
Potentiation of NMDA Receptors by AT1 Angiotensin Receptor Activation in Layer V Pyramidal Neurons of the Rat Prefrontal Cortex †

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. NMDA Induced Inward Currents in Layer V Pyramidal Neurons of the Rat PFC, Which Were Reproducible at Lower Concentrations
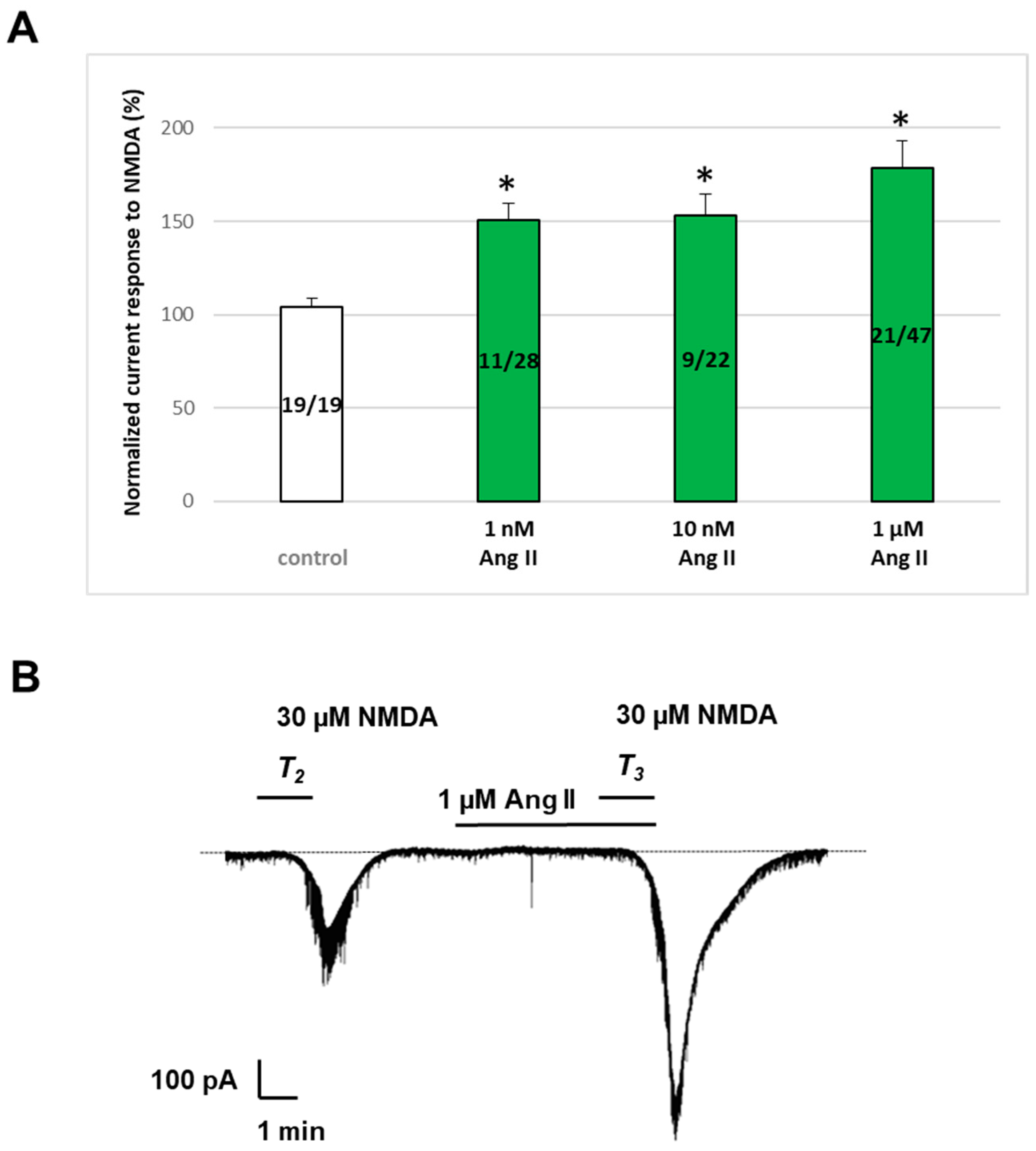
2.2. Ang II Potentiated NMDA-Induced Inward Currents in Layer V Pyramidal Neurons of the Rat PFC
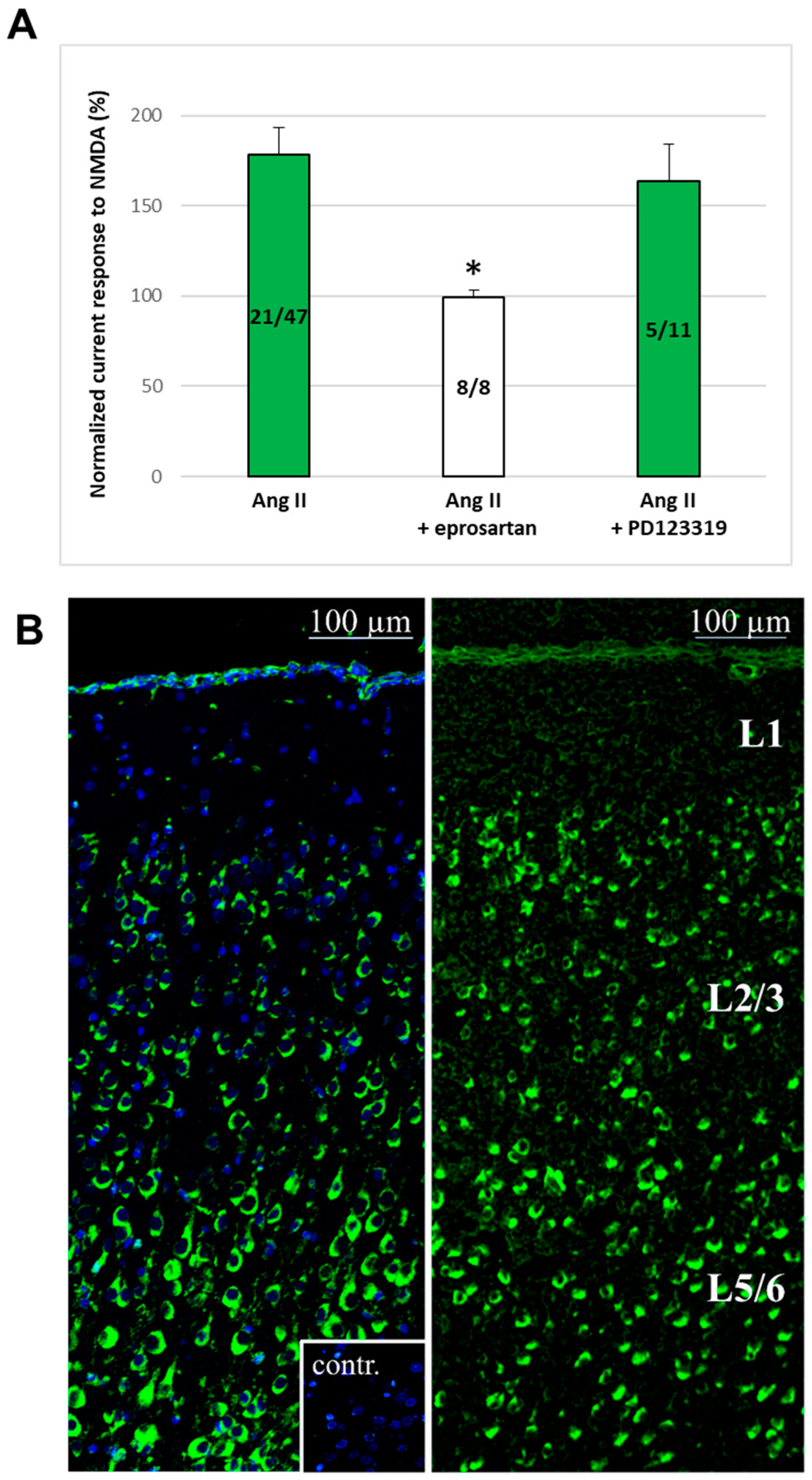
2.3. AT1 Receptors for Ang II Are Expressed in the Rat PFC, and the Ang II-Induced Potentiation Was Mediated by These Receptors
2.4. Synaptic Isolation of Pyramidal Neurons, as Well as Simultaneous D1 Antagonism, Abolished the Enhancement of NMDA Currents by Ang II
3. Discussion
4. Materials and Methods
4.1. Brain Slice Preparation
4.2. Tight Seal Whole-Cell Recording
4.3. Application of Drugs
4.4. Immunohistochemistry
4.5. In Situ Hybridization
4.6. Materials
4.7. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goldman-Rakic, P.S. Cellular basis of working memory. Neuron 1995, 14, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Kolb, B. Functions of the frontal cortex of the rat: A comparative review. Brain Res. 1984, 320, 65–98. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.M. The prefrontal cortex, mediator of cross-temporal contingencies. Hum. Neurobiol. 1985, 4, 169–179. [Google Scholar] [PubMed]
- Berger, B.; Thierry, A.M.; Tassin, J.P.; Moyne, M.A. Dopaminergic innervation of the rat prefrontal cortex: A fluorescence histochemical study. Brain Res. 1976, 106, 133–145. [Google Scholar] [CrossRef]
- Conde, F.; Audinat, E.; Maire-Lepoivre, E.; Crepel, F. Afferent connections of the medial frontal cortex of the rat. A study using retrograde transport of fluorescent dyes. I. Thalamic afferents. Brain Res. Bull. 1990, 24, 341–354. [Google Scholar] [CrossRef]
- Conde, F.; Maire-Lepoivre, E.; Audinat, E.; Crepel, F. Afferent connections of the medial frontal cortex of the rat. II. Cortical and subcortical afferents. J. Comp. Neurol. 1995, 352, 567–593. [Google Scholar] [CrossRef]
- Divac, I.; Bjorklund, A.; Lindvall, O.; Passingham, R.E. Converging projections from the mediodorsal thalamic nucleus and mesencephalic dopaminergic neurons to the neocortex in three species. J. Comp. Neurol. 1978, 180, 59–71. [Google Scholar] [CrossRef]
- Lewis, D.A.; Hayes, T.L.; Lund, J.S.; Oeth, K.M. Dopamine and the neural circuitry of primate prefrontal cortex: Implications for schizophrenia research. Neuropsychopharmacology 1992, 6, 127–134. [Google Scholar]
- Flynn, L.T.; Bouras, N.N.; Migovich, V.M.; Clarin, J.D.; Gao, W.J. The “psychiatric” neuron: The psychic neuron of the cerebral cortex, revisited. Front. Hum. Neurosci. 2024, 18, 1356674. [Google Scholar] [CrossRef]
- Goldman-Rakic, P.S. Working memory dysfunction in schizophrenia. J. Neuropsychiatry Clin. Neurosci. 1994, 6, 348–357. [Google Scholar] [CrossRef]
- Goldman-Rakic, P.S.; Castner, S.A.; Svensson, T.H.; Siever, L.J.; Williams, G.V. Targeting the dopamine D1 receptor in schizophrenia: Insights for cognitive dysfunction. Psychopharmacology 2004, 174, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Murakami, K.; Igarashi, H.; Okada, A. The convergence of axon terminals from the mediodorsal thalamic nucleus and ventral tegmental area on pyramidal cells in layer V of the rat prelimbic cortex. Eur. J. Neurosci. 1996, 8, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Sesack, S.R.; Pickel, V.M. Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J. Comp. Neurol. 1992, 320, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.R.; Seamans, J.K.; Gorelova, N. Electrophysiological and morphological properties of layers V-VI principal pyramidal cells in rat prefrontal cortex in vitro. J. Neurosci. 1996, 16, 1904–1921. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.; Deutch, A.Y. Dopaminergic mechanisms in the pathogenesis of schizophrenia. FASEB J. 1992, 6, 2413–2421. [Google Scholar] [CrossRef]
