
Innovative Insights into Single-Cell Technologies and Multi-Omics Integration in Livestock and Poultry

 ,
,
Abstract
:1. Introduction
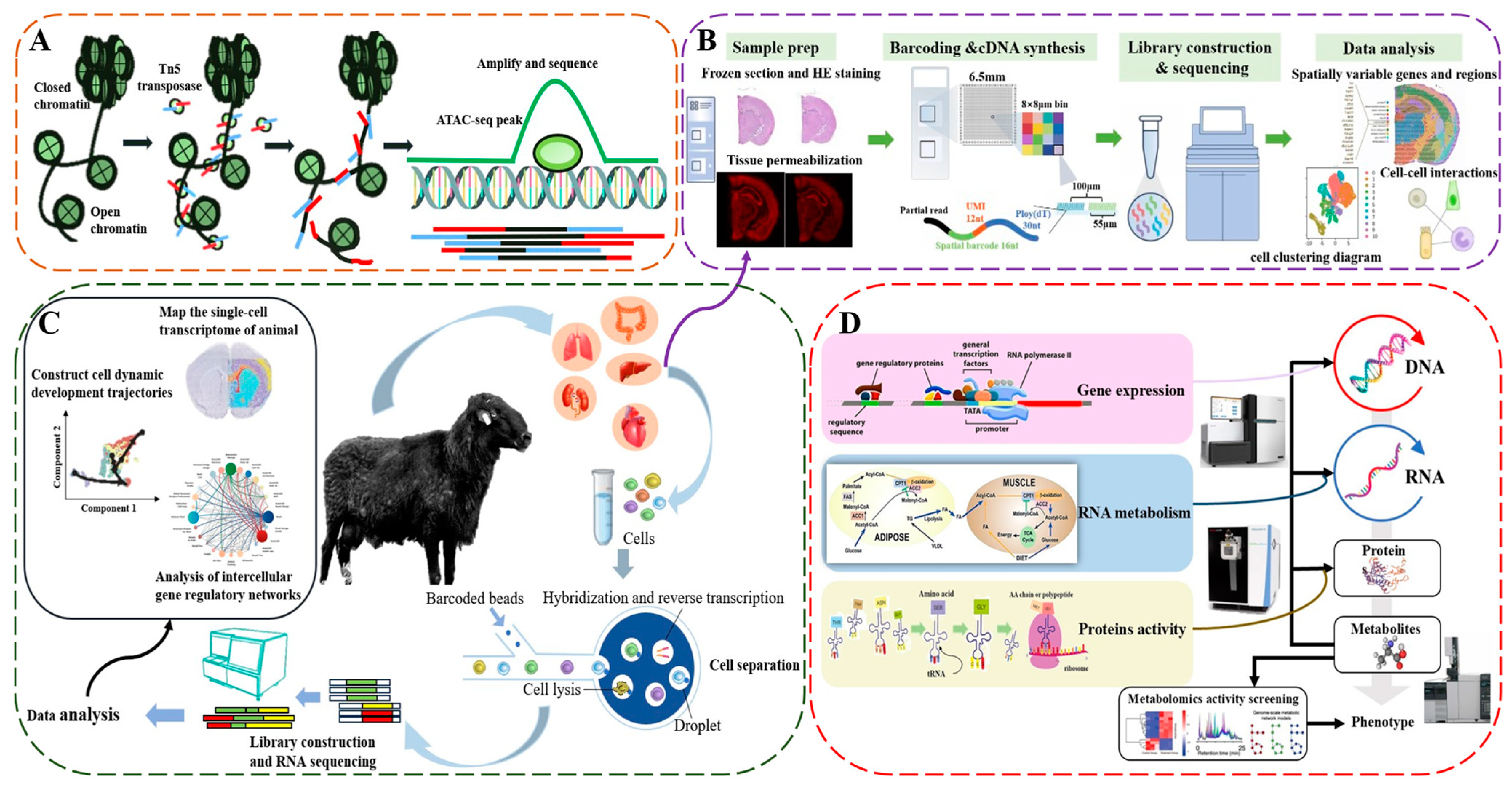
2. Fundamental Principles of scRNA-seq

{kind=link}
{kind=link}
{kind=link}
| Methods | Coverage Area | Read Depth | UMI | Amplification Method | Strand-Specific | Characteristics | Reference |
|---|---|---|---|---|---|---|---|
| Tang 2009 | Full length | 104–105 | No | Homopolymer tailing | No | High sensitivity and comprehensive transcriptome coverage | [46] |
| Strt-seq | 5′ end | 104–105 | Yes | Template switching | Yes | Captures complete transcript data, suitable for gene fusion studies | [47] |
| Smart-seq | Full length | 106 | No | Homopolymer tailing | No | High sensitivity, captures low-abundance transcripts | [48] |
| Drop-seq | 3′ end | 104–105 | Yes | In vitro transcription | Yes | High throughput, cost-effective for large sample sizes | [49] |
| Cel-seq | 3′ end | 104–105 | Yes | In vitro transcription | Yes | High sensitivity, low bias in expression quantification | [39] |
| Mars-seq | 3′ end | 104–105 | Yes | In vitro transcription | Yes | High throughput with robust data coverage | [50] |
| Cyto-seq | 3′ end | 103–104 | Yes | Designed primers | No | Ideal for cell surface labeling and functional studies | [51] |
| 10× Genomics | 3′ end | 104–105 | Yes | Template switching | Yes | Sensitive detection of low-abundance genes in heterogeneous cells | [43] |
| Scrb-seq | 3′ end | 104–105 | Yes | In vitro transcription | Yes | Suitable for studying cellular heterogeneity and dynamics | [52] |
3. Applications of scRNA-seq in Livestock and Poultry
3.1. Applications of scRNA-seq in Poultry
3.2. Application of scRNA-seq in Pigs
3.3. Application of scRNA-seq in Ruminants
4. Application of Multi-Omics Integration with scRNA-seq in Livestock and Poultry
4.1. Integrated Analysis of Single-Cell Transcriptomics and Epigenomics
4.2. Integrative Analysis of Single-Cell and Bulk RNA-seq
4.3. Integrated Analysis of Single-Cell Transcriptomics and Proteomics
4.4. Integrated Analysis of Single-Cell Transcriptomics and Spatial Transcriptomics
4.5. Integrated Analysis of Single-Cell Transcriptomics and Multi-Omics
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rowbury, R. Robert Hooke, 1635–1703. Sci. Prog. 2012, 95, 238–254. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, J.W.; Conchello, J.A. Fluorescence Microscopy. Nat. Methods 2005, 2, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Combs, C.A.; Shroff, H. Fluorescence Microscopy: A Concise Guide to Current Imaging Methods. Curr. Protoc. Neurosci. 2017, 79, 2.1.1–2.1.25. [Google Scholar] [CrossRef] [PubMed]
- Crosland-Taylor, P. A Device for Counting Small Particles Suspended in a Fluid through a Tube. Nature 1953, 171, 37–38. [Google Scholar] [CrossRef]
- Moldavan, A. A Modified Technic for the Detection of the Escherichia-Aerobacter Group in Milk. Am. J. Public Health Nations Health 1935, 25, 1032–1033. [Google Scholar] [CrossRef]
- Steinkamp, J.A.; Romero, A.; Dilla, M.A.V. Multiparameter Cell Sorting: Identification of Human Leukocytes by Acridine Orange Fluorescence. Acta Cytol. 1973, 17, 113–117. [Google Scholar]
- Li, R.; Gong, M.; Zhang, X.; Wang, F.; Liu, Z.; Zhang, L.; Yang, Q.; Xu, Y.; Xu, M.; Zhang, H.; et al. A Sheep Pangenome Reveals the Spectrum of Structural Variations and Their Effects on Tail Phenotypes. Genome Res. 2023, 33, 463–477. [Google Scholar] [CrossRef]
- Wang, M.; Ibeagha-Awemu, E.M. Impacts of Epigenetic Processes on the Health and Productivity of Livestock. Front. Genet. 2021, 11, 613636. [Google Scholar] [CrossRef]
- Sun, L.; Bai, M.; Xiang, L.; Zhang, G.; Ma, W.; Jiang, H. Comparative Transcriptome Profiling of Longissimus Muscle Tissues from Qianhua Mutton Merino and Small Tail Han Sheep. Sci. Rep. 2016, 6, 33586. [Google Scholar] [CrossRef]
