Advancements in the Application of scRNA-Seq in Breast Research: A Review

 , ,
, ,
Abstract
1. Introduction
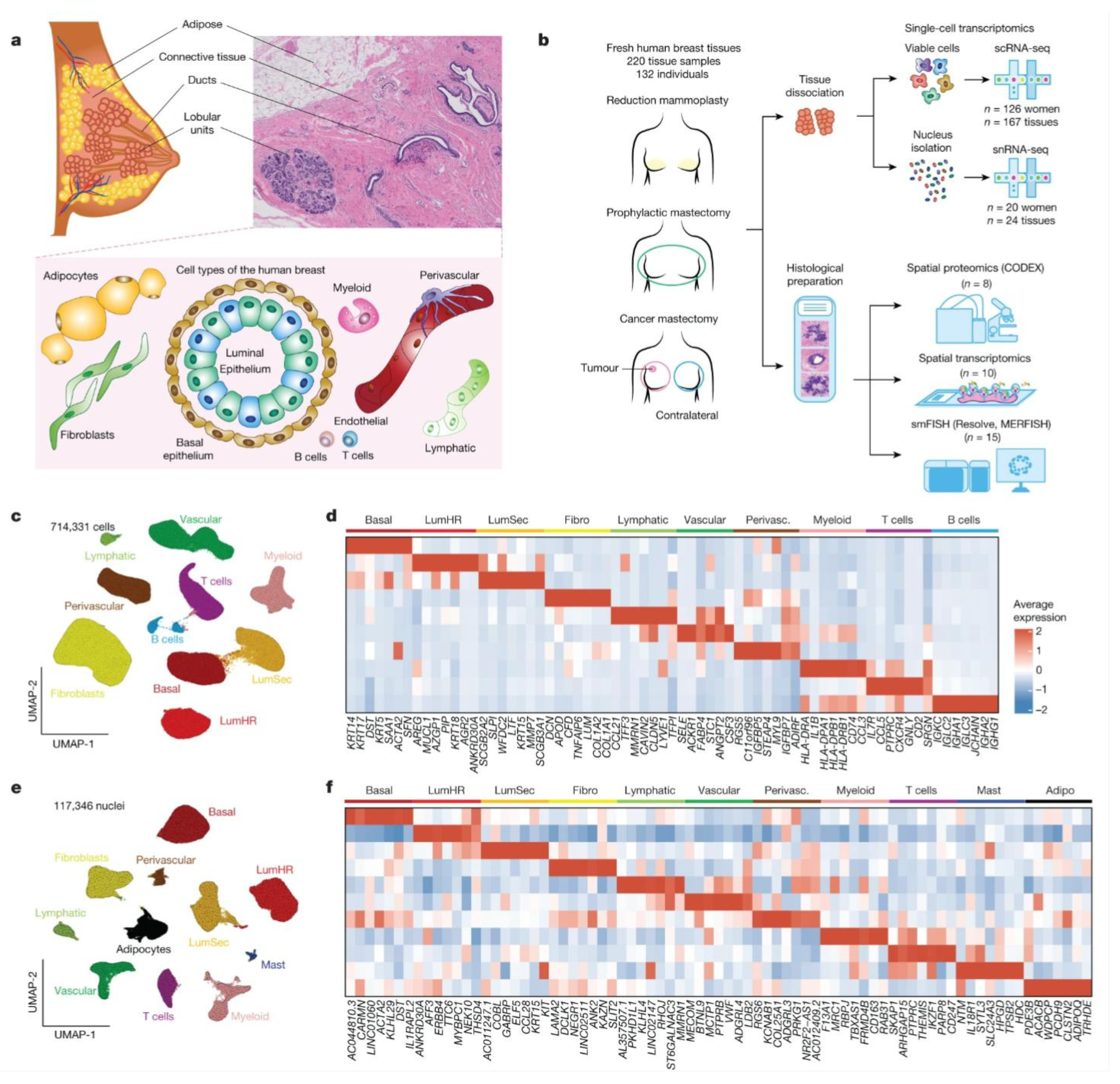
2. Different Cell Types in Breast
2.1. Luminal Hormone-Responsive Epithelial Cells
2.2. Luminal Secretory Epithelial Cells
2.3. Basal Myoepithelial Cells
2.4. Endothelial Cells
2.5. Immune Cells
2.6. Mesenchymal Cells
2.7. Perivascular Cells
2.8. Adipocyte and Mast Cells
3. scRNA-Seq Technology and Its Application in Breast

{kind=link}
| Cell Type | Marker Genes | Species Origin |
|---|---|---|
| Luminal hormone-responsive cells | AREG [93], MUCL1 [94], AZGP1 [95], PIP [96], KRT18 [97], AGR2 [97], ANKRD30A [27] | Human, Mouse |
| Luminal secretory epithelial cells | SCGB2A2 [94], SLP1 [98], WFDC2 [99], LTF [100], KRT15 [101], MMP7 [102], SCGB3A1 [103] | Human, Mouse |
| Basal myoepithelial cells | KTR14 [78], KRT17 [104], DST [103], KRT5 [105], SAA1 [106], ACTA2 [27], SFN [107] | Human, Mouse |
| Lymph endothelial cells | CCL21 [108], TFF3 [109], MMRN1 [110], CAVIN2 [111], CLDN5 [112], LYVE1 [96] | Human, Mouse |
| Vascular endothelial cells | SELE [113], ACKR1 [114], FABP4 [115], ANGPT2 [116], CSF3 [117] | Human, Mouse |
| T cells | IL7R [118], CCL5 [104], PTPRC [119], CXCR4 [120], GNLY [121], CD2 [122], SRGN [120] | Human, Mouse |
| B cells | IGKC [123], IGLC2 [124], IGHA1 [105], IGLC3 [105], JCHAIN [125], IGHA2 [126], IGHG1 [127] | Human, Mouse |
| Bone marrow cells | HLA-DRA [128], IL1B [129], HLA-DPA1 [130], HLA-DPB1 [131], HLA-DRB1 [132], CD74 [133], CCL3 [128] | Human, Mouse |
| Fibroblast cells | DCN [134], APOD [135], CFD [114], TNFAIP6 [136], LUM [137], COL1A2 [134], COL1A1 [138] | Human, Mouse |
| Perivascular cells | RGS5 [105], IGFBP5 [139], STEAP4 [140], MYL9 [141], IGFBP7 [142], ADIRF [143] | Human, Mouse |
| Mast cells | CMA1 [104], CPA3 [104], CTSG [144], KIT [145], TPSD1 [134], tryptase [146] | Human, Mouse |
| Adipocyte cells | APOD [104], CFD [147], DLK1 [148], SCARA5 [104] | Human, Mouse |
4. Advances in scRNA-Seq and Combined Multi-Omics Strategies in Breast Tumor Research
5. The Prospective Outlook of scRNA-Seq Technology in Breast Development
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blum, J.W.; Baumrucker, C.R. Colostral and milk insulin-like growth factors and related substances: Mammary gland and neonatal (intestinal and systemic) targets. Domest Anim. Endocrinol. 2002, 23, 101–110. [Google Scholar] [CrossRef]
- McNally, S.; Stein, T. Overview of Mammary Gland Development: A Comparison of Mouse and Human. Methods Mol. Biol. 2017, 1501, 1–17. [Google Scholar] [CrossRef]
- Ball, H.J.; Mackie, D.P. The ovine mammary gland as an experimental model to determine the virulence of animal ureaplasmas. J. Hyg. 1985, 95, 375–382. [Google Scholar] [CrossRef]
- Faulkin, L.J., Jr.; Deome, K.B. Regulation of growth and spacing of gland elements in the mammary fat pad of the C3H mouse. J. Natl. Cancer Inst. 1960, 24, 953–969. [Google Scholar]