- Grossberg, S. The imbalanced brain: From normal behavior to schizophrenia. Biol. Psychiatry 2000, 48, 81–98. [Google Scholar] [CrossRef]
- Verma, A.; Moghaddam, B. NMDA receptor antagonists impair prefrontal cortex function as assessed via spatial delayed alternation performance in rats: Modulation by dopamine. J. Neurosci. 1996, 16, 373–379. [Google Scholar] [CrossRef]
- Bader, M. Tissue renin-angiotensin-aldosterone systems: Targets for pharmacological therapy. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 439–465. [Google Scholar] [CrossRef]
- Wright, J.W.; Harding, J.W. Brain angiotensin receptor subtypes in the control of physiological and behavioral responses. Neurosci. Biobehav. Rev. 1994, 18, 21–53. [Google Scholar] [CrossRef]
- Saavedra, J.M. Emerging features of brain angiotensin receptors. Regul. Pept. 1999, 85, 31–45. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Eroli, F.; Garcia-Ptacek, S.; Maioli, S. Brain Renin-Angiotensin System as Novel and Potential Therapeutic Target for Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 139. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.W.; Harding, J.W. Contributions by the Brain Renin-Angiotensin System to Memory, Cognition, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 67, 469–480. [Google Scholar] [CrossRef] [PubMed]
- von Bohlen und Halbach, O.; Albrecht, D. The CNS renin-angiotensin system. Cell Tissue Res. 2006, 326, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, J.M. Brain angiotensin II: New developments, unanswered questions and therapeutic opportunities. Cell. Mol. Neurobiol. 2005, 25, 485–512. [Google Scholar] [CrossRef] [PubMed]
- Moffett, R.B.; Bumpus, F.M.; Husain, A. Cellular organization of the brain renin-angiotensin system. Life Sci. 1987, 41, 1867–1879. [Google Scholar] [CrossRef]
- Sernia, C.; Zeng, T.; Kerr, D.; Wyse, B. Novel perspectives on pituitary and brain angiotensinogen. Front. Neuroendocrinol. 1997, 18, 174–208. [Google Scholar] [CrossRef]
- Phillips, M.I.; Sumners, C. Angiotensin II in central nervous system physiology. Regul. Pept. 1998, 78, 1–11. [Google Scholar] [CrossRef]
- Stornetta, R.L.; Hawelu-Johnson, C.L.; Guyenet, P.G.; Lynch, K.R. Astrocytes synthesize angiotensinogen in brain. Science 1988, 242, 1444–1446. [Google Scholar] [CrossRef]
- Milsted, A.; Barna, B.P.; Ransohoff, R.M.; Brosnihan, K.B.; Ferrario, C.M. Astrocyte cultures derived from human brain tissue express angiotensinogen mRNA. Proc. Natl. Acad. Sci. USA 1990, 87, 5720–5723. [Google Scholar] [CrossRef]
- Hilgenfeldt, U. Angiotensinogen in rat cerebrospinal fluid. Clin. Exp. Hypertens. A 1984, 6, 1815–1824. [Google Scholar] [CrossRef]