- Ren, W.B.; Hou, X.Y.; Wang, Y.Q.; Badgery, W.; Li, X.; Ding, Y.; Guo, H.; Wu, Z.; Hu, N.N.; Kong, L.Q.; et al. Overgrazing Induces Alterations in the Hepatic Proteome of Sheep (Ovis Aries): An iTRAQ-Based Quantitative Proteomic Analysis. Proteome Sci. 2017, 15, 2. [Google Scholar] [CrossRef]
- Evans, H.C.; Dinh, T.T.N.; Ugur, M.R.; Hitit, M.; Sajeev, D.; Kaya, A.; Topper, E.; Nicodemus, M.C.; Smith, G.D.; Memili, E. Lipidomic Markers of Sperm Cryotolerance in Cattle. Sci. Rep. 2020, 10, 20192. [Google Scholar] [CrossRef] [PubMed]
- De Klerk, E.; ‘t Hoen, P.A. Alternative mRNA Transcription, Processing, and Translation: Insights from RNA Sequencing. Trends Genet. 2015, 31, 128–139. [Google Scholar] [CrossRef]
- Wang, J.; Gao, M.; Cheng, M.; Luo, J.; Lu, M.; Xing, X.; Sun, Y.; Lu, Y.; Li, X.; Shi, C.; et al. Single-Cell Transcriptional Analysis of Lamina Propria Lymphocytes in the Jejunum Reveals Innate Lymphoid Cell-like Cells in Pigs. J. Immunol. 2024, 212, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, H.; Li, C.; Yang, B.; Feng, X.; Cao, J.; Du, W.; Shahzad, M.; Khan, A.; Sun, S.C.; et al. Effects of Regulating Hippo and Wnt on the Development and Fate Differentiation of Bovine Embryo. Int. J. Mol. Sci. 2024, 25, 3912. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.E.; Klein, A.M. Lineage Tracing Meets Single-Cell Omics: Opportunities and Challenges. Nat. Rev. Genet. 2020, 21, 410–427. [Google Scholar] [CrossRef]
- Yuan, G.C.; Cai, L.; Elowitz, M.; Enver, T.; Fan, G. Challenges and Emerging Directions in Single-Cell Analysis. Genome Biol. 2017, 18, 84. [Google Scholar] [CrossRef]
- Pearson, H. Genetics: What Is a Gene? Nature 2006, 441, 398–401. [Google Scholar] [CrossRef]
- Scherrer, K. Is RNA the Working Genome in Eukaryotes? The 60 Year Evolution of a Conceptual Challenge. Exp. Cell Res. 2023, 424, 113493. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Regev, A.; Teichmann, S.A.; Lander, E.S.; Amit, I.; Human Cell Atlas Meeting Participants. Science Forum: The Human Cell Atlas. eLife 2017, 6, e27041. [Google Scholar] [CrossRef] [PubMed]
- Qiu, K.; Xu, D.D.; Wang, L.Q.; Zhang, X.; Jiao, N.; Gong, L.; Yin, J.D. Association Analysis of Single-Cell Rna Sequencing and Proteomics Reveals a Vital Role of Ca2+ Signaling in the Determination of Skeletal Muscle Development Potential. Cells 2020, 9, 1045. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Zhu, S.; Gu, F.; Valencak, T.G.; Liu, J.X.; Sun, H.Z. Cross-Tissue Single-Cell Transcriptomic Landscape Reveals the Key Cell Subtypes and Their Potential Roles in the Nutrient Absorption and Metabolism in Dairy Cattle. J. Adv. Res. 2021, 37, 1–18. [Google Scholar] [CrossRef]
- Yang, H.; Ma, J.; Wan, Z.; Wang, Q.; Wang, Z.; Zhao, J.; Wang, F.; Zhang, Y. Characterization of Sheep Spermatogenesis through Single-Cell RNA Sequencing. FASEB J. 2020, 35, e21187. [Google Scholar] [CrossRef] [PubMed]
- Estermann, M.A.; Williams, S.; Hirst, C.E.; Roly, Z.Y.; Serralbo, O.; Adhikari, D.; Powell, D.; Major, A.T.; Smith, C.A. Insights into Gonadal Sex Differentiation Provided by Single-Cell Transcriptomics in the Chicken Embryo. Cell Rep. 2020, 31, 107491. [Google Scholar] [CrossRef]
- Du, X.; Lai, S.; Zhao, W.; Xu, X.; Xu, W.; Zeng, T.; Tian, Y.; Lu, L. Single-Cell RNA Sequencing Revealed the Liver Heterogeneity between Egg-Laying Duck and Ceased-Laying Duck. BMC Genom. 2022, 23, 857. [Google Scholar] [CrossRef]
- Zhou, B.Y.; Wang, L. Reading the Heart at Single-Cell Resolution. J. Mol. Cell. Cardiol. 2020, 148, 34–45. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-Cell RNA Sequencing Technologies and Bioinformatics Pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Slovin, S.; Carissimo, A.; Panariello, F.; Grimaldi, A.; Bouché, V.; Gambardella, G.; Cacchiarelli, D. Single-Cell Rna Sequencing Analysis: A Step-by-Step Overview. In RNA Bioinformatics; Methods in Molecular Biology; Humana: New York, NY, USA, 2021; Volume 2284, pp. 343–365. [Google Scholar] [CrossRef]