- Sternlicht, M.D.; Kouros-Mehr, H.; Lu, P.; Werb, Z. Hormonal and local control of mammary branching morphogenesis. Differentiation 2006, 74, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Richert, M.M.; Schwertfeger, K.L.; Ryder, J.W.; Anderson, S.M. An atlas of mouse mammary gland development. J. Mammary Gland Biol. Neoplasia 2000, 5, 227–241. [Google Scholar] [CrossRef]
- Brisken, C. Hormonal control of alveolar development and its implications for breast carcinogenesis. J. Mammary Gland Biol. Neoplasia 2002, 7, 39–48. [Google Scholar] [CrossRef]
- Walton, K.D.; Wagner, K.U.; Rucker, E.B., 3rd; Shillingford, J.M.; Miyoshi, K.; Hennighausen, L. Conditional deletion of the bcl-x gene from mouse mammary epithelium results in accelerated apoptosis during involution but does not compromise cell function during lactation. Mech. Dev. 2001, 109, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.D.; Huynh, H.T. Role of tissue remodeling in mammary epithelial cell proliferation and morphogenesis. J. Dairy Sci. 1991, 74, 2801–2807. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Maeda, T.; Kudo, K. Endogenous Serotonin and Milk Production Regulation in the Mammary Gland. Yakugaku Zasshi 2018, 138, 829–836. [Google Scholar] [CrossRef]
- Dai, W.T.; Zou, Y.X.; White, R.R.; Liu, J.X.; Liu, H.Y. Transcriptomic profiles of the bovine mammary gland during lactation and the dry period. Funct. Integr. Genom. 2018, 18, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Capuco, A.V.; Wood, D.L.; Baldwin, R.; McLeod, K.; Paape, M.J. Mammary cell number, proliferation, and apoptosis during a bovine lactation: Relation to milk production and effect of bST. J. Dairy Sci. 2001, 84, 2177–2187. [Google Scholar] [CrossRef]
- Atabai, K.; Sheppard, D.; Werb, Z. Roles of the innate immune system in mammary gland remodeling during involution. J. Mammary Gland Biol. Neoplasia 2007, 12, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; He, L.; Cai, L. Transcriptome Sequencing: RNA-Seq. Methods Mol. Biol. 2018, 1754, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.C.; Cai, L.; Elowitz, M.; Enver, T.; Fan, G.; Guo, G.; Irizarry, R.; Kharchenko, P.; Kim, J.; Orkin, S.; et al. Challenges and emerging directions in single-cell analysis. Genome Biol. 2017, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, S.T.; Zhang, X.Y.; Ding, H.R.; Yuan, Y.; He, J.J.; Wang, M.S.; Yang, B.; Li, Y.B. The Evolution of Single-Cell RNA Sequencing Technology and Application: Progress and Perspectives. Int. J. Mol. Sci. 2023, 24, 2943. [Google Scholar] [CrossRef]
- Inman, J.L.; Robertson, C.; Mott, J.D.; Bissell, M.J. Mammary gland development: Cell fate specification, stem cells and the microenvironment. Development 2015, 142, 1028–1042. [Google Scholar] [CrossRef]