- Yang, G.; Gray, T.S.; Sigmund, C.D.; Cassell, M.D. The angiotensinogen gene is expressed in both astrocytes and neurons in murine central nervous system. Brain Res. 1999, 817, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J.; Ingelfinger, J.; Pratt, R.E.; Ellison, K.E. Identification of renin and angiotensinogen messenger RNA sequences in mouse and rat brains. Hypertension 1986, 8, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Cassell, M.D.; Sigmund, C.D. The brain renin-angiotensin system in transgenic mice carrying a highly regulated human renin transgene. Circ. Res. 2002, 90, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Cuadra, A.E.; Shan, Z.; Sumners, C.; Raizada, M.K. A current view of brain renin-angiotensin system: Is the (pro)renin receptor the missing link? Pharmacol. Ther. 2010, 125, 27–38. [Google Scholar] [CrossRef] [PubMed]
- de Kloet, A.D.; Liu, M.; Rodriguez, V.; Krause, E.G.; Sumners, C. Role of neurons and glia in the CNS actions of the renin-angiotensin system in cardiovascular control. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R444–R458. [Google Scholar] [CrossRef]
- Chai, S.Y.; Mendelsohn, F.A.; Paxinos, G. Angiotensin converting enzyme in rat brain visualized by quantitative in vitro autoradiography. Neuroscience 1987, 20, 615–627. [Google Scholar] [CrossRef]
- de Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar]
- Horiuchi, M.; Lehtonen, J.Y.; Daviet, L. Signaling Mechanism of the AT2 Angiotensin II Receptor: Crosstalk between AT1 and AT2 Receptors in Cell Growth. Trends Endocrinol. Metab. 1999, 10, 391–396. [Google Scholar] [CrossRef]
- Kang, J.; Richards, E.M.; Posner, P.; Sumners, C. Modulation of the delayed rectifier K+ current in neurons by an angiotensin II type 2 receptor fragment. Am. J. Physiol. 1995, 268 Pt 1, C278–C282. [Google Scholar] [CrossRef]
- Turu, G.; Varnai, P.; Gyombolai, P.; Szidonya, L.; Offertaler, L.; Bagdy, G.; Kunos, G.; Hunyady, L. Paracrine transactivation of the CB1 cannabinoid receptor by AT1 angiotensin and other Gq/11 protein-coupled receptors. J. Biol. Chem. 2009, 284, 16914–16921. [Google Scholar] [CrossRef]
- Rowe, B.P.; Saylor, D.L.; Speth, R.C. Analysis of angiotensin II receptor subtypes in individual rat brain nuclei. Neuroendocrinology 1992, 55, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Allen, A.M.; Paxinos, G.; Mendelsohn, F.A. Mapping of angiotensin II receptor subtype heterogeneity in rat brain. J. Comp. Neurol. 1992, 316, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, K.; Saavedra, J.M. Characterization and development of angiotensin II receptor subtypes (AT1 and AT2) in rat brain. Am. J. Physiol. 1991, 261 Pt 2, R209–R216. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, K.; Saavedra, J.M. Quantitative autoradiography reveals different angiotensin II receptor subtypes in selected rat brain nuclei. J. Neurochem. 1991, 56, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Obermuller, N.; Unger, T.; Culman, J.; Gohlke, P.; de Gasparo, M.; Bottari, S.P. Distribution of angiotensin II receptor subtypes in rat brain nuclei. Neurosci. Lett. 1991, 132, 11–15. [Google Scholar] [CrossRef]
- Lenkei, Z.; Palkovits, M.; Corvol, P.; Llorens-Cortes, C. Distribution of angiotensin II type-2 receptor (AT2) mRNA expression in the adult rat brain. J. Comp. Neurol. 1996, 373, 322–339. [Google Scholar] [CrossRef]
- Wright, J.W.; Harding, J.W. Brain angiotensin receptor subtypes AT1, AT2, and AT4 and their functions. Regul. Pept 1995, 59, 269–295. [Google Scholar] [CrossRef]
- Chai, S.Y.; Bastias, M.A.; Clune, E.F.; Matsacos, D.J.; Mustafa, T.; Lee, J.H.; McDowall, S.G.; Paxinos, G.; Mendelsohn, F.A.; Albiston, A.L. Distribution of angiotensin IV binding sites (AT4 receptor) in the human forebrain, midbrain and pons as visualised by in vitro receptor autoradiography. J. Chem. Neuroanat. 2000, 20, 339–348. [Google Scholar] [CrossRef]