- Yokoyama, W.M.; Christensen, M.; Santos, G.D.; Miller, D.; Ho, J.; Wu, T.; Dziegelewski, M.; Neethling, F.A. Production of Monoclonal Antibodies. Curr. Protoc. Immunol. 2013, 102, 2.5.1–2.5.29. [Google Scholar] [CrossRef]
- Antoniadi, I.; Skalick, V.; Sun, G.; Ma, W.; Galbraith, D.W.; Novák, O.; Ljung, K. Fluorescence Activated Cell Sorting—A Selective Tool for Plant Cell Isolation and Analysis. Cytom. Part A 2022, 101, 725–736. [Google Scholar] [CrossRef]
- Frhlich, J.; Knig, H. New Techniques for Isolation of Single Prokaryotic Cells. FEMS Microbiol. Rev. 2000, 24, 567–572. [Google Scholar] [CrossRef]
- Lecault, V.; White, A.K.; Singhal, A.; Hansen, C.L. Microfluidic Single Cell Analysis: From Promise to Practice. Curr. Opin. Chem. Biol. 2012, 16, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.L.; Jiang, Y.Z.; Tang, Q.Q.; He, X.D.; Shen, Z.J.; Zhang, B.Y. Detection of Circulating Tumor Cells Using a Novel Immunomagnetic Bead Method in Lung Cancer Patients. J. Clin. Lab. Anal. 2016, 30, 656–662. [Google Scholar] [CrossRef]
- Emmert-Buck, M.R.; Bonner, R.F.; Smith, P.D.; Chuaqui, R.F.; Zhuang, Z.; Goldstein, S.R.; Weiss, R.A.; Liotta, L.A. Laser Capture Microdissection. Science 1996, 274, 998–1001. [Google Scholar] [CrossRef]
- Gross, A.; Schoendube, J.; Zimmermann, S.; Steeb, M.; Zengerle, R.; Koltay, P. Technologies for Single-Cell Isolation. Int. J. Mol. Sci. 2015, 16, 16897–16919. [Google Scholar] [CrossRef] [PubMed]
- Kind, D.; Baskaran, P.; Ramirez, F.; Giner, M.; Hayes, M.; Santacruz, D.; Koss, C.K.; Kasmi, K.C.E.; Wijayawardena, B.; Viollet, C. Automation Enables High-Throughput and Reproducible Single-Cell Transcriptomics Library Preparation. SLAS Technol. 2021, 27, 135–142. [Google Scholar] [CrossRef]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative Analysis of Single-Cell Rna Sequencing Methods. Mol. Cell 2017, 65, 631–643. [Google Scholar] [CrossRef]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-Cell RNA-Seq by Multiplexed Linear Amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef]
- Wang, Y.; Yan, Y.; Thompson, K.N.; Bae, S.; Huttenhower, C. Whole Microbial Community Viability Is Not Quantitatively Reflected by Propidium Monoazide Sequencing Approach. Microbiome 2021, 9, 17. [Google Scholar] [CrossRef]
- Wang, Y.; Navin, N.E. Advances and Applications of Single-Cell Sequencing Technologies. Mol. Cell 2015, 58, 598–609. [Google Scholar] [CrossRef]
- Lee, J.; Hyeon, D.Y.; Hwang, D. Single-Cell Multiomics: Technologies and Data Analysis Methods. Exp. Mol. Med. 2020, 52, 1428–1442. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.S.; Li, L.; Ye, C.; Peng, M.; Zhang, Y. Hybrid Assembly of Ultra-Long Nanopore Reads Augmented with 10x-Genomics Contigs: Demonstrated with a Human Genome. Genomics 2019, 111, 1896–1901. [Google Scholar] [CrossRef] [PubMed]
- Olsen, T.K.; Baryawno, N. Introduction to Single-cell Rna Sequencing. Curr. Protoc. Mol. Biol. 2018, 122, e57. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, E.; Deng, Q. Single-Cell RNA Sequencing: Technical Advancements and Biological Applications. Mol. Asp. Med. 2017, 59, 36–46. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Nordman, E.; Li, B.; Xu, N.; Bashkirov, V.I.; Lao, K.; Surani, M.A. RNA-Seq Analysis to Capture the Transcriptome Landscape of a Single Cell. Nat. Protoc. 2010, 5, 516–535. [Google Scholar] [CrossRef]
- Natarajan, K.N. Single-Cell Tagged Reverse Transcription (Strt-Seq). In Single Cell Methods; Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 1979, pp. 133–153. [Google Scholar] [CrossRef]
- Ramsköld, D.; Luo, S.; Wang, Y.C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C. Full-Length mRNA-Seq from Single-Cell Levels of RNA and Individual Circulating Tumor Cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef]