- Twigger, A.J.; Khaled, W.T. Mammary gland development from a single cell ‘omics view. Semin. Cell Dev. Biol. 2021, 114, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Wang, R.; Jiang, W.; Yin, Q.; Peng, G.; Yang, R.; Yu, Q.C.; Chen, J.; Li, J.; Cheung, T.H.; et al. Hormones induce the formation of luminal-derived basal cells in the mammary gland. Cell Res. 2019, 29, 206–220. [Google Scholar] [CrossRef]
- Jena, M.K.; Khan, F.B.; Ali, S.A.; Abdullah, A.; Sharma, A.K.; Yadav, V.; Kancharla, S.; Kolli, P.; Mandadapu, G.; Sahoo, A.K.; et al. Molecular complexity of mammary glands development: A review of lactogenic differentiation in epithelial cells. Artif. Cells Nanomed. Biotechnol. 2023, 51, 491–508. [Google Scholar] [CrossRef] [PubMed]
- Pierson, E.; Althoff, T.; Thomas, D.; Hillard, P.; Leskovec, J. Daily, weekly, seasonal and menstrual cycles in women’s mood, behaviour and vital signs. Nat. Hum. Behav. 2021, 5, 716–725. [Google Scholar] [CrossRef]
- Pervolarakis, N.; Nguyen, Q.H.; Williams, J.; Gong, Y.; Gutierrez, G.; Sun, P.; Jhutty, D.; Zheng, G.X.Y.; Nemec, C.M.; Dai, X.; et al. Integrated Single-Cell Transcriptomics and Chromatin Accessibility Analysis Reveals Regulators of Mammary Epithelial Cell Identity. Cell Rep. 2020, 33, 108273. [Google Scholar] [CrossRef]
- Alex, A.; Bhandary, E.; McGuire, K.P. Anatomy and Physiology of the Breast during Pregnancy and Lactation. Adv. Exp. Med. Biol. 2020, 1252, 3–7. [Google Scholar] [CrossRef]
- Dai, W.; White, R.; Liu, J.; Liu, H. Organelles coordinate milk production and secretion during lactation: Insights into mammary pathologies. Prog. Lipid Res. 2022, 86, 101159. [Google Scholar] [CrossRef] [PubMed]
- Slepicka, P.F.; Somasundara, A.V.H.; Dos Santos, C.O. The molecular basis of mammary gland development and epithelial differentiation. Semin. Cell Dev. Biol. 2021, 114, 93–112. [Google Scholar] [CrossRef]
- Jassim, A.; Rahrmann, E.P.; Simons, B.D.; Gilbertson, R.J. Cancers make their own luck: Theories of cancer origins. Nat. Rev. Cancer 2023, 23, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.H.; Pervolarakis, N.; Blake, K.; Ma, D.; Davis, R.T.; James, N.; Phung, A.T.; Willey, E.; Kumar, R.; Jabart, E.; et al. Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nat. Commun. 2018, 9, 2028. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Jiang, X.; Li, G.; Zhu, Y.; Zhang, H. Junctional complexes in epithelial cells: Sentinels for extracellular insults and intracellular homeostasis. FEBS J. 2022, 289, 7314–7333. [Google Scholar] [CrossRef] [PubMed]
- Faraldo, M.M.; Taddei-De La Hosseraye, I.; Teulière, J.; Deugnier, M.A.; Moumen, M.; Thiery, J.P.; Glukhova, M.A. Mammary gland development: Role of basal myoepithelial cells. J. Soc. Biol. 2006, 200, 193–198. [Google Scholar] [CrossRef]
- Sopel, M. The myoepithelial cell: Its role in normal mammary glands and breast cancer. Folia Morphol. 2010, 69, 1–14. [Google Scholar]