- Vanderheyden, P.M. From angiotensin IV binding site to AT4 receptor. Mol. Cell. Endocrinol. 2009, 302, 159–166. [Google Scholar] [CrossRef]
- Becker, L.K.; Etelvino, G.M.; Walther, T.; Santos, R.A.; Campagnole-Santos, M.J. Immunofluorescence localization of the receptor Mas in cardiovascular-related areas of the rat brain. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1416–H1424. [Google Scholar] [CrossRef]
- Santos, R.A.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, M.; Heinemann, S. Cloned glutamate receptors. Annu. Rev. Neurosci. 1994, 17, 31–108. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, S.; Sucher, N.J.; Bradley, D.; Tafazzoli, A.; Trinh, D.; Hetrick, W.P.; Potkin, S.G.; Sandman, C.A.; Bunney, W.E., Jr.; Jones, E.G. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J. Neurosci. 1996, 16, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Scherzer, C.R.; Landwehrmeyer, G.B.; Kerner, J.A.; Counihan, T.J.; Kosinski, C.M.; Standaert, D.G.; Daggett, L.P.; Velicelebi, G.; Penney, J.B.; Young, A.B. Expression of N-methyl-D-aspartate receptor subunit mRNAs in the human brain: Hippocampus and cortex. J. Comp. Neurol. 1998, 390, 75–90. [Google Scholar] [CrossRef]
- Koles, L.; Wirkner, K.; Illes, P. Modulation of ionotropic glutamate receptor channels. Neurochem. Res. 2001, 26, 925–932. [Google Scholar] [CrossRef]
- Paoletti, P.; Neyton, J. NMDA receptor subunits: Function and pharmacology. Curr. Opin. Pharmacol. 2007, 7, 39–47. [Google Scholar] [CrossRef]
- Nakanishi, S. Molecular diversity of glutamate receptors and implications for brain function. Science 1992, 258, 597–603. [Google Scholar] [CrossRef]
- Gebre, A.K.; Altaye, B.M.; Atey, T.M.; Tuem, K.B.; Berhe, D.F. Targeting Renin-Angiotensin System Against Alzheimer’s Disease. Front. Pharmacol. 2018, 9, 440. [Google Scholar] [CrossRef]
- Saavedra, J.M. Beneficial effects of Angiotensin II receptor blockers in brain disorders. Pharmacol. Res. 2017, 125 Pt A, 91–103. [Google Scholar] [CrossRef]
- Braszko, J.J. Dopamine D4 receptor antagonist L745,870 abolishes cognitive effects of intracerebroventricular angiotensin IV and des-Phe(6)-Ang IV in rats. Eur. Neuropsychopharmacol. 2009, 19, 85–91. [Google Scholar] [CrossRef]
- Gelband, C.H.; Sumners, C.; Lu, D.; Raizada, M.K. Angiotensin receptors and norepinephrine neuromodulation: Implications of functional coupling. Regul. Pept. 1997, 72, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Kobiec, T.; Otero-Losada, M.; Chevalier, G.; Udovin, L.; Bordet, S.; Menendez-Maissonave, C.; Capani, F.; Perez-Lloret, S. The Renin-Angiotensin System Modulates Dopaminergic Neurotransmission: A New Player on the Scene. Front. Synaptic. Neurosci. 2021, 13, 638519. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.L.; Munn, C.; Ross, R.C.; Harding, J.W.; Wright, J.W. The role of the AT4 and cholinergic systems in the Nucleus Basalis Magnocellularis (NBM): Effects on spatial memory. Brain Res. 2009, 1272, 25–31. [Google Scholar] [CrossRef] [PubMed]
- de Miranda, A.S.; Macedo, D.S.; Rocha, N.P.; Teixeira, A.L. Targeting the Renin-Angiotensin System (RAS) for Neuropsychiatric Disorders. Curr. Neuropharmacol. 2024, 22, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Labandeira-Garcia, J.L.; Labandeira, C.M.; Guerra, M.J.; Rodriguez-Perez, A.I. The role of the brain renin-angiotensin system in Parkinson s disease. Transl. Neurodegener. 2024, 13, 22. [Google Scholar] [CrossRef]