- Zhang, X.; Li, T.; Liu, F.; Chen, Y.; Yao, J.; Li, Z.; Huang, Y.; Wang, J. Comparative Analysis of Droplet-Based Ultra-High-Throughput Single-Cell RNA-Seq Systems. Mol. Cell 2019, 73, 130–142.e5. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef]
- Fan, H.C.; Fu, G.K.; Fodor, S.P.A. Expression profiling. Combinatorial Labeling of Single Cells for Gene Expression Cytometry. Science 2015, 347, 1258367. [Google Scholar] [CrossRef]
- Ni, J.; Hu, C.; Li, H.; Li, X.; Fu, Q.; Czajkowsky, D.M.; Guo, Y.; Shao, Z. Significant Improvement in Data Quality with Simplified SCRB-Seq. Acta Biochim. Biophys. Sin. 2020, 52, 457–459. [Google Scholar] [CrossRef]
- Blanca, P.S.; Griffiths, J.A.; Carolina, G.; Hiscock, T.W.; Wajid, J.; Calero-Nieto, F.J.; Carla, M.; Ximena, I.S.; Tyser, R.C.V.; Ho, D.L.L.; et al. A Single-Cell Molecular Map of Mouse Gastrulation and Early Organogenesis. Nature 2019, 566, 490–495. [Google Scholar] [CrossRef]
- Davie, K.; Janssens, J.; Koldere, D.; Waegeneer, M.D.; Pech, U.; Kreft, U.; Aibar, S.; Makhzami, S.; Christiaens, V.; González-Blas, C.B.; et al. A Single-Cell Transcriptome Atlas of the Aging Drosophila Brain. Cell 2018, 174, 982–998.e20. [Google Scholar] [CrossRef] [PubMed]
- Cosacak, M.I.; Bhattarai, P.; Reinhardt, S.; Petzold, A.; Dahl, A.; Zhang, Y.; Kizil, C. Single-Cell Transcriptomics Analyses of Neural Stem Cell Heterogeneity and Contextual Plasticity in a Zebrafish Brain Model of Amyloid Toxicity. Cell Rep. 2019, 27, 1307–1318.e3. [Google Scholar] [CrossRef] [PubMed]
- Mottet, A.; Tempio, G. Global Poultry Production: Current State and Future Outlook and Challenges. World’s Poult. Sci. J. 2016, 73, 245–256. [Google Scholar] [CrossRef]
- Korver, D.R. Review: Current Challenges in Poultry Nutrition, Health, and Welfare. Animal 2023, 17, 100755. [Google Scholar] [CrossRef]
- Capel, B. Vertebrate Sex Determination: Evolutionary Plasticity of a Fundamental Switch. Nat. Rev. Genet. 2017, 18, 675–689. [Google Scholar] [CrossRef]
- Defalco, T.; Capel, B. Gonad Morphogenesis in Vertebrates: Divergent Means to a Convergent End. Annu. Rev. Cell Dev. Biol. 2009, 25, 457–482. [Google Scholar] [CrossRef]
- Feregrino, C.; Sacher, F.; Parnas, O.; Tschopp, P. A Single-Cell Transcriptomic Atlas of the Developing Chicken Limb. BMC Genom. 2019, 20, 401. [Google Scholar] [CrossRef]
- Yeboah, R.L.; Pira, C.U.; Shankel, M.; Cooper, A.M.; Haro, E.; Ly, V.D.; Wysong, K.; Zhang, M.; Sandoval, N.; Oberg, K.C. Sox, Fox, and Lmx1b Binding Sites Differentially Regulate a Gdf5-Associated Regulatory Region during Elbow Development. Front. Cell Dev. Biol. 2023, 11, 1215406. [Google Scholar] [CrossRef]
- Li, J.; Xing, S.; Zhao, G.; Zheng, M.; Yang, X.; Sun, J.; Wen, J.; Liu, R. Identification of Diverse Cell Populations in Skeletal Muscles and Biomarkers for Intramuscular Fat of Chicken by Single-Cell RNA Sequencing. BMC Genom. 2020, 21, 752. [Google Scholar] [CrossRef]
- Thiery, A.P.; Buzzi, A.L.; Hamrud, E.; Cheshire, C.; Luscombe, N.M.; Briscoe, J.; Streit, A. scRNA-Sequencing in Chick Suggests a Probabilistic Model for Cell Fate Allocation at the Neural Plate Border. eLife 2023, 12, e82717. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Jensen, J.D. Sustainability Implications of Rising Global Pork Demand. Anim. Front. 2022, 12, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.D.; Wan, B.Y.; Qiu, K.; Wang, Y.B.; Zhang, X.; Jiao, N.; Yan, E.F.; Wu, J.W.; Yu, R.; Gao, S.; et al. Single-Cell Rna-Sequencing Provides Insight into Skeletal Muscle Evolution during the Selection of Muscle Characteristics. Adv. Sci. 2023, 10, e2305080. [Google Scholar] [CrossRef]
- Ma, L.; Meng, Y.; An, Y.; Han, P.; Zhang, C.; Yue, Y.; Wen, C.; Shi, X.E.; Jin, J.; Yang, G.; et al. Single-cell RNA-seqreveals Novel Interaction between Muscle Satellite Cells and Fibro-adipogenic Progenitors Mediated with FGF7 Signalling. J. Cachexia Sarcopenia Muscle 2024, 15, 1388–1403. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhang, Y.; Tian, Y.; Wu, S.; Gao, F.; Yuan, X. Deciphering Cellular Heterogeneity and Communication Patterns in Porcine Antral Follicles by Single-Cell Rna Sequencing. Animals 2023, 13, 3019. [Google Scholar] [CrossRef]
