Tomioka, H.; Kawano, T.; Shimamura, M.; Morishita, R.; et al. The CD153 vaccine is a senotherapeutic option for preventing the accumulation of senescent T cells in mice. Nat. Commun. 2020, 11, 2482. [Google Scholar] [CrossRef]
- Deng, E.Z.; Fleishman, R.H.; Xie, Z.; Marino, G.B.; Clarke, D.J.B.; Ma’ayan, A. Computational screen to identify potential targets for immunotherapeutic identification and removal of senescence cells. Aging Cell 2023, 22, e13809. [Google Scholar] [CrossRef]
- Ureña-Bailén, G.; Dobrowolski, J.-M.; Hou, Y.; Dirlam, A.; Roig-Merino, A.; Schleicher, S.; Atar, D.; Seitz, C.; Feucht, J.; Antony, J.S.; et al. Preclinical Evaluation of CRISPR-Edited CAR-NK-92 Cells for Off-the-Shelf Treatment of AML and B-ALL. Int. J. Mol. Sci. 2022, 23, 12828. [Google Scholar] [CrossRef]
- Kawai, Y.; Kawana-Tachikawa, A.; Kitayama, S.; Ueda, T.; Miki, S.; Watanabe, A.; Kaneko, S. Generation of highly proliferative, rejuvenated cytotoxic T cell clones through pluripotency reprogramming for adoptive immunotherapy. Mol. Ther. 2021, 29, 3027–3041. [Google Scholar] [CrossRef]
- Weng, J.; Moriarty, K.E.; Baio, F.E.; Chu, F.; Kim, S.D.; He, J.; Jie, Z.; Xie, X.; Ma, W.; Qian, J.; et al. IL-15 enhances the antitumor effect of human antigen-specific CD8+ T cells by cellular senescence delay. OncoImmunology 2016, 5, e1237327. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; O Butler, M.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Marton, C.; Minaud, A.; Coupet, C.A.; Chauvin, M.; Dhiab, J.; Vallet, H.; Boddaert, J.; Kehrer, N.; Bastien, B.; Inchauspe, G.; et al. IL-7 producing immunotherapy improves ex vivo T cell functions of immunosenescent patients, especially post hip fracture. Hum. Vaccin. Immunother. 2023, 19, 2232247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pfannenstiel, L.W.; Bolesta, E.; Montes, C.L.; Zhang, X.; Chapoval, A.I.; Gartenhaus, R.B.; Strome, S.E.; Gastman, B.R. Interleukin-7 inhibits tumor-induced CD27-CD28-suppressor T cells: Implications for cancer immunotherapy. Clin. Cancer Res. 2011, 17, 4975–4986. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Yao, J.; Wu, D.; Huang, X.; Wang, Y.; Li, X.; Lu, Q.; Qiu, Y. Type 2 cytokine signaling in macrophages protects from cellular senescence and organismal aging. Immunity 2024, 57, 513–527.e6. [Google Scholar] [CrossRef]
- Tang, X.; Deng, B.; Zang, A.; He, X.; Zhou, Y.; Wang, D.; Li, D.; Dai, X.; Chen, J.; Zhang, X.; et al. Characterization of age-related immune features after autologous NK cell infusion: Protocol for an open-label and randomized controlled trial. Front. Immunol. 2022, 13, 940577. [Google Scholar] [CrossRef]
- Chelyapov, N.; Nguyen, T.T.; Gonzalez, R. Autologous NK cells propagated and activated ex vivo decrease senescence markers in human PBMCs. Biochem. Biophys. Rep. 2022, 32, 101380. [Google Scholar] [CrossRef]
- Park, M.D.; Le Berichel, J.; Hamon, P.; Wilk, M.; Belabed, M.; Yatim, N.; Saffon, A.; Boumelha, J.; Falcomata, C.; Tepper, A.; et al. Hematopoietic aging promotes cancer by fueling IL-1⍺-driven emergency myelopoiesis. Science 2024, 386, eadn0327. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Wu, J.; Xiao, Y.; Sun, J.; Sun, H.; Chen, H.; Zhu, Y.; Fu, H.; Yu, C.; E., W.; Lai, S.; et al. A single-cell survey of cellular hierarchy in acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 128. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Liu, L. Senescent T Cells: The Silent Culprit in Acute Myeloid Leukemia Progression? Int. J. Mol. Sci. 2024, 25, 12550. https://doi.org/10.3390/ijms252312550
Zhang X, Liu L. Senescent T Cells: The Silent Culprit in Acute Myeloid Leukemia Progression? International Journal of Molecular Sciences. 2024; 25(23):12550. https://doi.org/10.3390/ijms252312550
Chicago/Turabian StyleZhang, Xiaolan, and Lingbo Liu. 2024. "Senescent T Cells: The Silent Culprit in Acute Myeloid Leukemia Progression?" International Journal of Molecular Sciences 25, no. 23: 12550. https://doi.org/10.3390/ijms252312550
APA StyleZhang, X., & Liu, L. (2024). Senescent T Cells: The Silent Culprit in Acute Myeloid Leukemia Progression? International Journal of Molecular Sciences, 25(23), 12550. https://doi.org/10.3390/ijms252312550