- Bondy, B.; Baghai, T.C.; Zill, P.; Schule, C.; Eser, D.; Deiml, T.; Zwanzger, P.; Ella, R.; Rupprecht, R. Genetic variants in the angiotensin I-converting-enzyme (ACE) and angiotensin II receptor (AT1) gene and clinical outcome in depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2005, 29, 1094–1099. [Google Scholar] [CrossRef]
- Wright, J.W.; Harding, J.W. The brain angiotensin system and extracellular matrix molecules in neural plasticity, learning, and memory. Prog. Neurobiol. 2004, 72, 263–293. [Google Scholar] [CrossRef]
- Schelman, W.R.; Andres, R.; Ferguson, P.; Orr, B.; Kang, E.; Weyhenmeyer, J.A. Angiotensin II attenuates NMDA receptor-mediated neuronal cell death and prevents the associated reduction in Bcl-2 expression. Brain Res. Mol. Brain Res. 2004, 128, 20–29. [Google Scholar] [CrossRef]
- Schelman, W.R.; Kurth, J.L.; Berdeaux, R.L.; Norby, S.W.; Weyhenmeyer, J.A. Angiotensin II type-2 (AT2) receptor-mediated inhibition of NMDA receptor signalling in neuronal cells. Brain Res. Mol. Brain Res. 1997, 48, 197–205. [Google Scholar] [CrossRef]
- Davis, C.J.; Kramar, E.A.; De, A.; Meighan, P.C.; Simasko, S.M.; Wright, J.W.; Harding, J.W. AT4 receptor activation increases intracellular calcium influx and induces a non-N-methyl-D-aspartate dependent form of long-term potentiation. Neuroscience 2006, 137, 1369–1379. [Google Scholar] [CrossRef]
- Hellner, K.; Walther, T.; Schubert, M.; Albrecht, D. Angiotensin-(1–7) enhances LTP in the hippocampus through the G-protein-coupled receptor Mas. Mol. Cell. Neurosci. 2005, 29, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Tchekalarova, J.; Albrecht, D. Angiotensin II suppresses long-term depression in the lateral amygdala of mice via L-type calcium channels. Neurosci. Lett. 2007, 415, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Marshall, K.C. Angiotensin II depresses glutamate depolarizations and excitatory postsynaptic potentials in locus coeruleus through angiotensin II subtype 2 receptors. Neuroscience 1994, 62, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.G.; Marshall, K.C. Angiotensin II modulation of glutamate excitation of locus coeruleus neurons. Neurosci. Lett. 1990, 118, 261–264. [Google Scholar] [CrossRef]
- Wirkner, K.; Koles, L.; Thummler, S.; Luthardt, J.; Poelchen, W.; Franke, H.; Furst, S.; Illes, P. Interaction between P2Y and NMDA receptors in layer V pyramidal neurons of the rat prefrontal cortex. Neuropharmacology 2002, 42, 476–488. [Google Scholar] [CrossRef]
- Wirkner, K.; Gunther, A.; Weber, M.; Guzman, S.J.; Krause, T.; Fuchs, J.; Koles, L.; Norenberg, W.; Illes, P. Modulation of NMDA receptor current in layer V pyramidal neurons of the rat prefrontal cortex by P2Y receptor activation. Cereb. Cortex 2007, 17, 621–631. [Google Scholar] [CrossRef]
- Koles, L.; Kato, E.; Hanuska, A.; Zadori, Z.S.; Al-Khrasani, M.; Zelles, T.; Rubini, P.; Illes, P. Modulation of excitatory neurotransmission by neuronal/glial signalling molecules: Interplay between purinergic and glutamatergic systems. Purinergic. Signal. 2016, 12, 1–24. [Google Scholar] [CrossRef]
- Krugel, U.; Koles, L.; Illes, P. Integration of neuronal and glial signalling by pyramidal cells of the rat prefrontal cortex; control of cognitive functions and addictive behaviour by purinergic mechanisms. Neuropsychopharmacol. Hung 2013, 15, 206–213. [Google Scholar]
- Wirkner, K.; Krause, T.; Koles, L.; Thummler, S.; Al-Khrasani, M.; Illes, P. D1 but not D2 dopamine receptors or adrenoceptors mediate dopamine-induced potentiation of N-methyl-d-aspartate currents in the rat prefrontal cortex. Neurosci. Lett. 2004, 372, 89–93. [Google Scholar] [CrossRef]