- Han, P.P.; Guo, Y.P.; Zhang, W.; Wang, D.Y.; Wu, Y.L.; Li, X.Y.; Zhu, M.J. Single-Cell RNA-Sequencing Reveals Heterogeneity and Transcriptional Dynamics in Porcine Circulating CD8+ T Cells. Cells 2024, 13, 692. [Google Scholar] [CrossRef]
- Li, J.B.; Wang, L.G.; Yu, D.W.; Hao, J.F.; Zhang, Y.P.; Zhang, L.C.; Adeola, A.C.; Mao, B.Y.; Gao, Y.; Wu, S.F.; et al. Single-Cell RNA-Sequencing Reveals Thoracolumbar Vertebra Heterogeneity and Rib-Genesis in Pigs. Genom. Proteom. Bioinform. 2021, 19, 423–436. [Google Scholar] [CrossRef]
- Wang, F.; Ding, P.; Liang, X.; Ding, X.; Brandt, C.B.; Sjstedt, E.; Zhu, J.; Bolund, S.; Zhang, L.; Rooij, L.P.M.H. Endothelial Cell Heterogeneity and Microglia Regulons Revealed by a Pig Cell Landscape at Single-Cell Level. Nat. Commun. 2022, 13, 3620. [Google Scholar] [CrossRef]
- Peng, Y.; Qiao, H. The Application of Single-Cell RNA Sequencing in Mammalian Meiosis Studies. Front. Cell Dev. Biol. 2021, 9, 673642. [Google Scholar] [CrossRef]
- Latorraca, L.B.; Galvão, A.; Rabaglino, M.B.; D’Augero, J.M.; Kelsey, G.; Fair, T. Single-Cell Profiling Reveals Transcriptome Dynamics during Bovine Oocyte Growth. BMC Genom. 2024, 25, 335. [Google Scholar] [CrossRef]
- Ge, T.; Wen, Y.; Li, B.; Huang, X.; Jiang, S.; Zhang, E. Singlecell Sequencing Reveals the Reproductive Variations between Primiparous and Multiparous Hu Ewes. J. Anim. Sci. Biotechnol. 2023, 14, 144. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, J.; Zhang, P.; Huang, X.; Yang, X.; Liu, R.; Sun, Q.; Lu, Y.; Zhang, M.; Fu, Q. Single-Cell RNA Sequencing Uncovers Dynamic Roadmap and Cell-Cell Communication during Buffalo Spermatogenesis. iScience 2022, 26, 105733. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pei, J.; Xiong, L.; Guo, S.; Cao, M.; Kang, Y.; Ding, Z.; La, Y.; Liang, C.; Yan, P.; et al. Single-Cell Rna Sequencing Reveals Atlas of Yak Testis Cells. Int. J. Mol. Sci. 2023, 24, 7982. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.W.; Li, T.T.; Du, X.M.; Shen, Q.Y.; Zhang, M.F.; Wei, Y.D.; Yang, D.H.; Xu, W.J.; Chen, W.B.; Bai, C.L. Single-Cell RNA Sequencing Reveals Atlas of Dairy Goat Testis Cells. Zool. Res. 2021, 42, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Zhong, L.; Zhang, S.; Mu, H.; Xiang, J.; Yue, L.; Dai, Y.; Han, J. Bovine Lineage Specification Revealed by Single-Cell Gene Expression Analysis from Zygote to Blastocyst. Biol. Reprod. 2017, 97, 5–17. [Google Scholar] [CrossRef]
- Zhao, L.; Long, C.; Zhao, G.; Su, J.; Ren, J.; Sun, W.; Wang, Z.; Zhang, J.; Liu, M.; Hao, C.; et al. Reprogramming Barriers in Bovine Cells Nuclear Transfer Revealed by Single-Cell RNA-Seq Analysis. J. Cell. Mol. Med. 2022, 26, 4792–4804. [Google Scholar] [CrossRef]
- Lyu, P.; Qi, Y.; Tu, Z.J.; Jiang, H. Single-Cell Rna Sequencing Reveals Heterogeneity of Cultured Bovine Satellite Cells. Front. Genet. 2021, 12, 742077. [Google Scholar] [CrossRef]
- Potter, S.S. Single-Cell RNA Sequencing for the Study of Development, Physiology and Disease. Nat. Rev. Nephrol. 2018, 14, 479–492. [Google Scholar] [CrossRef]
- Vandereyken, K.; Sifrim, A.; Thienpont, B.; Voet, T. Methods and Applications for Single-Cell and Spatial Multi-Omics. Nat. Rev. Genet. 2023, 24, 494–515. [Google Scholar] [CrossRef]
- Yan, Y.; Zhu, S.; Jia, M.; Chen, X.; Qi, W.; Gu, F.; Valencak, T.G.; Liu, J.X.; Sun, H.Z. Advances in Single-Cell Transcriptomics in Animal Research. J. Anim. Sci. Biotechnol. 2024, 15, 102. [Google Scholar] [CrossRef]