- Gudjonsson, T.; Adriance, M.C.; Sternlicht, M.D.; Petersen, O.W.; Bissell, M.J. Myoepithelial cells: Their origin and function in breast morphogenesis and neoplasia. J. Mammary Gland Biol. Neoplasia 2005, 10, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Ryman, V.E.; Packiriswamy, N.; Sordillo, L.M. Role of endothelial cells in bovine mammary gland health and disease. Anim. Health Res. Rev. 2015, 16, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Aitken, S.L.; Corl, C.M.; Sordillo, L.M. Immunopathology of mastitis: Insights into disease recognition and resolution. J. Mammary Gland Biol. Neoplasia 2011, 16, 291–304. [Google Scholar] [CrossRef]
- Hannan, F.M.; Elajnaf, T.; Vandenberg, L.N.; Kennedy, S.H.; Thakker, R.V. Hormonal regulation of mammary gland development and lactation. Nat. Rev. Endocrinol. 2023, 19, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, W.; Yang, R.; Li, C.; Wu, T.; Dong, X.B.; Zhou, B.; Guo, X.; Chen, J.; Liu, Z.; et al. Endothelial Wnts control mammary epithelial patterning via fibroblast signaling. Cell Rep. 2021, 34, 108897. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Chen, Z.; Geng, L.; Wang, J.; Liang, H.; Cao, Y.; Chen, H.; Huang, W.; Su, M.; Wang, H.; et al. Mucosal Profiling of Pediatric-Onset Colitis and IBD Reveals Common Pathogenics and Therapeutic Pathways. Cell 2019, 179, 1160–1176.e1124. [Google Scholar] [CrossRef] [PubMed]
- Loftus, R.M.; Finlay, D.K. Immunometabolism: Cellular Metabolism Turns Immune Regulator. J. Biol. Chem. 2016, 291, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.L.; Danforth, D.N. The Role of Immune Cells in Breast Tissue and Immunotherapy for the Treatment of Breast Cancer. Clin. Breast Cancer 2021, 21, e63–e73. [Google Scholar] [CrossRef] [PubMed]
- Okabe, Y.; Medzhitov, R. Tissue biology perspective on macrophages. Nat. Immunol. 2016, 17, 9–17. [Google Scholar] [CrossRef]
- Hilligan, K.L.; Ronchese, F. Antigen presentation by dendritic cells and their instruction of CD4+ T helper cell responses. Cell Mol. Immunol. 2020, 17, 587–599. [Google Scholar] [CrossRef]
- Saibil, S.D.; Ohashi, P.S. Targeting T cell activation in immuno-oncology. Curr. Oncol. 2020, 27, S98–S105. [Google Scholar] [CrossRef]
- Sun, W.; Zhu, C.; Li, Y.; Wu, X.; Shi, X.; Liu, W. B cell activation and autoantibody production in autoimmune diseases. Best Pract. Res. Clin. Rheumatol. 2024, 38, 101936. [Google Scholar] [CrossRef] [PubMed]
- Saadh, M.J.; Rasulova, I.; Khalil, M.; Farahim, F.; Sârbu, I.; Ciongradi, C.I.; Omar, T.M.; Alhili, A.; Jawad, M.J.; Hani, T.; et al. Natural killer cell-mediated immune surveillance in cancer: Role of tumor microenvironment. Pathol. Res. Pract. 2024, 254, 155120. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, T.; Zhu, Z.; Yang, Y. The association between immune cells and breast cancer: Insights from mendelian randomization and meta-analysis. Int. J. Surg. 2024, 10, 1097. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, X.; Shi, Y.; Li, J.; Zheng, L.; Cui, L.; Zhang, J.; Wang, L.; Han, Z.; Han, Y.; et al. In vivo tracking and comparison of the therapeutic effects of MSCs and HSCs for liver injury. PLoS ONE 2013, 8, e62363. [Google Scholar] [CrossRef] [PubMed]