- Oliveira, J.F.; Krugel, U.; Koles, L.; Illes, P.; Wirkner, K. Blockade of glutamate transporters leads to potentiation of NMDA receptor current in layer V pyramidal neurons of the rat prefrontal cortex via group II metabotropic glutamate receptor activation. Neuropharmacology 2008, 55, 447–453. [Google Scholar] [CrossRef]
- Luthardt, J.; Borvendeg, S.J.; Sperlagh, B.; Poelchen, W.; Wirkner, K.; Illes, P. P2Y(1) receptor activation inhibits NMDA receptor-channels in layer V pyramidal neurons of the rat prefrontal and parietal cortex. Neurochem. Int. 2003, 42, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Goldman-Rakic, P.S.; Muly, E.C., 3rd; Williams, G.V. D(1) receptors in prefrontal cells and circuits. Brain Res. Brain Res. Rev. 2000, 31, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Colwell, C.S.; Levine, M.S. Excitatory synaptic transmission in neostriatal neurons: Regulation by cyclic AMP-dependent mechanisms. J. Neurosci. 1995, 15 Pt 1, 1704–1713. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, C.; Andre, V.M.; Jocoy, E.L.; Levine, M.S. NMDA and Dopamine: Diverse Mechanisms Applied to Interacting Receptor Systems. In Biology of the NMDA Receptor; Van Dongen, A.M., Ed.; Frontiers in Neuroscience; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009. [Google Scholar]
- Cepeda, C.; Levine, M.S. Dopamine-NMDA receptor interactions: Twenty years later. Dev. Neurosci. 2012, 34, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, C.; Radisavljevic, Z.; Peacock, W.; Levine, M.S.; Buchwald, N.A. Differential modulation by dopamine of responses evoked by excitatory amino acids in human cortex. Synapse 1992, 11, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Lidow, M.S.; Wang, F.; Cao, Y.; Goldman-Rakic, P.S. Layer V neurons bear the majority of mRNAs encoding the five distinct dopamine receptor subtypes in the primate prefrontal cortex. Synapse 1998, 28, 10–20. [Google Scholar] [CrossRef]
- Seeman, P. Cloned dopamine receptors: Targets in therapy of drug abuse. NIDA Res. Monogr. 1992, 126, 34–47. [Google Scholar]
- Brown, D.C.; Steward, L.J.; Ge, J.; Barnes, N.M. Ability of angiotensin II to modulate striatal dopamine release via the AT1 receptor in vitro and in vivo. Br. J. Pharmacol. 1996, 118, 414–420. [Google Scholar] [CrossRef]
- Mendelsohn, F.A.; Jenkins, T.A.; Berkovic, S.F. Effects of angiotensin II on dopamine and serotonin turnover in the striatum of conscious rats. Brain Res. 1993, 613, 221–229. [Google Scholar] [CrossRef]
- Kuzhikandathil, E.V.; Oxford, G.S. Classic D1 dopamine receptor antagonist R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH23390) directly inhibits G protein-coupled inwardly rectifying potassium channels. Mol. Pharmacol. 2002, 62, 119–126. [Google Scholar] [CrossRef]
- Inderwiedenstrasse, L.; Kienitz, M.C. Angiotensin receptors and alpha(1B)-adrenergic receptors regulate native IK((ACh)) and phosphorylation-deficient GIRK4 (S418A) channels through different PKC isoforms. Pflugers. Arch. 2024, 476, 1041–1064. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kubota, Y. GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb. Cortex 1997, 7, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Lamour, Y.; Dutar, P.; Jobert, A. Effects of neuropeptides on rat cortical neurons: Laminar distribution and interaction with the effect of acetylcholine. Neuroscience 1983, 10, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Phillis, J.W.; Limacher, J.J. Excitation of cerebral cortical neurons by various polypeptides. Exp. Neurol. 1974, 43, 414–423. [Google Scholar] [CrossRef]
- Kawaguchi, Y. Groupings of nonpyramidal and pyramidal cells with specific physiological and morphological characteristics in rat frontal cortex. J. Neurophysiol. 1993, 69, 416–431. [Google Scholar] [CrossRef]
- Seong, H.J.; Carter, A.G. D1 receptor modulation of action potential firing in a subpopulation of layer 5 pyramidal neurons in the prefrontal cortex. J. Neurosci. 2012, 32, 10516–10521. [Google Scholar] [CrossRef]
- Anastasiades, P.G.; Boada, C.; Carter, A.G. Cell-Type-Specific D1 Dopamine Receptor Modulation of Projection Neurons and Interneurons in the Prefrontal Cortex. Cereb. Cortex 2019, 29, 3224–3242. [Google Scholar] [CrossRef]
- Fischer-Ferraro, C.; Nahmod, V.E.; Goldstein, D.J.; Finkielman, S. Angiotensin and renin in rat and dog brain. J. Exp. Med. 1971, 133, 353–361. [Google Scholar] [CrossRef]
- Phillips, M.I.; Stenstrom, B. Angiotensin II in rat brain comigrates with authentic angiotensin II in high pressure liquid chromatography. Circ. Res. 1985, 56, 212–219. [Google Scholar] [CrossRef]
- Grobe, J.L.; Xu, D.; Sigmund, C.D. An intracellular renin-angiotensin system in neurons: Fact, hypothesis, or fantasy. Physiology 2008, 23, 187–193. [Google Scholar] [CrossRef]
- Jackson, L.; Eldahshan, W.; Fagan, S.C.; Ergul, A. Within the Brain: The Renin Angiotensin System. Int. J. Mol. Sci. 2018, 19, 876. [Google Scholar] [CrossRef] [PubMed]
- Labandeira-Garcia, J.L.; Valenzuela, R.; Costa-Besada, M.A.; Villar-Cheda, B.; Rodriguez-Perez, A.I. The intracellular renin-angiotensin system: Friend or foe. Some light from the dopaminergic neurons. Prog. Neurobiol. 2021, 199, 101919. [Google Scholar] [CrossRef] [PubMed]
- Sasamura, H.; Dzau, V.J.; Pratt, R.E. Desensitization of angiotensin receptor function. Kidney Int. 1994, 46, 1499–1501. [Google Scholar] [CrossRef] [PubMed]
- Gebke, E.; Muller, A.R.; Jurzak, M.; Gerstberger, R. Angiotensin II-induced calcium signalling in neurons and astrocytes of rat circumventricular organs. Neuroscience 1998, 85, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Hunyady, L.; Catt, K.J.; Clark, A.J.; Gaborik, Z. Mechanisms and functions of AT(1) angiotensin receptor internalization. Regul. Pept 2000, 91, 29–44. [Google Scholar] [CrossRef]
- Abdul-Muneer, P.M.; Bhowmick, S.; Briski, N. Angiotensin II Causes Neuronal Damage in Stretch-Injured Neurons: Protective Effects of Losartan, an Angiotensin T(1) Receptor Blocker. Mol. Neurobiol. 2018, 55, 5901–5912. [Google Scholar] [CrossRef]
- Min, L.J.; Mogi, M.; Iwanami, J.; Sakata, A.; Jing, F.; Tsukuda, K.; Ohshima, K.; Horiuchi, M. Angiotensin II and aldosterone-induced neuronal damage in neurons through an astrocyte-dependent mechanism. Hypertens. Res. 2011, 34, 773–778. [Google Scholar] [CrossRef]
- Yamamoto, E.; Tamamaki, N.; Nakamura, T.; Kataoka, K.; Tokutomi, Y.; Dong, Y.F.; Fukuda, M.; Matsuba, S.; Ogawa, H.; Kim-Mitsuyama, S. Excess salt causes cerebral neuronal apoptosis and inflammation in stroke-prone hypertensive rats through angiotensin II-induced NADPH oxidase activation. Stroke 2008, 39, 3049–3056. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Mucke, L. A network dysfunction perspective on neurodegenerative diseases. Nature 2006, 443, 768–773. [Google Scholar] [CrossRef]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- ProteinAtlas.org. The Open Access Resource for Human Proteins. 2024. Available online: https://www.proteinatlas.org (accessed on 1 August 2024).