- Ku, C.S.; Naidoo, N.; Wu, M.; Soong, R. Studying the Epigenome Using next Generation Sequencing. J. Med. Genet. 2011, 48, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Moshitch-Moshkovitz, S.; Dominissini, D.; Rechavi, G. The Epitranscriptome Toolbox. Cell 2022, 185, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Marjani, S.L.; Jiang, Z. The Epigenetics of Gametes and Early Embryos and Potential Long-Range Consequences in Livestock Species—Filling in the Picture With Epigenomic Analyses. Front. Genet. 2021, 12, 557934. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of Native Chromatin for Fast and Sensitive Epigenomic Profiling of Open Chromatin, DNA-Binding Proteins and Nucleosome Position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-Seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21.29.1–21.29.9. [Google Scholar] [CrossRef]
- Reznikoff, W. The Tn5 Transposon. Annu. Rev. Microbiol. 1993, 47, 945–963. [Google Scholar] [CrossRef]
- Cai, C.; Wan, P.; Wang, H.; Cai, X.; Wang, J.; Chai, Z.; Wang, J.; Wang, H.; Zhang, M.; Yang, N. Transcriptional and Open Chromatin Analysis of Bovine Skeletal Muscle Development by Single-Cell Sequencing. Cell Prolif. 2023, 56, e13430. [Google Scholar] [CrossRef]
- Li, J.; Zhang, X.; Wang, X.; Wang, Z.; Li, X.; Zheng, J.; Li, J.; Xu, G.; Sun, C.; Yi, G.; et al. Single-Nucleus Transcriptional and Chromatin Accessible Profiles Reveal Critical Cell Types and Molecular Architecture Underlying Chicken Sex Determination. J. Adv. Res. 2024, S2090-1232, 00185-1. [Google Scholar] [CrossRef]
- Hino, S.; Sato, T.; Nakao, M. Chromatin Immunoprecipitation Sequencing (ChIP-Seq) for Detecting Histone Modifications and Modifiers. In Epigenomics; Methods in Molecular Biology; Humana: New York, NY, USA, 2023; Volume 2577, pp. 55–64. [Google Scholar] [CrossRef]
- Nakato, R.; Sakata, T. Methods for ChIP-Seq Analysis: A Practical Workflow and Advanced Applications. Methods 2021, 187, 44–53. [Google Scholar] [CrossRef]
- Park, P.J. ChIP-Seq: Advantages and Challenges of a Maturing Technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef]
- Koay, T.W.; Osterhof, C.; Orlando, I.M.C.; Keppner, A.; Andre, D.; Yousefian, S.; Alonso, M.S.; Correia, M.; Markworth, R.; Schödel, J.; et al. Androglobin Gene Expression Patterns and FOXJ1-Dependent Regulation Indicate Its Functional Association with Ciliogenesis. J. Biol. Chem. 2021, 296, 100291. [Google Scholar] [CrossRef]
- Li, D.; Ning, C.; Zhang, J.; Wang, Y.; Tang, Q.; Kui, H.; Wang, T.; He, M.; Jin, L.; Li, J.; et al. Dynamic Transcriptome and Chromatin Architecture in Granulosa Cells during Chicken Folliculogenesis. Nat. Commun. 2022, 13, 131. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.C. From Bulk, Single-Cell to Spatial RNA Sequencing. Int. J. Oral Sci. 2022, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Bogdanoff, D.; Wang, Y.; Hartoularos, G.C.; Woo, J.M.; Mowery, C.T.; Nisonoff, H.M.; Lee, D.S.; Sun, Y.; Lee, J.; et al. XYZeq: Spatially Resolved Single-Cell RNA Sequencing Reveals Expression Heterogeneity in the Tumor Microenvironment. Sci. Adv. 2021, 7, eabg4755. [Google Scholar] [CrossRef]
- Zhang, L.; Li, F.; Lei, P.; Guo, M.; Liu, R.; Wang, L.; Yu, T.; Lv, Y.; Zhang, T.; Zeng, W.; et al. Single-Cell RNA-Sequencing Reveals the Dynamic Process and Novel Markers in Porcine Spermatogenesis. J. Anim. Sci. Biotechnol. 2021, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Cai, J.; Rong, L.; Yang, S.; Li, S. Transcriptome Identification of Genes Associated with Uterus–Vagina Junction Epithelial Folds Formation in Chicken Hens. Poult. Sci. 2023, 102, 102624. [Google Scholar] [CrossRef]
- Hinkel, R.; Ramanujam, D.; Kaczmarek, V.; Howe, A.; Klett, K.; Beck, C.; Dueck, A.; Thum, T.; Laugwitz, K.L.; Maegdefessel, L.; et al. Antimir-21 Prevents Myocardial Dysfunction in a Pig Model of Ischemia/Reperfusion Injury. J. Am. Coll. Cardiol. 2020, 75, 1788–1800. [Google Scholar] [CrossRef]
- Calió, M.; Gantenbein, B.; Egli, M.; Poveda, L.; Ille, F. The Cellular Composition of Bovine Coccygeal Intervertebral Discs: A Comprehensive Single-Cell Rnaseq Analysis. Int. J. Mol. Sci. 2021, 22, 4917. [Google Scholar] [CrossRef]