- Costela-Ruiz, V.J.; Melguizo-Rodríguez, L.; Bellotti, C.; Illescas-Montes, R.; Stanco, D.; Arciola, C.R.; Lucarelli, E. Different Sources of Mesenchymal Stem Cells for Tissue Regeneration: A Guide to Identifying the Most Favorable One in Orthopedics and Dentistry Applications. Int. J. Mol. Sci. 2022, 23, 6356. [Google Scholar] [CrossRef]
- Si, Z.; Wang, X.; Sun, C.; Kang, Y.; Xu, J.; Wang, X.; Hui, Y. Adipose-derived stem cells: Sources, potency, and implications for regenerative therapies. Biomed. Pharmacother. 2019, 114, 108765. [Google Scholar] [CrossRef]
- Kemp, K.C.; Hows, J.; Donaldson, C. Bone marrow-derived mesenchymal stem cells. Leuk. Lymphoma 2005, 46, 1531–1544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. The Role of Mesenchymal Stem Cells in Modulating the Breast Cancer Microenvironment. Cell Transplt. 2023, 32, 9636897231220073. [Google Scholar] [CrossRef]
- Dou, T.; Li, J.; Zhang, Y.; Pei, W.; Zhang, B.; Wang, B.; Wang, Y.; Jia, H. The cellular composition of the tumor microenvironment is an important marker for predicting therapeutic efficacy in breast cancer. Front. Immunol. 2024, 15, 1368687. [Google Scholar] [CrossRef] [PubMed]
- Kadri, N.; Amu, S.; Iacobaeus, E.; Boberg, E.; Le Blanc, K. Current perspectives on mesenchymal stromal cell therapy for graft versus host disease. Cell Mol. Immunol. 2023, 20, 613–625. [Google Scholar] [CrossRef]
- Eirin, A.; Zhu, X.Y.; Puranik, A.S.; Woollard, J.R.; Tang, H.; Dasari, S.; Lerman, A.; van Wijnen, A.J.; Lerman, L.O. Comparative proteomic analysis of extracellular vesicles isolated from porcine adipose tissue-derived mesenchymal stem/stromal cells. Sci. Rep. 2016, 6, 36120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lin, R.; Wu, H.; Jiang, X.; Gao, J. Mesenchymal stem cells: A living carrier for active tumor-targeted delivery. Adv. Drug Deliv. Rev. 2022, 185, 114300. [Google Scholar] [CrossRef] [PubMed]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients With Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef]
- Zhang, C.; Du, Z.; Gao, Y.; Lim, K.S.; Zhou, W.; Huang, H.; He, H.; Xiao, J.; Xu, D.; Li, Q. Methionine secreted by tumor-associated pericytes supports cancer stem cells in clear cell renal carcinoma. Cell Metab. 2024, 36, 778–792.e710. [Google Scholar] [CrossRef]
- Figueiredo, A.M.; Villacampa, P.; Diéguez-Hurtado, R.; José Lozano, J.; Kobialka, P.; Cortazar, A.R.; Martinez-Romero, A.; Angulo-Urarte, A.; Franco, C.A.; Claret, M.; et al. Phosphoinositide 3-Kinase-Regulated Pericyte Maturation Governs Vascular Remodeling. Circulation 2020, 142, 688–704. [Google Scholar] [CrossRef]