- Tayler, H.M.; MacLachlan, R.; Guzel, O.; Fisher, R.A.; Skrobot, O.A.; Abulfadl, M.A.; Kehoe, P.G.; Miners, J.S. Altered Gene Expression Within the Renin-Angiotensin System in Normal Aging and Dementia. J. Gerontol. A Biol. Sci. Med. Sci. 2024, 79, glad241. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.X.; Zhang, Y.Q.; Hu, B.; Zhang, J.; Zhao, Q. Association of AT1R polymorphism with hypertension risk: An update meta-analysis based on 28,952 subjects. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.; Speth, R.C.; Trivedi, M. Renin-angiotensin system gene expression and neurodegenerative diseases. J. Renin-Angiotensin-Aldosterone Syst. 2016, 17, 1470320316666750. [Google Scholar] [CrossRef] [PubMed]
- Zettergren, A.; Kern, S.; Gustafson, D.; Gudmundsson, P.; Sigstrom, R.; Ostling, S.; Eriksson, E.; Zetterberg, H.; Blennow, K.; Skoog, I. The ACE Gene Is Associated with Late-Life Major Depression and Age at Dementia Onset in a Population-Based Cohort. Am. J. Geriatr. Psychiatry 2017, 25, 170–177. [Google Scholar] [CrossRef]
- Edwards, F.A.; Konnerth, A.; Sakmann, B.; Takahashi, T. A thin slice preparation for patch clamp recordings from neurones of the mammalian central nervous system. Pflugers. Arch. 1989, 414, 600–612. [Google Scholar] [CrossRef]
- Barry, P.H. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J. Neurosci. Methods 1994, 51, 107–116. [Google Scholar] [CrossRef]
- Gage, G.J.; Kipke, D.R.; Shain, W. Whole animal perfusion fixation for rodents. J. Vis. Exp. 2012, e3564. [Google Scholar] [CrossRef]
- Durst, M.; Konczol, K.; Ocskay, K.; Sipos, K.; Varnai, P.; Szilvasy-Szabo, A.; Fekete, C.; Toth, Z.E. Hypothalamic Nesfatin-1 Resistance May Underlie the Development of Type 2 Diabetes Mellitus in Maternally Undernourished Non-obese Rats. Front. Neurosci. 2022, 16, 828571. [Google Scholar] [CrossRef]
- Köles, L.; Hanuska, A.; Kollár, K.; Zelles, T.; Kató, E. Angiotensin II potentiates NMDA currents in layer V pyramidal cells of rat prefrontal cortex. In Proceedings of the Intrinsic Activity meeting abstracts, 22nd Scientific Symposium of the Austrian Pharmacological Society. Joint meeting with the Hungarian Society for Experimental and Clinical Pharmacology, Vienna, Austria, 8–10 September 2016. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanuska, A.; Ribiczey, P.; Kató, E.; Papp, Z.T.; Varga, Z.V.; Giricz, Z.; Tóth, Z.E.; Könczöl, K.; Zsembery, Á.; Zelles, T.; et al. Potentiation of NMDA Receptors by AT1 Angiotensin Receptor Activation in Layer V Pyramidal Neurons of the Rat Prefrontal Cortex. Int. J. Mol. Sci. 2024, 25, 12644. https://doi.org/10.3390/ijms252312644
Hanuska A, Ribiczey P, Kató E, Papp ZT, Varga ZV, Giricz Z, Tóth ZE, Könczöl K, Zsembery Á, Zelles T, et al. Potentiation of NMDA Receptors by AT1 Angiotensin Receptor Activation in Layer V Pyramidal Neurons of the Rat Prefrontal Cortex. International Journal of Molecular Sciences. 2024; 25(23):12644. https://doi.org/10.3390/ijms252312644
Chicago/Turabian StyleHanuska, Adrienn, Polett Ribiczey, Erzsébet Kató, Zsolt Tamás Papp, Zoltán V. Varga, Zoltán Giricz, Zsuzsanna E. Tóth, Katalin Könczöl, Ákos Zsembery, Tibor Zelles, and et al. 2024. "Potentiation of NMDA Receptors by AT1 Angiotensin Receptor Activation in Layer V Pyramidal Neurons of the Rat Prefrontal Cortex" International Journal of Molecular Sciences 25, no. 23: 12644. https://doi.org/10.3390/ijms252312644
APA StyleHanuska, A., Ribiczey, P., Kató, E., Papp, Z. T., Varga, Z. V., Giricz, Z., Tóth, Z. E., Könczöl, K., Zsembery, Á., Zelles, T., Harsing, L. G., Jr., & Köles, L. (2024). Potentiation of NMDA Receptors by AT1 Angiotensin Receptor Activation in Layer V Pyramidal Neurons of the Rat Prefrontal Cortex. International Journal of Molecular Sciences, 25(23), 12644. https://doi.org/10.3390/ijms252312644