- Bonatto, D.; Revers, L.F.; Brendel, M.; Denoya, C.D. The Eukaryotic Pso2/Snm1/Artemis Proteins and Their Function as Genomic and Cellular Caretakers. Braz. J. Med. Biol. Res. 2005, 38, 321–334. [Google Scholar] [CrossRef]
- Wang, X.; He, C. Dynamic RNA Modifications in Posttranscriptional Regulation. Mol. Cell 2014, 56, 5–12. [Google Scholar] [CrossRef]
- Lorenz, M.; Lawson, F.; Owens, D.; Raccah, D.; Roy-Duval, L.; Lehmann, A.; Perfetti, R.; Blonde, L. Differential Effects of Glucagon-like Peptide-1 Receptor Agonists on Heart Rate. Cardiovasc. Diabetol. 2017, 16, 6. [Google Scholar] [CrossRef]
- Zhu, Y.; Piehowski, P.D.; Zhao, R.; Chen, J.; Shen, Y.; Moore, R.J.; Shukla, A.K.; Petyuk, V.A.; Campbell-Thompson, M.; Mathews, C.E. Nanodroplet Processing Platform for Deep and Quantitative Proteome Profiling of 10–100 Mammalian Cells. Nat. Commun. 2018, 9, 882. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Scheibinger, M.; Ellwanger, D.C.; Krey, J.F.; Choi, D.; Kelly, R.T.; Heller, S.; Barr-Gillespie, P.G. Single-Cell Proteomics Reveals Changes in Expression during Hair-Cell Development. eLife 2019, 8, e50777. [Google Scholar] [CrossRef] [PubMed]
- Vickovic, S.; Eraslan, G.; Salmén, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Äijö, T.; Bonneau, R.; Bergenstråhle, L.; Navarro, J.F. High-Definition Spatial Transcriptomics for in Situ Tissue Profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Salmén, F.; Ståhl, P.L.; Mollbrink, A.; Navarro, J.F.; Vickovic, S.; Frisén, J.; Lundeberg, J. Barcoded Solid-Phase RNA Capture for Spatial Transcriptomics Profiling in Mammalian Tissue Sections. Nat. Protoc. 2018, 13, 2501–2534. [Google Scholar] [CrossRef] [PubMed]
- Kerzel, T.; Beretta, S.; Naldini, L.; Squadrito, M.L. VisualZoneR: A Computational Protocol to Identify Compartmental Zones from Single Cell Spatial Transcriptomics Using R. STAR Protoc. 2024, 5, 103196. [Google Scholar] [CrossRef]
- Moffitt, J.R.; Zhuang, X. RNA Imaging with Multiplexed Error-Robust Fluorescence In Situ Hybridization (MERFISH). In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 572, pp. 1–49. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, Y.; Ni, G.; Li, S.; Balderson, B.; Zou, Q.; Liu, H.; Jiang, Y.; Sun, J.; Ding, X. Integrating Single-cell and Spatial Transcriptomics Reveals Heterogeneity of Early Pig Skin Development and a Subpopulation with Hair Placode Formation. Adv. Sci. 2024, 11, e2306703. [Google Scholar] [CrossRef]
- Cai, S.F.; Hu, B.; Wang, X.Y.; Liu, T.N.; Lin, Z.H.; Tong, X.; Xu, R.; Chen, M.L.; Duo, T.Q.; Zhu, Q.; et al. Integrative Single-Cell RNA-Seq and ATAC-Seq Analysis of Myogenic Differentiation in Pig. BMC Biol. 2023, 21, 19. [Google Scholar] [CrossRef]
- Mantri, M.; Scuderi, G.J.; Abedini-Nassab, R.; Wang, M.F.Z.; McKellar, D.; Shi, H.; Grodner, B.; Butcher, J.T.; Vlaminck, I.D. Spatiotemporal Single-Cell RNA Sequencing of Developing Chicken Hearts Identifies Interplay between Cellular Differentiation and Morphogenesis. Nat. Commun. 2021, 12, 1771. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Peng, R.; Liang, J.; Jahejo, A.R.; Zhang, L.; Lan, M.; Liu, X.; Ran, M.; Yang, X.; Lu, Y. Spatiotemporal Single-Cell RNA Sequencing Reveals the Role of Steroid Hormone Pathway during Chicken Primordial Follicle Formation. Poult. Sci. 2024, 103, 104090. [Google Scholar] [CrossRef]
- Tolonen, A.C.; Xavier, R.J. Dissecting the Human Microbiome with Single-Cell Genomics. Genome Med. 2017, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.J.; Lukowski, J.K.; Anderton, C.R. Spatially Resolved Mass Spectrometry at the Single Cell: Recent Innovations in Proteomics and Metabolomics. J. Am. Soc. Mass Spectrom. 2021, 32, 872–894. [Google Scholar] [CrossRef]
- Han, B.; Tian, D.; Li, X.; Liu, S.; Tian, F.; Liu, D.; Wang, S.; Zhao, K. Multiomics Analyses Provide New Insight into Genetic Variation of Reproductive Adaptability in Tibetan Sheep. Mol. Biol. Evol. 2024, 41, msae058. [Google Scholar] [CrossRef]