- Wu, H.; Zhou, X.; Gong, H.; Ni, Z.; Xu, Q. Perivascular tissue stem cells are crucial players in vascular disease. Free Radic. Biol. Med. 2021, 165, 324–333. [Google Scholar] [CrossRef]
- Avolio, E.; Alvino, V.V.; Ghorbel, M.T.; Campagnolo, P. Perivascular cells and tissue engineering: Current applications and untapped potential. Pharmacol. Ther. 2017, 171, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, D.; Khel, M.I.; Pütz, T.; Zinke, J.; Jia, X.; Sommer, K.; Filipski, K.; Thorsen, F.; Freiman, T.M.; Günther, S.; et al. A flow cytometry-based protocol for syngenic isolation of neurovascular unit cells from mouse and human tissues. Nat. Protoc. 2023, 18, 1510–1542. [Google Scholar] [CrossRef]
- Lee, J.; Henderson, K.; Massidda, M.W.; Armenta-Ochoa, M.; Im, B.G.; Veith, A.; Lee, B.K.; Kim, M.; Maceda, P.; Yoon, E.; et al. Mechanobiological conditioning of mesenchymal stem cells for enhanced vascular regeneration. Nat. Biomed. Eng. 2021, 5, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Wanjare, M.; Kusuma, S.; Gerecht, S. Defining Differences among Perivascular Cells Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2014, 2, 746. [Google Scholar] [CrossRef] [PubMed]
- Brock, C.K.; Hebert, K.L.; Artiles, M.; Wright, M.K.; Cheng, T.; Windsor, G.O.; Nguyen, K.; Alzoubi, M.S.; Collins-Burow, B.M.; Martin, E.C.; et al. A Role for Adipocytes and Adipose Stem Cells in the Breast Tumor Microenvironment and Regenerative Medicine. Front. Physiol. 2021, 12, 751239. [Google Scholar] [CrossRef]
- Kothari, C.; Diorio, C.; Durocher, F. The Importance of Breast Adipose Tissue in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 5760. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Weaver, B.; Choi, H.W.; Abraham, S.N.; Staats, H.F. Mast cell activators as novel immune regulators. Curr. Opin. Pharmacol. 2018, 41, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Hanes, M.R.; Giacomantonio, C.A.; Marshall, J.S. Mast Cells and Skin and Breast Cancers: A Complicated and Microenvironment-Dependent Role. Cells 2021, 10, 986. [Google Scholar] [CrossRef] [PubMed]
- Cimpean, A.M.; Tamma, R.; Ruggieri, S.; Nico, B.; Toma, A.; Ribatti, D. Mast cells in breast cancer angiogenesis. Crit. Rev. Oncol. Hematol. 2017, 115, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar] [CrossRef]
- Adil, A.; Kumar, V.; Jan, A.T.; Asger, M. Single-Cell Transcriptomics: Current Methods and Challenges in Data Acquisition and Analysis. Front. Neurosci. 2021, 15, 591122. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Slovin, S.; Carissimo, A.; Panariello, F.; Grimaldi, A.; Bouché, V.; Gambardella, G.; Cacchiarelli, D. Single-Cell RNA Sequencing Analysis: A Step-by-Step Overview. Methods Mol. Biol. 2021, 2284, 343–365. [Google Scholar] [CrossRef]
- Manohar, S.M.; Shah, P.; Nair, A. Flow cytometry: Principles, applications and recent advances. Bioanalysis 2021, 13, 181–198. [Google Scholar] [CrossRef]
- Wang, C.; Qiu, J.; Liu, M.; Wang, Y.; Yu, Y.; Liu, H.; Zhang, Y.; Han, L. Microfluidic Biochips for Single-Cell Isolation and Single-Cell Analysis of Multiomics and Exosomes. Adv. Sci. 2024, 11, e2401263. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, Y.; Zhang, Q.; Ren, X.; Zhang, Z. Direct Comparative Analyses of 10X Genomics Chromium and Smart-seq2. Genom. Proteom. Bioinform. 2021, 19, 253–266. [Google Scholar] [CrossRef]
- Vermeersch, L.; Jariani, A.; Helsen, J.; Heineike, B.M.; Verstrepen, K.J. Single-Cell RNA Sequencing in Yeast Using the 10× Genomics Chromium Device. Methods Mol. Biol. 2022, 2477, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Lao, K.; Surani, M.A. Development and applications of single-cell transcriptome analysis. Nat. Methods 2011, 8, S6–S11. [Google Scholar] [CrossRef]
- Nguyen, V.; Griss, J. scAnnotatR: Framework to accurately classify cell types in single-cell RNA-sequencing data. BMC Bioinform. 2022, 23, 44. [Google Scholar] [CrossRef]
- Hyman, L.B.; Christopher, C.R.; Romero, P.A. Single-cell nucleic acid profiling in droplets (SNAPD) enables high-throughput analysis of heterogeneous cell populations. Nucleic Acids Res. 2021, 49, e103. [Google Scholar] [CrossRef]
- Bach, K.; Pensa, S.; Grzelak, M.; Hadfield, J.; Adams, D.J.; Marioni, J.C.; Khaled, W.T. Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA sequencing. Nat. Commun. 2017, 8, 2128. [Google Scholar] [CrossRef]
- Twigger, A.J.; Engelbrecht, L.K.; Bach, K.; Schultz-Pernice, I.; Pensa, S.; Stenning, J.; Petricca, S.; Scheel, C.H.; Khaled, W.T. Transcriptional changes in the mammary gland during lactation revealed by single cell sequencing of cells from human milk. Nat. Commun. 2022, 13, 562. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.L.; Smalley, M.J. Integrating single-cell RNA-sequencing and functional assays to decipher mammary cell states and lineage hierarchies. NPJ Breast. Cancer 2020, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Saeki, K.; Chang, G.; Kanaya, N.; Wu, X.; Wang, J.; Bernal, L.; Ha, D.; Neuhausen, S.L.; Chen, S. Mammary cell gene expression atlas links epithelial cell remodeling events to breast carcinogenesis. Commun. Biol. 2021, 4, 660. [Google Scholar] [CrossRef]
- Poliwoda, S.; Noor, N.; Downs, E.; Schaaf, A.; Cantwell, A.; Ganti, L.; Kaye, A.D.; Mosel, L.I.; Carroll, C.B.; Viswanath, O.; et al. Stem cells: A comprehensive review of origins and emerging clinical roles in medical practice. Orthop. Rev. 2022, 14, 37498. [Google Scholar] [CrossRef] [PubMed]
- Giraddi, R.R.; Chung, C.Y.; Heinz, R.E.; Balcioglu, O.; Novotny, M.; Trejo, C.L.; Dravis, C.; Hagos, B.M.; Mehrabad, E.M.; Rodewald, L.W.; et al. Single-Cell Transcriptomes Distinguish Stem Cell State Changes and Lineage Specification Programs in Early Mammary Gland Development. Cell Rep. 2018, 24, 1653–1666.e1657. [Google Scholar] [CrossRef] [PubMed]
- Pal, B.; Chen, Y.; Vaillant, F.; Jamieson, P.; Gordon, L.; Rios, A.C.; Wilcox, S.; Fu, N.; