
Endothelial Dysfunction and Liver Cirrhosis: Unraveling of a Complex Relationship

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
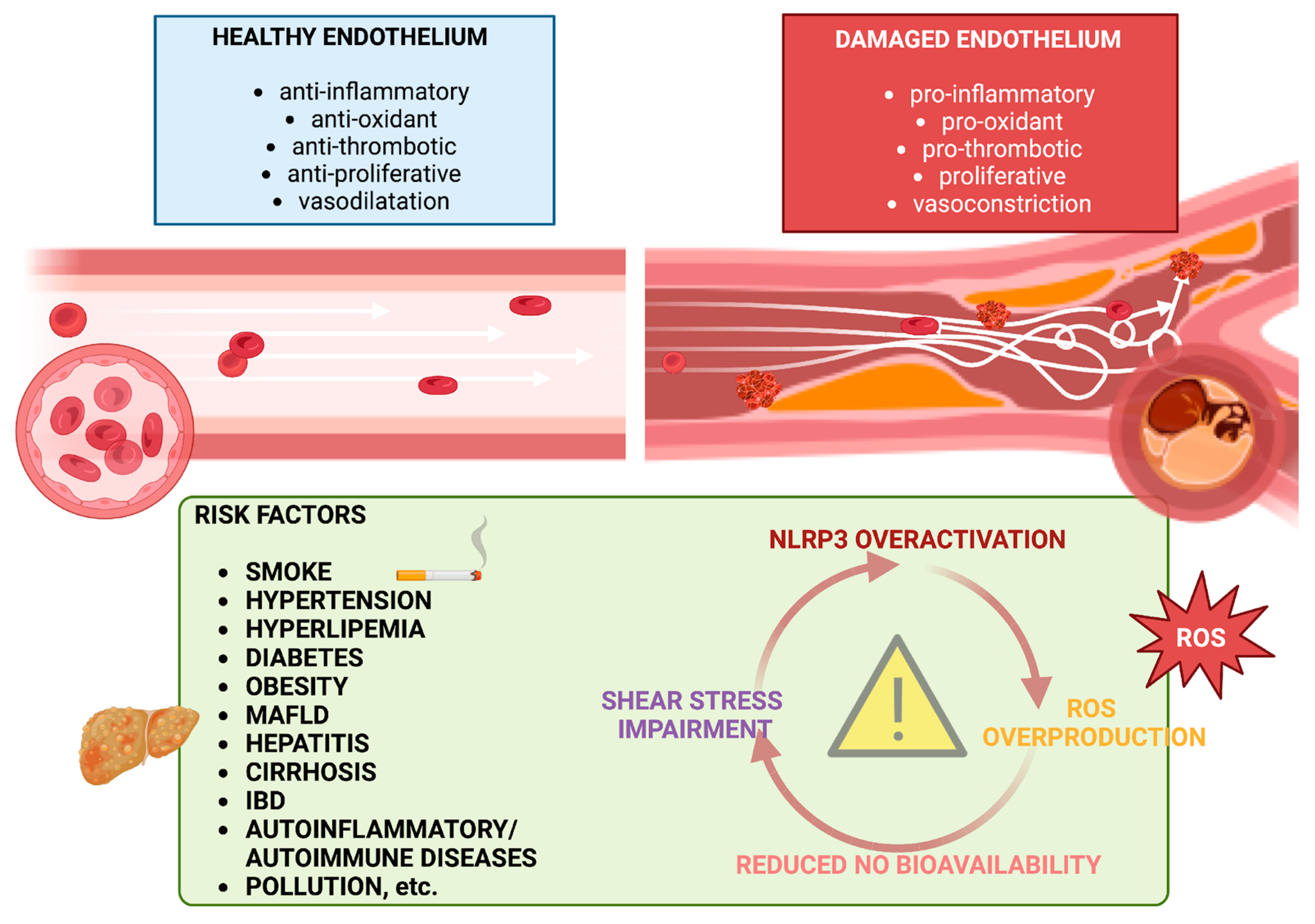
2. Pathogenesis of Endothelial Dysfunction
2.1. NO, ROS, and Inflammation
2.2. Flow Shear Stress
3. Endothelial Dysfunction in Liver Disease
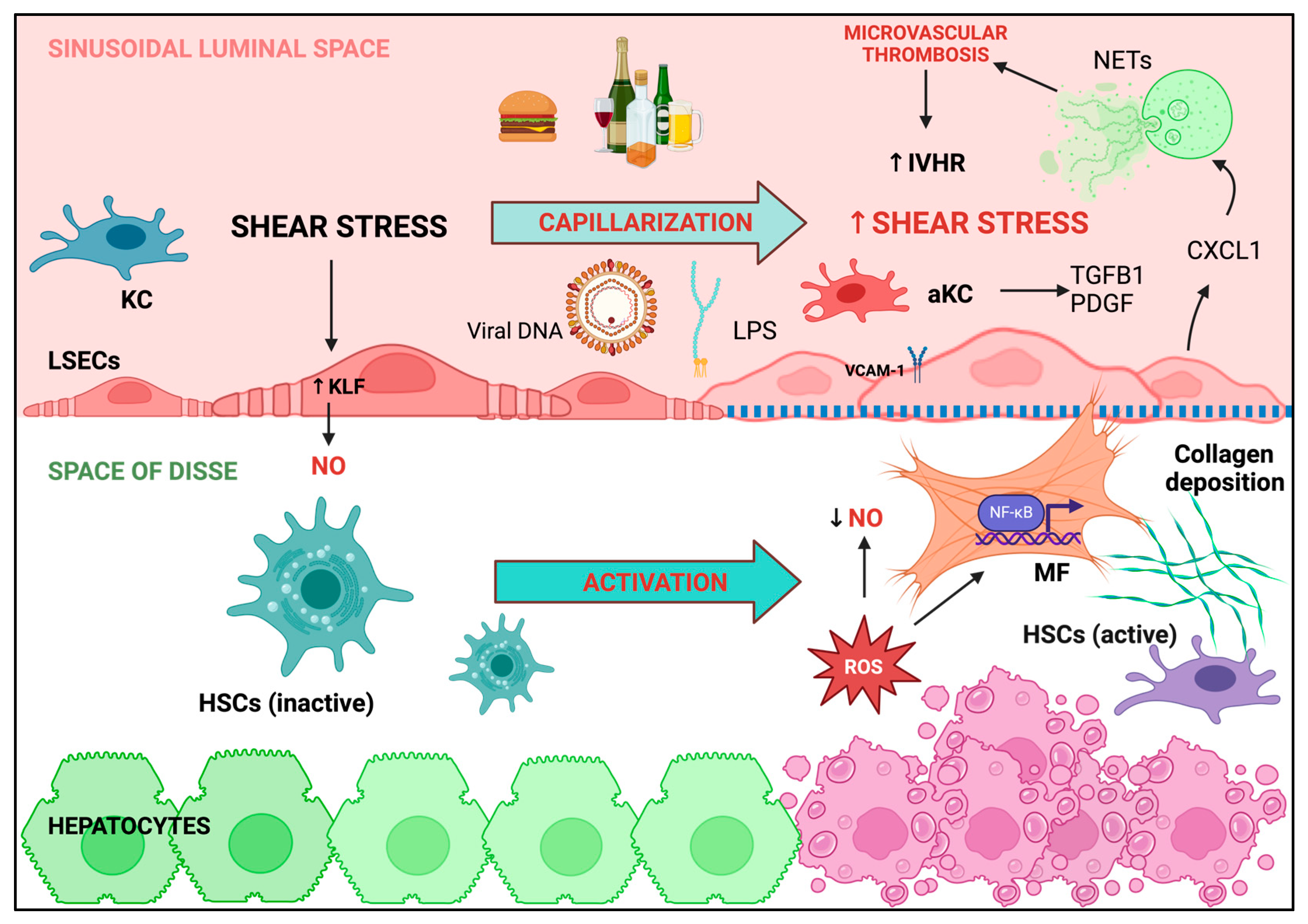
3.1. Liver Sinusoidal Endothelial Cells: Key Players in Maintaining Intrahepatic Endothelial Function
3.2. Hepatic Macrophages Trigger the Inflammatory Pathways Involved in LSECs Dysfunction and Liver Disease Progression
3.3. ROS Overproduction Favors Intrahepatic Thrombosis
3.4. Shear Stress and Neutrophils Regulate of LSECs Function
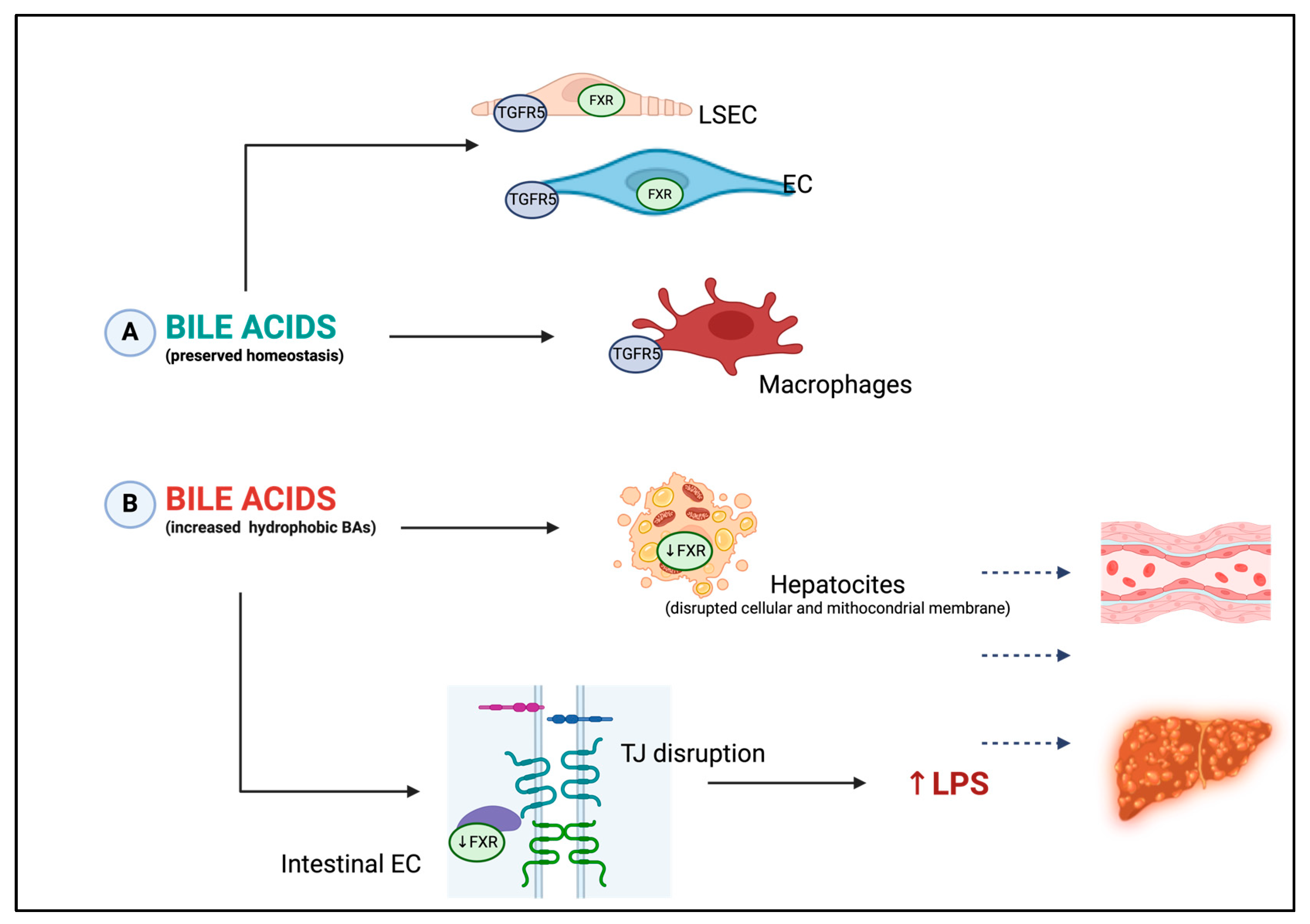
3.5. Bile Acids: New Key Players in Endothelial Dysfunction Occurrence
3.6. Nitric Oxide in the Pathogenesis of Portal Hypertension
4. Assessment of Endothelial Dysfunction: The Role of Flow-Mediated Dilation
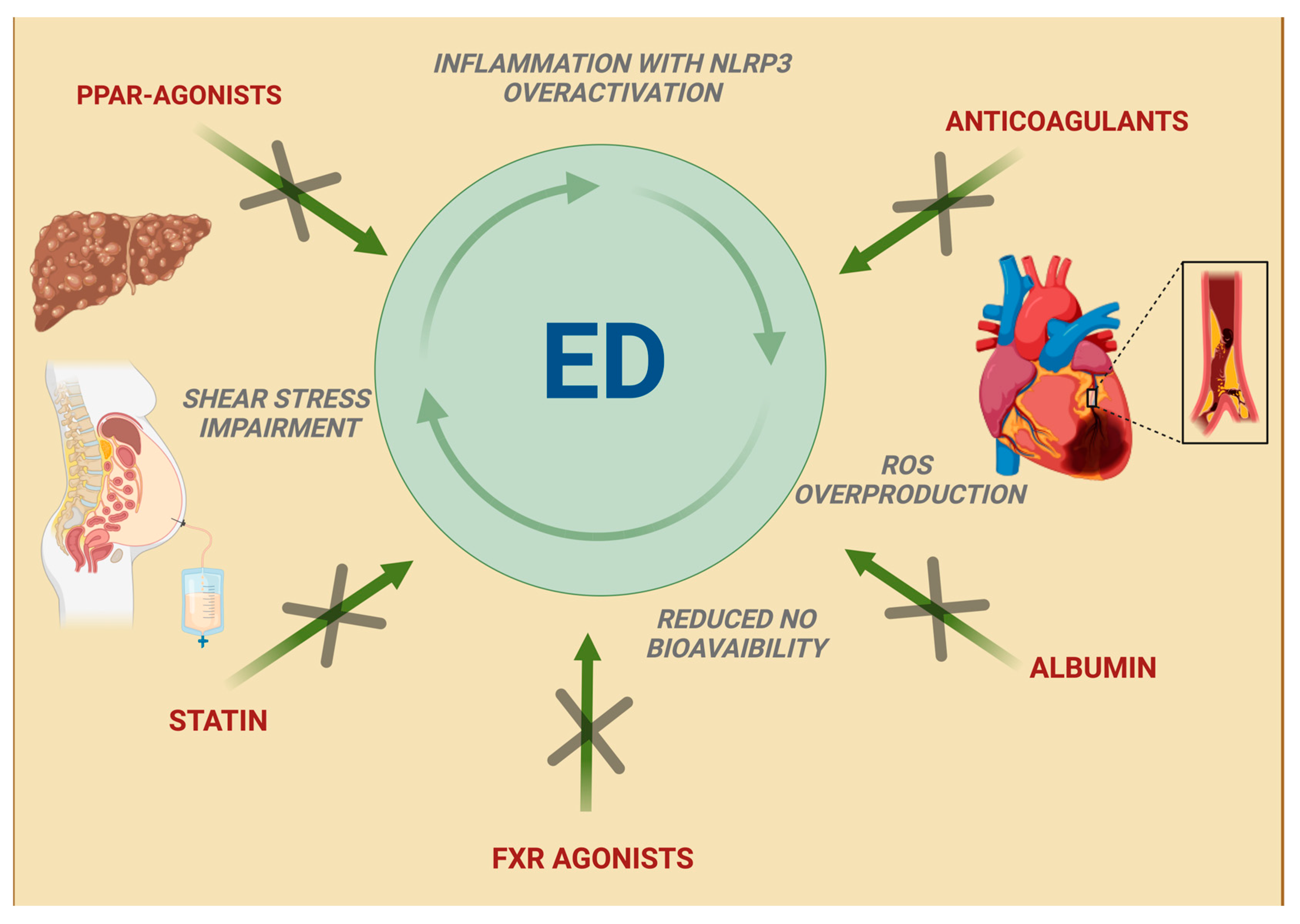
5. New Therapeutic Approaches in Liver Cirrhosis: An Emerging Relationship with Improved Endothelial Function
5.1. Albumin
5.2. Statins
5.3. Peroxisome Proliferator-Activated Receptor Agonists
5.4. Anticoagulants
5.5. Farnesoid X Receptor Agonists
6. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Iwakiri, Y.; Groszmann, R.J. The hyperdynamic circulation of chronic liver diseases: From the patient to the molecule. Hepatology 2006, 43 (Suppl. S1), S121–S131. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J.; Groszmann, R.J.; Shah, V.H. Evolution in the understanding of the pathophysiological basis of portal hypertension: How changes in paradigm are leading to successful new treatments. J. Hepatol. 2015, 62 (Suppl. S1), S121–S130. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bernardi, M.; Moreau, R.; Angeli, P.; Schnabl, B.; Arroyo, V. Mechanisms of decompensation and organ failure in cirrhosis: From peripheral arterial vasodilation to systemic inflammation hypothesis. J. Hepatol. 2015, 63, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Jalan, R.; D’Amico, G.; Trebicka, J.; Moreau, R.; Angeli, P.; Arroyo, V. New clinical and pathophysiological perspectives defining the trajectory of cirrhosis. J. Hepatol. 2021, 75 (Suppl. S1), S14–S26. [Google Scholar] [CrossRef] [PubMed]
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hunt, B.J.; Jurd, K.M. Endothelial cell activation. A central pathophysiological process. BMJ 1998, 316, 1328–1329. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moncada, S.; Higgs, E.A. The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol. 2006, 147 (Suppl. S1), S193–S201. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P. Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. Int. J. Mol. Sci. 2017, 18, 2563. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kvietys, P.R.; Granger, D.N. Role of reactive oxygen and nitrogen species in the vascular responses to inflammation. Free Radic. Biol. Med. 2012, 52, 556–592. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lim, S.; Park, S. Role of vascular smooth muscle cell in the inflammation of atherosclerosis. BMB Rep. 2014, 47, 1–7, Erratum in BMB Rep. 2016, 49, 134. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Staiculescu, M.C.; Foote, C.; Meininger, G.A.; Martinez-Lemus, L.A. The role of reactive oxygen species in microvascular remodeling. Int. J. Mol. Sci. 2014, 15, 23792–23835. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.F.; Yu, T.; Chu, X.M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, W.; Zhao, M.; Zhao, S.; Lu, Q.; Ni, L.; Zou, C.; Lu, L.; Xu, X.; Guan, H.; Zheng, Z.; et al. Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: A novel inhibitory effect of minocycline. Inflamm. Res. 2017, 66, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; He, X.; Wu, L.M.; Zhang, R.Y.; Li, L.M.; Wu, C.M.; Lu, Y.B.; Hu, B.; Shi, C.; Lu, Z.F.; et al. MLKL Aggravates Ox-LDL-Induced Cell Pyroptosis via Activation of NLRP3 Inflammasome in Human Umbilical Vein Endothelial Cells. Inflammation 2020, 43, 2222–2231. [Google Scholar] [CrossRef] [PubMed]
- Li, X.X.; Ling, S.K.; Hu, M.Y.; Ma, Y.; Li, Y.; Huang, P.L. Protective effects of acarbose against vascular endothelial dysfunction through inhibiting Nox4/NLRP3 inflammasome pathway in diabetic rats. Free Radic. Biol. Med. 2019, 145, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cau, S.B.; Bruder-Nascimento, A.; Silva, M.B.; Ramalho, F.N.Z.; Mestriner, F.; Alves-Lopes, R.; Ferreira, N.; Tostes, R.C.; Bruder-Nascimento, T. Angiotensin-II activates vascular inflammasome and induces vascular damage. Vasc. Pharmacol. 2021, 139, 106881. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Knorr, J.; Wree, A.; Tacke, F.; Feldstein, A.E. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin. Liver Dis. 2020, 40, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.-H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, B.; Jiang, M.; Chu, Y.; Wang, W.; Chen, D.; Li, X.; Zhang, Z.; Zhang, D.; Fan, D.; Nie, Y.; et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2018, 68, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, S.; Wan, T.; Huang, Y.; Pang, N.; Jiang, X.; Gu, Y.; Zhang, Z.; Luo, J.; Yang, L. Cyanidin-3-O-β-glucoside inactivates NLRP3 inflammasome and alleviates alcoholic steatohepatitis via SirT1/NF-κB signaling pathway. Free Radic. Biol. Med. 2020, 160, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Iracheta-Vellve, A.; Petrasek, J.; Satishchandran, A.; Gyongyosi, B.; Saha, B.