- Gao, X.; Wang, S.; Wang, Y.F.; Li, S.; Wu, S.X.; Yan, R.G.; Zhang, Y.W.; Wan, R.D.; He, Z.; Song, R.D.; et al. Long Read Genome Assemblies Complemented by Single Cell RNA-Sequencing Reveal Genetic and Cellular Mechanisms Underlying the Adaptive Evolution of Yak. Nat. Commun. 2022, 13, 4887. [Google Scholar] [CrossRef] [PubMed]
- Lloréns-Rico, V.; Simcock, J.A.; Huys, G.R.B.; Raes, J. Single-Cell Approaches in Human Microbiome Research. Cell 2022, 185, 2725–2738. [Google Scholar] [CrossRef]
- Shen, Y.; Qian, Q.; Ding, L.; Qu, W.; Zhang, T.; Song, M.; Huang, Y.; Wang, M.; Xu, Z.; Chen, J.; et al. High-Throughput Single-Microbe RNA Sequencing Reveals Adaptive State Heterogeneity and Host-Phage Activity Associations in Human Gut Microbiome. Protein Cell 2024, PWAE027. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Zhu, S.; Xue, M.Y.; Chen, H.; Xu, J.; Song, M.; Tang, Y.; Liu, X.; Tao, Y.; Zhang, T.; et al. Single-Cell Transcriptomics across 2,534 Microbial Species Reveals Functional Heterogeneity in the Rumen Microbiome. Nat. Microbiol. 2024, 9, 1884–1898. [Google Scholar] [CrossRef]
- Wu, J.J.; Zhu, S.; Tang, Y.F.; Gu, F.F.; Liu, J.X.; Sun, H.Z. Microbiota-Host Crosstalk in the Newborn and Adult Rumen at Single-Cell Resolution. BMC Biol. 2022, 20, 280. [Google Scholar] [CrossRef]
- Gu, F.; Zhu, S.; Tang, Y.; Liu, X.; Jia, M.; Malmuthuge, N.; Valencak, T.G.; McFadden, J.W.; Liu, J.; Sun, H. Gut Microbiome Is Linked to Functions of Peripheral Immune Cells in Transition Cows during Excessive Lipolysis. Microbiome 2023, 11, 40. [Google Scholar] [CrossRef]


| Methods | Number of Cells | Separation Mechanism | Speed | Advantages | Disadvantages |
|---|---|---|---|---|---|
| Serial Dilution | Large | Poor concentration control | Slow | Simple, cost-effective | Low purity, risk of contamination, inefficient for multi-cell isolation |
| FACS | Millions | Fluorescence detection, Light Scattering Measurement | Fast | High efficiency, accurate, multi-cell compatible | Expensive, high operational demand, some cell damage |
| Micromanipulation | Low | Cell visualization | Slow | High precision, direct observation | Time-intensive, complex, low throughput |
| Drop-Seq | Hundreds or thousands | Microfluidics | Fast | High throughput, suitable for multiple samples | Specialized equipment and technical support required |
| Immunomagnetic separation | Millions | Antibody-bound magnetic beads | Fast | High efficiency, high specificity | Potential for cell growth impact, risk of cell damage |
| LCM | Low | Visualization-based | Slow | High precision, maintains cell integrity | Expensive, complex, low throughput |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Li, M.; Gao, Z.; Ma, H.; Chong, Y.; Hong, J.; Wu, J.; Wu, D.; Xi, D.; Deng, W. Innovative Insights into Single-Cell Technologies and Multi-Omics Integration in Livestock and Poultry. Int. J. Mol. Sci. 2024, 25, 12940. https://doi.org/10.3390/ijms252312940
Lu Y, Li M, Gao Z, Ma H, Chong Y, Hong J, Wu J, Wu D, Xi D, Deng W. Innovative Insights into Single-Cell Technologies and Multi-Omics Integration in Livestock and Poultry. International Journal of Molecular Sciences. 2024; 25(23):12940. https://doi.org/10.3390/ijms252312940
Chicago/Turabian StyleLu, Ying, Mengfei Li, Zhendong Gao, Hongming Ma, Yuqing Chong, Jieyun Hong, Jiao Wu, Dongwang Wu, Dongmei Xi, and Weidong Deng. 2024. "Innovative Insights into Single-Cell Technologies and Multi-Omics Integration in Livestock and Poultry" International Journal of Molecular Sciences 25, no. 23: 12940. https://doi.org/10.3390/ijms252312940
APA StyleLu, Y., Li, M., Gao, Z., Ma, H., Chong, Y., Hong, J., Wu, J., Wu, D., Xi, D., & Deng, W. (2024). Innovative Insights into Single-Cell Technologies and Multi-Omics Integration in Livestock and Poultry. International Journal of Molecular Sciences, 25(23), 12940. https://doi.org/10.3390/ijms252312940