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Inhibition of sterile danger signals, uric acid and ATP, prevents inflammasome activation and protects from alcoholic steatohepatitis in mice. J. Hepatol. 2015, 63, 1147–1155. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Petrasek, J.; Iracheta-Vellve, A.; Saha, B.; Satishchandran, A.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Metabolic danger signals, uric acid and ATP, mediate inflammatory cross-talk between hepatocytes and immune cells in alcoholic liver disease. J. Leukoc. Biol. 2015, 98, 249–256. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hoyt, L.R.; Randall, M.J.; Ather, J.L.; DePuccio, D.P.; Landry, C.C.; Qian, X.; Janssen-Heininger, Y.M.; van der Vliet, A.; Dixon, A.E.; Amiel, E.; et al. Mitochondrial ROS induced by chronic ethanol exposure promote hyper-activation of the NLRP3 inflammasome. Redox Biol. 2017, 12, 883–896. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Heo, M.J.; Kim, T.H.; You, J.S.; Blaya, D.; Sancho-Bru, P.; Kim, S.G. Alcohol dysregulates miR-148a in hepatocytes through FoxO1, facilitating pyroptosis via TXNIP overexpression. Gut 2019, 68, 708–720. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Molyvdas, A.; Georgopoulou, U.; Lazaridis, N.; Hytiroglou, P.; Dimitriadis, A.; Foka, P.; Vassiliadis, T.; Loli, G.; Phillipidis, A.; Zebekakis, P.; et al. The role of the NLRP3 inflammasome and the activation of IL-1β in the pathogenesis of chronic viral hepatic inflammation. Cytokine 2018, 110, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.H.; Ding, J.; Xie, X.X.; Yang, X.H.; Wu, X.F.; Chen, Z.X.; Guo, Q.L.; Gao, W.Y.; Wang, X.Z.; Li, D. Hepatitis B virus X protein promotes liver cell pyroptosis under oxidative stress through NLRP3 inflammasome activation. Inflamm. Res. 2020, 69, 683–696. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Russo, F.P.; Zanetto, A.; Pinto, E.; Battistella, S.; Penzo, B.; Burra, P.; Farinati, F. Hepatocellular Carcinoma in Chronic Viral Hepatitis: Where Do We Stand? Int. J. Mol. Sci. 2022, 23, 500. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yu, X.; Lan, P.; Hou, X.; Han, Q.; Lu, N.; Li, T.; Jiao, C.; Zhang, J.; Zhang, C.; Tian, Z. HBV inhibits LPS-induced NLRP3 inflammasome activation and IL-1β production via suppressing the NF-κB pathway and ROS production. J. Hepatol. 2017, 66, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Lei, Q.; Li, T.; Li, L.; Qin, B. Hepatitis B core antigen can regulate NLRP3 inflammasome pathway in HepG2 cells. J. Med. Virol. 2019, 91, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- Kofahi, H.M.; Taylor, N.G.; Hirasawa, K.; Grant, M.D.; Russell, R.S. Hepatitis C Virus Infection of Cultured Human Hepatoma Cells Causes Apoptosis and Pyroptosis in Both Infected and Bystander Cells. Sci. Rep. 2016, 6, 37433. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Negash, A.A.; Olson, R.M.; Griffin, S.; Gale, M., Jr. Modulation of calcium signaling pathway by hepatitis C virus core protein stimulates NLRP3 inflammasome activation. PLoS Pathog. 2019, 15, e1007593. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gordon, E.; Schimmel, L.; Frye, M. The Importance of Mechanical Forces for in vitro Endothelial Cell Biology. Front. Physiol. 2020, 11, 684. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chatzizisis, Y.S.; Coskun, A.U.; Jonas, M.; Edelman, E.R.; Feldman, C.L.; Stone, P.H. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: Molecular, cellular, and vascular behavior. J. Am. Coll. Cardiol. 2007, 49, 2379–2393. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, Y.; Shang, M.; Liu, X.; Munn, L.L. Endothelial mechanobiology in atherosclerosis. Cardiovasc. Res. 2023, 119, 1656–1675. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, K.K.; McCourt, P.; Berg, T.; Crossley, C.; Le Couteur, D.; Wake, K.; Smedsrød, B. The scavenger endothelial cell: A new player in homeostasis and immunity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 12, 1217–1230. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.H.; Kim, Y.K.; Kim, M.R.; Jang, J.H.; Lee, S. Emerging Roles of Vascular Cell Adhesion Molecule-1 (VCAM-1) in Immunological Disorders and Cancer. Int. J. Mol. Sci. 2018, 19, 1057. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gracia-Sancho, J.; Marrone, G.; Fernández-Iglesias, A. Hepatic microcirculation and mechanisms of portal hypertension. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Marrone, G.; Shah, V.H.; Gracia-Sancho, J. Sinusoidal communication in liver fibrosis and regeneration. J. Hepatol. 2016, 65, 608–617. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gracia-Sancho, J.; Caparrós, E.; Fernández-Iglesias, A.; Francés, R. Role of liver sinusoidal endothelial cells in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 411–431. [Google Scholar] [CrossRef] [PubMed]
- Kalucka, J.; Bierhansl, L.; Conchinha, N.V.; Missiaen, R.; Elia, I.; Brüning, U.; Scheinok, S.; Treps, L.; Cantelmo, A.R.; Dubois, C.; et al. Quiescent Endothelial Cells Upregulate Fatty Acid β-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018, 28, 881–894. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Furuta, K.; Islam, S.; Caporarello, N.; Kostallari, E.; Dielis, K.; Tschumperlin, D.J.; Hirsova, P.; Ibrahim, S.H. Liver sinusoidal endothelial cell expressed vascular cell adhesion molecule 1 promotes liver fibrosis. Front. Immunol. 2022, 13, 983255. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bitto, N.; Ghigliazza, G.; Lavorato, S.; Caputo, C.; La Mura, V. Improving Management of Portal Hypertension: The Potential Benefit of Non-Etiological Therapies in Cirrhosis. J. Clin. Med. 2023, 12, 934. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kamm, D.R.; McCommis, K.S. Hepatic stellate cells in physiology and pathology. J. Physiol. 2022, 600, 1825–1837. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bocca, C.; Protopapa, F.; Foglia, B.; Maggiora, M.; Cannito, S.; Parola, M.; Novo, E. Hepatic Myofibroblasts: A Heterogeneous and Redox-Modulated Cell Population in Liver Fibrogenesis. Antioxidants 2022, 11, 1278. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cheng, D.; Chai, J.; Wang, H.; Fu, L.; Peng, S.; Ni, X. Hepatic macrophages: Key players in the development and progression of liver fibrosis. Liver Int. 2021, 41, 2279–2294. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.C.; Bai, J.; Han, H.; Qin, H.Y. The versatility of macrophage heterogeneity in liver fibrosis. Front. Immunol. 2022, 13, 968879. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Novo, E.; Cannito, S.; Paternostro, C.; Bocca, C.; Miglietta, A.; Parola, M. Cellular and molecular mechanisms in liver fibrogenesis. Arch. Biochem. Biophys. 2014, 548, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y.; Rho, H.S.; Kim, A.; Kim, T.H.; Jang, K.; Jun, D.W.; Kim, J.W.; Kim, B.; Kim, S.G. FXR Inhibits Endoplasmic Reticulum Stress-Induced NLRP3 Inflammasome in Hepatocytes and Ameliorates Liver Injury. Cell Rep. 2018, 24, 2985–2999. [Google Scholar] [CrossRef] [PubMed]
- García-Calderó, H.; Rodríguez-Vilarrupla, A.; Gracia-Sancho, J.; Diví, M.; Laviña, B.; Bosch, J.; García-Pagán, J.C. Tempol administration, a superoxide dismutase mimetic, reduces hepatic vascular resistance and portal pressure in cirrhotic rats. J. Hepatol. 2011, 54, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Bouabout, G.; Ayme-Dietrich, E.; Jacob, H.; Champy, M.F.; Birling, M.C.; Pavlovic, G.; Madeira, L.; Fertak, L.E.; Petit-Demoulière, B.; Sorg, T.; et al. Nox4 genetic inhibition in experimental hypertension and metabolic syndrome. Arch. Cardiovasc. Dis. 2018, 111, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Laviña, B.; Rodríguez-Vilarrupla, A.; Brandes, R.P.; Fernández, M.; Bosch, J.; García-Pagán, J.C. Evidence against a role for NADPH oxidase modulating hepatic vascular tone in cirrhosis. Gastroenterology 2007, 133, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Aspects Med. 2019, 65, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, Y.; Trebicka, J. Portal hypertension in cirrhosis: Pathophysiological mechanisms and therapy. JHEP Rep. 2021, 3, 100316. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bellanti, F.; Mangieri, D.; Vendemiale, G. Redox Biology and Liver Fibrosis. Int. J. Mol. Sci. 2023, 25, 410. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ahmadian, E.; Pennefather, P.S.; Eftekhari, A.; Heidari, R.; Eghbal, M.A. Role of renin-angiotensin system in liver diseases: An outline on the potential therapeutic points of intervention. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Andueza, A.; Garde, N.; García-Garzón, A.; Ansorena, E.; López-Zabalza, M.J.; Iraburu, M.J.; Zalba, G.; Martínez-Irujo, J.J. NADPH oxidase 5 promotes proliferation and fibrosis in human hepatic stellate cells. Free Radic. Biol. Med. 2018, 126, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Limmer, A.; Ohl, J.; Kurts, C.; Ljunggren, H.G.; Reiss, Y.; Groettrup, M.; Momburg, F.; Arnold, B.; Knolle, P.A. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat. Med. 2000, 6, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Weston, C.J.; Oo, Y.H.; Westerlund, N.; Stamataki, Z.; Youster, J.; Hubscher, S.G.; Salmi, M.; Jalkanen, S.; Lalor, P.F.; et al. Common lymphatic endothelial and vascular endothelial receptor-1 mediates the transmigration of regulatory T cells across human hepatic sinusoidal endothelium. J. Immunol. 2011, 186, 4147–4155. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Airola, C.; Pallozzi, M.; Cerrito, L.; Santopaolo, F.; Stella, L.; Gasbarrini, A.; Ponziani, F.R. Microvascular Thrombosis and Liver Fibrosis Progression: Mechanisms and Clinical Applications. Cells 2023, 12, 1712. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Goel, A.; Hegarty, R.; Dixit, S.; Tucker, B.; Douiri, A.; Kyrana, E.; Jain, V.; Dhawan, A.; Grammatikopoulos, T. Transient elastography and von Willebrand factor as predictors of portal hypertension and decompensation in children. JHEP Rep. 2023, 12, 100935. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Islek, A.; Ilhan, D.; Ozturk, N.; Guven, B.; Sag, E. Altered von Willebrand Factor and ADAMTS13 Levels in Children with Cirrhosis and Extrahepatic Portal Hypertension. J. Pediatr. Hematol. Oncol. 2021, 43, e951–e956. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Yan, X.; Li, C.; Li, X.; Ma, X.; Zhang, C.; Ju, S.; Tian, J.; Qi, X. von Willebrand factor as a biomarker of clinically significant portal hypertension and severe portal hypertension: A systematic review and meta-analysis. BMJ Open 2019, 9, e025656. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Felli, E.; Selicean, S.; Guixé-Muntet, S.; Wang, C.; Bosch, J.; Berzigotti, A.; Gracia-Sancho, J. Mechanobiology of portal hypertension. JHEP Rep. 2023, 5, 100869. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gracia-Sancho, J.; Russo, L.; García-Calderó, H.; García-Pagán, J.C.; García-Cardeña, G.; Bosch, J. Endothelial expression of transcription factor Kruppel-like factor 2 and its vasoprotective target genes in the normal and cirrhotic rat liver. Gut. 2011, 60, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; Heo, W.; Park, J.I.; Kim, K.M.; Oh, H.T.; Yoo, G.D.; Park, J.; Shin, S.; Do, Y.; Jeong, M.G.; et al. Endothelial TAZ inhibits capillarization of liver sinusoidal endothelium and damage-induced liver fibrosis via nitric oxide production. Theranostics 2023, 13, 4182–4196. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guixé-Muntet, S.; de Mesquita, F.C.; Vila, S.; Hernández-Gea, V.; Peralta, C.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. Cross-talk between autophagy and KLF2 determines endothelial cell phenotype and microvascular function in acute liver injury. J. Hepatol. 2017, 66, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, P.; Zhang, R.; Watanabe, I.; Chang, E.; Vinayachandran, V.; Nayak, L.; Lapping, S.; Liao, S.; Madera, A.; et al. KLF2 regulates neutrophil activation and thrombosis in cardiac hypertrophy and heart failure progression. J. Clin. Investig. 2022, 132, e147191. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hilscher, M.B.; Sehrawat, T.; Arab, J.P.; Zeng, Z.; Gao, J.; Liu, M.; Kostallari, E.; Gao, Y.; Simonetto, D.A.; Yaqoob, U.; et al. Mechanical Stretch Increases Expression of CXCL1 in Liver Sinusoidal Endothelial Cells to Recruit Neutrophils, Generate Sinusoidal Microthombi, and Promote Portal Hypertension. Gastroenterology 2019, 157, 193–209.e9. [Google Scholar] [CrossRef] [PubMed]
- van der Windt, D.J.; Sud, V.; Zhang, H.; Varley, P.R.; Goswami, J.; Yazdani, H.O.; Tohme, S.; Loughran, P.; O’Doherty, R.M.; Minervini, M.I.; et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 2018, 68, 1347–1360. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, W.W.; Wu, L.; Lu, W.; Chen, W.; Yan, W.; Qi, C.; Xuan, S.; Shang, A. Lipopolysaccharides increase the risk of colorectal cancer recurrence and metastasis due to the induction of neutrophil extracellular traps after curative resection. J. Cancer Res. Clin. Oncol. 2021, 147, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Martin-Mateos, R.M.; Shah, V.H. Gut-liver axis, cirrhosis and portal hypertension: The chicken and the egg. Hepatol. Int. 2018, 12 (Suppl. S1), 24–33. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Charitos, I.A.; Aliani, M.; Tondo, P.; Venneri, M.; Castellana, G.; Scioscia, G.; Castellaneta, F.; Lacedonia, D.; Carone, M. Biomolecular Actions by Intestinal Endotoxemia in Metabolic Syndrome. Int. J. Mol. Sci. 2024, 25, 2841. [Google Scholar] [CrossRef]
- Philips, C.A.; Augustine, P. Gut Barrier and Microbiota in Cirrhosis. J. Clin. Exp. Hepatol. 2022, 12, 625–638. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Y.; Zhang, Y.; Liu, Y.; Xu, J.; Liu, Y. Gut-Liver Axis: Liver Sinusoidal Endothelial Cells Function as the Hepatic Barrier in Colitis-Induced Liver Injury. Front. Cell Dev. Biol. 2021, 9, 702890. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, Y.Y.; Lin, H.C. Alteration of intrahepatic microcirculation in cirrhotic livers. J. Chin. Med. Assoc. 2015, 78, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Wang, S.; Wang, P.; Tang, C.; Wang, Z.; Chen, L.; Luo, G.; Chen, H.; Liu, Y.; Feng, B.; et al. Bile acids and their receptors in regulation of gut health and diseases. Prog. Lipid Res. 2023, 89, 101210. [Google Scholar] [CrossRef] [PubMed]
- Bilson, J.; Scorletti, E.; Swann, J.R.; Byrne, C.D. Bile Acids as Emerging Players at the Intersection of Steatotic Liver Disease and Cardiovascular Diseases. Biomolecules 2024, 14, 841. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pablo Arab, J.; Barrera, F.; Arrese, M. Bile Acids and Portal Hypertension. Ann. Hepatol. 2017, 16 (Suppl. S1), S83–S86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lv, T.; Wang, X.; Wu, M.; Zhang, R.; Yang, X.; Fu, Y.; Liu, Z. Role of the microbiota-gut-heart axis between bile acids and cardiovascular disease. Biomed. Pharmacother. 2024, 174, 116567. [Google Scholar] [CrossRef] [PubMed]
- Manilla, V.; Santopaolo, F.; Gasbarrini, A.; Ponziani, F.R. Type 2 Diabetes Mellitus and Liver Disease: Across the Gut-Liver Axis from Fibrosis to Cancer. Nutrients 2023, 15, 2521. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Klindt, C.; Reich, M.; Hellwig, B.; Stindt, J.; Rahnenführer, J.; Hengstler, J.G.; Köhrer, K.; Schoonjans, K.; Häussinger, D.; Keitel, V. The G Protein-Coupled Bile Acid Receptor TGR5 (Gpbar1) Modulates Endothelin-1 Signaling in Liver. Cells 2019, 8, 1467. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Keitel, V.; Häussinger, D. Role of TGR5 (GPBAR1) in Liver Disease. Semin. Liver Dis. 2018, 38, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Chen, W.D.; Wang, Y.D. TGR5, Not Only a Metabolic Regulator. Front. Physiol. 2016, 7, 646. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Panzitt, K.; Wagner, M. FXR in liver physiology: Multiple faces to regulate liver metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166133. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Cao, L.; Jiang, C.; Che, Y.; Zhang, S.; Takahashi, S.; Wang, G.; Gonzalez, F.J. Farnesoid X Receptor Regulation of the NLRP3 Inflammasome Underlies Cholestasis-Associated Sepsis. Cell Metab. 2017, 25, 856–867.e5. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sauerbruch, T.; Hennenberg, M.; Trebicka, J.; Beuers, U. Bile Acids, Liver Cirrhosis, and Extrahepatic Vascular Dysfunction. Front. Physiol. 2021, 12, 718783. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pryymachuk, G.; El-Awaad, E.; Piekarek, N.; Drebber, U.; Maul, A.C.; Hescheler, J.; Wodarz, A.; Pfitzer, G.; Neiss, W.F.; Pietsch, M.; et al. Angiotensin II type 1 receptor localizes at the blood-bile barrier in humans and pigs. Histochem. Cell Biol. 2022, 157, 513–524. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moris, D.; Giaginis, C.; Tsourouflis, G.; Theocharis, S. Farnesoid-X Receptor (FXR) as a Promising Pharmaceutical Target in Atherosclerosis. Curr. Med. Chem. 2017, 24, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yang, J.; Wang, Y.; Qi, Y.; Yang, W.; Li, Y. Farnesoid X Receptor Agonists as Therapeutic Target for Cardiometabolic Diseases. Front. Pharmacol. 2020, 11, 1247. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sturm, L.; Bettinger, D.; Roth, L.; Zoldan, K.; Stolz, L.; Gahm, C.; Huber, J.P.; Reincke, M.; Kaeser, R.; Boettler, T.; et al. Plasma Cyclic Guanosine Monophosphate Is a Promising Biomarker of Clinically Significant Portal Hypertension in Patients with Liver Cirrhosis. Front. Med 2022, 8, 803119. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kreisel, W.; Lazaro, A.; Trebicka, J.; Grosse Perdekamp, M.; Schmitt-Graeff, A.; Deibert, P. Cyclic GMP in Liver Cirrhosis-Role in Pathophysiology of Portal Hypertension and Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 10372. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bahadoran, Z.; Mirmiran, P.; Kashfi, K.; Ghasemi, A. Vascular nitric oxide resistance in type 2 diabetes. Cell Death Dis. 2023, 14, 410. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zuchi, C.; Tritto, I.; Carluccio, E.; Mattei, C.; Cattadori, G.; Ambrosio, G. Role of endothelial dysfunction in heart failure. Heart Fail. Rev. 2020, 25, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.; Lv, D.Y.; Yu, H.L.; Chen, H.; Luo, S.X. iNOS regulates activation of the NLRP3 inflammasome through the sGC/cGMP/PKG/TACE/TNF-α axis in response to cigarette smoke resulting in aortic endothelial pyroptosis and vascular dysfunction. Int. Immunopharmacol. 2021, 101 Pt B, 108334. [Google Scholar] [CrossRef] [PubMed]
- Balzer, M.S.; Pavkovic, M.; Frederick, J.; Abedini, A.; Freyberger, A.; Vienenkötter, J.; Mathar, I.; Siudak, K.; Eitner, F.; Sandner, P.; et al. Treatment effects of soluble guanylate cyclase modulation on diabetic kidney disease at single-cell resolution. Cell Rep. Med. 2023, 4, 100992. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lombardi, C.M.; Cimino, G.; Pagnesi, M.; Dell’Aquila, A.; Tomasoni, D.; Ravera, A.; Inciardi, R.; Carubelli, V.; Vizzardi, E.; Nodari, S.; et al. Vericiguat for Heart Failure with Reduced Ejection Fraction. Curr. Cardiol. Rep. 2021, 23, 144. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thijssen, D.H.J.; Bruno, R.M.; van Mil, A.C.C.M.; Holder, S.M.; Faita, F.; Greyling, A.; Zock, P.L.; Taddei, S.; Deanfield, J.E.; Luscher, T.; et al. Expert consensus and evidence-based recommendations for the assessment of flow-mediated dilation in humans. Eur. Heart J. 2019, 40, 2534–2547. [Google Scholar] [CrossRef] [PubMed]
- Papagiouvanni, I.; Sarafidis, P.; Theodorakopoulou, M.P.; Sinakos, E.; Goulis, I. Endothelial and microvascular function in liver cirrhosis: An old concept that needs re-evaluation? Ann. Gastroenterol. 2022, 35, 471–482. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Armentano, R.L.; Arbeitman, C.R.; Cymberknop, L.J.; Farro, I.; Viotti, R.; Cardelino, J. Flow Mediated Dilation in Cirrhosis: A Pilot Study in Different Stages of the Disease. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2018, 2018, 4564–4566. [Google Scholar] [CrossRef] [PubMed]
- Sárközi, A.; Cseh, D.; Gerlei, Z.; Kollai, M. Reduced neural baroreflex sensitivity is related to enhanced endothelial function in patients with end-stage liver disease. Scand. J. Gastroenterol. 2018, 53, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Marcacci, M.; Fiorini, M.; Lattanzi, A.; Venturelli, G.; Roveri, G.; Boni, C.; Zappia, F.; Pietrangelo, A.; Rossi, R.; Ventura, P. Is flow-mediated dilatation (fmd) assessment a reliable marker of endothelial dysfunction in liver cirrhosis? J. Hepatol. 2013, 58, S93. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Funaro, B.; Lupascu, A.; Ainora, M.E.; Garcovich, M.; Caracciolo, G.; Quadarella, A.; Nesci, A.; Riccardi, L.; Gasbarrini, A.; et al. Minimal Hepatic Encephalopathy is Associated with Increased Cerebral Vascular Resistance. A Transcranial Doppler Ultrasound Study. Sci. Rep. 2019, 9, 15373. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Berzigotti, A.; Erice, E.; Gilabert, R.; Reverter, E.; Abraldes, J.G.; García-Pagan, J.C.; Bosch, J. Cardiovascular risk factors and systemic endothelial function in patients with cirrhosis. Am. J. Gastroenterol. 2013, 108, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Gbaruko, U.K.; Slyvka, N.O.; Bojchuk, T.M.; Ivashchuk, O.I.; Plesh, I.A.; Cherevatenko, V.O. Value of Endothelial Dysfunction in the Pathogenesis of Portal Hypertension. Int. J. Collab. Res. Intern. Med. Public Health 2012, 4, 1040. [Google Scholar]
- Hagström, H.; Adams, L.A.; Allen, A.M.; Byrne, C.D.; Chang, Y.; Duseja, A.; Grønbæk, H.; Ismail, M.H.; Jepsen, P.; Kanwal, F.; et al. The future of International Classification of Diseases coding in steatotic liver disease: An expert panel Delphi consensus statement. Hepatol. Commun. 2024, 8, e0386. [Google Scholar] [CrossRef] [PubMed]
- Mellemkjær, A.; Kjær, M.B.; Haldrup, D.; Grønbæk, H.; Thomsen, K.L. Management of cardiovascular risk in patients with metabolic dysfunction-associated steatotic liver disease. Eur. J. Intern. Med. 2024, 122, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Theofilis, P.; Vordoni, A.; Nakas, N.; Kalaitzidis, R.G. Endothelial Dysfunction in Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Life 2022, 12, 718. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yinzhi, D.; Jianhua, H.; Hesheng, L. The roles of liver sinusoidal endothelial cells in liver ischemia/reperfusion injury. J. Gastroenterol. Hepatol. 2024, 39, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Qu, K.; Yan, Z.; Wu, Y.; Chen, Y.; Qu, P.; Xu, X.; Yuan, P.; Huang, X.; Xing, J.; Zhang, H.; et al. Transarterial chemoembolization aggravated peritumoral fibrosis via hypoxia-inducible factor-1α dependent pathway in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2015, 30, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, M.; Angeli, P.; Claria, J.; Moreau, R.; Gines, P.; Jalan, R.; Caraceni, P.; Fernandez, J.; Gerbes, A.L.; O’Brien, A.J.; et al. Albumin in decompensated cirrhosis: New concepts and perspectives. Gut 2020, 69, 1127–1138. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tufoni, M.; Baldassarre, M.; Zaccherini, G.; Antognoli, A.; Caraceni, P. Hemodynamic and Systemic Effects of Albumin in Patients with Advanced Liver Disease. Curr. Hepatol. Rep. 2020, 19, 147–158. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fernández, J.; Clària, J.; Amorós, A.; Aguilar, F.; Castro, M.; Casulleras, M.; Acevedo, J.; Duran-Güell, M.; Nuñez, L.; Costa, M.; et al. Effects of Albumin Treatment on Systemic and Portal Hemodynamics and Systemic Inflammation in Patients with Decompensated Cirrhosis. Gastroenterology 2019, 159, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Fagan, A.; Gavis, E.A.; Gallagher, M.L.; Mousel, T.; Davis, B.; Puri, P.; Sterling, R.K.; Luketic, V.A.; Lee, H.; Matherly, S.C.; et al. A double-blind randomized placebo-controlled trial of albumin in outpatients with hepatic encephalopathy: HEAL study. J. Hepatol. 2023, 78, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Meurer, L.; Cohen, S.M. Drug-Induced Liver Injury from Statins. Clin. Liver Dis. 2020, 24, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Marrone, G.; Maeso-Díaz, R.; García-Cardena, G.; Abraldes, J.G.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: Behind the molecular mechanisms of statins. Gut 2015, 64, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- La Mura, V.; Gagliano, N.; Arnaboldi, F.; Sartori, P.; Procacci, P.; Denti, L.; Liguori, E.; Bitto, N.; Ristagno, G.; Latini, R.; et al. Simvastatin Prevents Liver Microthrombosis and Sepsis Induced Coagulopathy in a Rat Model of Endotoxemia. Cells 2022, 11, 1148. [Google Scholar] [CrossRef]
- Abraldes, J.G.; Albillos, A.; Bañares, R.; Turnes, J.; González, R.; García-Pagán, J.C.; Bosch, J. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: A randomized controlled trial. Gastroenterology 2009, 136, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Pose, E.; Napoleone, L.; Amin, A.; Campion, D.; Jimenez, C.; Piano, S.; Roux, O.; Uschner, F.E.; de Wit, K.; Zaccherini, G.; et al. Safety of two different doses of simvastatin plus rifaximin in decompensated cirrhosis (LIVERHOPE-SAFETY): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Gastroenterol. Hepatol. 2020, 5, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Pose, E.; Solà, E.; Lozano, J.J.; Juanola, A.; Sidorova, J.; Zaccherini, G.; de Wit, K.; Uschner, F.; Tonon, M.; Kazankov, K.; et al. Treatment with Simvastatin and Rifaximin Restores the Plasma Metabolomic Profile in Patients with Decompensated Cirrhosis. Hepatol. Commun. 2022, 6, 1100–1112. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, A.N.; Xu, C.F.; Liu, Y.R.; Sun, D.Q.; Jiang, L.; Tang, L.J.; Zhu, P.W.; Chen, S.D.; Liu, W.Y.; Wang, X.D.; et al. Secondary bile acids improve risk prediction for non-invasive identification of mild liver fibrosis in nonalcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2023, 57, 872–885. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kaplan, D.E.; Mehta, R.; Garcia-Tsao, G.; Albrecht, J.; Aytaman, A.; Baffy, G.; Bajaj, J.; Hernaez, R.; Hunt, K.; Ioannou, G.; et al. SACRED: Effect of simvastatin on hepatic decompensation and death in subjects with high-risk compensated cirrhosis: Statins and Cirrhosis: Reducing Events of Decompensation. Contemp. Clin. Trials. 2021, 104, 106367. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schwabl, P.; Payer, B.A.; Grahovac, J.; Klein, S.; Horvatits, T.; Mitterhauser, M.; Stift, J.; Boucher, Y.; Trebicka, J.; Trauner, M.; et al. Pioglitazone decreases portosystemic shunting by modulating inflammation and angiogenesis in cirrhotic and non-cirrhotic portal hypertensive rats. J. Hepatol. 2014, 60, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, G.; Celsa, C.; Enea, M.; Vaccaro, M.; Di Marco, V.; Ciccioli, C.; Infantino, G.; La Mantia, C.; Parisi, S.; Vernuccio, F.; et al. Effect of pharmacological interventions and placebo on liver Histology in nonalcoholic steatohepatitis: A network meta-analysis. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vilarrupla, A.; Laviña, B.; García-Calderó, H.; Russo, L.; Rosado, E.; Roglans, N.; Bosch, J.; García-Pagán, J.C. PPARα activation improves endothelial dysfunction and reduces fibrosis and portal pressure in cirrhotic rats. J. Hepatol. 2012, 56, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Miyauchi, T.; Sakai, S.; Irukayama-Tomobe, Y.; Goto, K.; Yamaguchi, I. Stimulation of peroxisome-proliferator-activated receptor alpha (PPAR alpha) attenuates cardiac fibrosis and endothelin-1 production in pressure-overloaded rat hearts. Clin. Sci. 2002, 103 (Suppl. S48), 284S–288S. [Google Scholar] [CrossRef] [PubMed]
- Faccia, M.; Ainora, M.E.; Ponziani, F.R.; Riccardi, L.; Garcovich, M.; Gasbarrini, A.; Pompili, M.; Zocco, M.A. Portal vein thrombosis in cirrhosis: Why a well-known complication is still matter of debate. World J. Gastroenterol. 2019, 25, 4437–4451. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Faccia, M.; Santopaolo, F.; Gasbarrini, A.; Pompili, M.; Zocco, M.A.; Ponziani, F.R. Risk factors for portal vein thrombosis or venous thromboembolism in a large cohort of hospitalized cirrhotic patients. Intern. Emerg. Med. 2022, 17, 1327–1334. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, J.; Dang, X.; Zhang, L.; Li, W. A pilot study of safety and efficacy comparison of low molecular heparin calcium sequential oral anticoagulants in the treatment of cirrhotic portal vein thrombosis. Eur. J. Gastroenterol. Hepatol. 2024, 36, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Semmler, G.; Pomej, K.; Bauer, D.J.M.; Balcar, L.; Simbrunner, B.; Binter, T.; Hartl, L.; Becker, J.; Pinter, M.; Quehenberger, P.; et al. Safety of direct oral anticoagulants in patients with advanced liver disease. Liver Int. 2021, 41, 2159–2170. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- De Maria, C.; Galante, A.; Fasoli, A.; De Gottardi, A. When and how to use direct oral anticoagulants in patients with advanced chronic liver disease? Curr. Opin. Pharmacol. 2021, 60, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Eichholz, J.C.; Wedemeyer, H.; Maasoumy, B. The Challenge of Anticoagulation in Liver Cirrhosis. Visc. Med. 2024, 39, 169–176. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pasta, A.; Calabrese, F.; Labanca, S.; Marenco, S.; Pieri, G.; Plaz Torres, M.C.; Intagliata, N.M.; Caldwell, S.H.; Giannini, E.G. Safety and efficacy of venous thromboembolism prophylaxis in patients with cirrhosis: A systematic review and meta-analysis. Liver Int. 2023, 43, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Cammà, C.; Marietta, M.; Luongo, M.; Critelli, R.; Colopi, S.; Tata, C.; Zecchini, R.; Gitto, S.; Petta, S.; et al. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology 2012, 143, 1253–1260.e4. [Google Scholar] [CrossRef] [PubMed]
- Cerini, F.; Vilaseca, M.; Lafoz, E.; García-Irigoyen, O.; García-Calderó, H.; Tripathi, D.M.; Avila, M.; Reverter, J.C.; Bosch, J.; Gracia-Sancho, J.; et al. Enoxaparin reduces hepatic vascular resistance and portal pressure in cirrhotic rats. J. Hepatol. 2016, 64, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Yagüe, S.; Alvarez, A.V.; Castilla, A.; González, P.F.R.; Llamas, P.; Caramelo, C. Modulation of the effect of vascular endothelial growth factor on endothelial cells by heparin: Critical role of nitric oxide-mediated mechanisms. J. Nephrol. 2005, 18, 234–242. [Google Scholar] [PubMed]
- Chandrabalan, A.; Ramachandran, R. Molecular mechanisms regulating Proteinase-Activated Receptors (PARs). FEBS J. 2021, 288, 2697–2726. [Google Scholar] [CrossRef] [PubMed]
- Fortea, J.I.; Zipprich, A.; Fernandez-Mena, C.; Puerto, M.; Bosoi, C.; Almagro, J.; Hollenbach, M.; Bañares, J.; Rodríguez-Sánchez, B.; Cercenado, E.; et al. Enoxaparin does not ameliorate liver fibrosis or portal hypertension in rats with advanced cirrhosis. Liver Int. 2018, 38, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Maurin, N. Heparinresistenz und Antithrombinmangel [Heparin resistance and antithrombin deficiency]. Med. Klin. 2009, 104, 441–449. (In German) [Google Scholar] [CrossRef] [PubMed]
- Vilaseca, M.; García-Calderó, H.; Lafoz, E.; García-Irigoyen, O.; Avila, M.A.; Reverter, J.C.; Bosch, J.; Hernández-Gea, V.; Gracia-Sancho, J.; García-Pagán, J.C. The anticoagulant rivaroxaban lowers portal hypertension in cirrhotic rats mainly by deactivating hepatic stellate cells. Hepatology 2017, 65, 2031–2044. [Google Scholar] [CrossRef] [PubMed]
- Pereira Portela, C.; Gautier, L.A.; Zermatten, M.G.; Fraga, M.; Moradpour, D.; Bertaggia Calderara, D.; Aliotta, A.; Veuthey, L.; De Gottardi, A.; Stirnimann, G.; et al. Direct oral anticoagulants in cirrhosis: Rationale and current evidence. JHEP Rep. 2024, 6, 101116. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Verbeke, L.; Mannaerts, I.; Schierwagen, R.; Govaere, O.; Klein, S.; Vander Elst, I.; Windmolders, P.; Farre, R.; Wenes, M.; Mazzone, M.; et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci. Rep. 2016, 6, 33453. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fuchs, C.D.; Claudel, T.; Mlitz, V.; Riva, A.; Menz, M.; Brusilovskaya, K.; Haller, F.; Baumgartner, M.; Königshofer, P.; Unger, L.W.; et al. GLP-2 Improves Hepatic Inflammation and Fibrosis in Mdr2−/− Mice Via Activation of NR4a1/Nur77 in Hepatic Stellate Cells and Intestinal FXR Signaling. Cell Mol. Gastroenterol. Hepatol. 2023, 16, 847–856. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, X.X.; Xie, C.; Libby, A.E.; Ranjit, S.; Levi, J.; Myakala, K.; Bhasin, K.; Jones, B.A.; Orlicky, D.J.; Takahashi, S.; et al. The role of FXR and TGR5 in reversing and preventing progression of Western diet-induced hepatic steatosis, inflammation, and fibrosis in mice. J. Biol. Chem. 2022, 298, 102530. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ratziu, V.; Harrison, S.A.; Loustaud-Ratti, V.; Bureau, C.; Lawitz, E.; Abdelmalek, M.; Alkhouri, N.; Francque, S.; Girma, H.; Darteil, R.; et al. Hepatic and renal improvements with FXR agonist vonafexor in individuals with suspected fibrotic NASH. J. Hepatol. 2023, 78, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Vuppalanchi, R.; Levy, C.; Floreani, A.; Andreone, P.; LaRusso, N.F.; Shrestha, R.; Trotter, J.; Goldberg, D.; Rushbrook, S.; et al. AESOP Study Investigators. A randomized, placebo-controlled, phase II study of obeticholic acid for primary sclerosing cholangitis. J. Hepatol. 2020, 73, 94–101. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bowlus, C.L.; Pockros, P.J.; Kremer, A.E.; Parés, A.; Forman, L.M.; Drenth, J.P.H.; Ryder, S.D.; Terracciano, L.; Jin, Y.; Liberman, A.; et al. Long-Term Obeticholic Acid Therapy Improves Histological Endpoints in Patients with Primary Biliary Cholangitis. Clin. Gastroenterol. Hepatol. 2020, 18, 1170–1178.e6. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Harrison, S.A.; Elkhashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.E.; Kayali, Z.; Wong, V.W.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a Nonsteroidal FXR Agonist, in Patients with Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology 2020, 72, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Schwabl, P.; Hambruch, E.; Seeland, B.A. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J. Hepatol. 2017, 66, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Schwabl, P.; Hambruch, E.; Budas, G.R.; Supper, P.; Burnet, M.; Liles, J.T.; Birkel, M.; Brusilovskaya, K.; Königshofer, P.; Peck-Radosavljevic, M.; et al. The Non-Steroidal FXR Agonist Cilofexor Improves Portal Hypertension and Reduces Hepatic Fibrosis in a Rat NASH Model. Biomedicines 2021, 9, 60. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Matyas, C.; Haskó, G.; Liaudet, L. Interplay of cardiovascular mediators, oxidative stress and inflammation in liver disease and its complications. Nat. Rev. Cardiol. 2021, 18, 117–135. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nesci, A.; Ruggieri, V.; Manilla, V.; Spinelli, I.; Santoro, L.; Di Giorgio, A.; Santoliquido, A.; Ponziani, F.R. Endothelial Dysfunction and Liver Cirrhosis: Unraveling of a Complex Relationship. Int. J. Mol. Sci. 2024, 25, 12859. https://doi.org/10.3390/ijms252312859
Nesci A, Ruggieri V, Manilla V, Spinelli I, Santoro L, Di Giorgio A, Santoliquido A, Ponziani FR. Endothelial Dysfunction and Liver Cirrhosis: Unraveling of a Complex Relationship. International Journal of Molecular Sciences. 2024; 25(23):12859. https://doi.org/10.3390/ijms252312859
Chicago/Turabian StyleNesci, Antonio, Vittorio Ruggieri, Vittoria Manilla, Irene Spinelli, Luca Santoro, Angela Di Giorgio, Angelo Santoliquido, and Francesca Romana Ponziani. 2024. "Endothelial Dysfunction and Liver Cirrhosis: Unraveling of a Complex Relationship" International Journal of Molecular Sciences 25, no. 23: 12859. https://doi.org/10.3390/ijms252312859
APA StyleNesci, A., Ruggieri, V., Manilla, V., Spinelli, I., Santoro, L., Di Giorgio, A., Santoliquido, A., & Ponziani, F. R. (2024). Endothelial Dysfunction and Liver Cirrhosis: Unraveling of a Complex Relationship. International Journal of Molecular Sciences, 25(23), 12859. https://doi.org/10.3390/ijms252312859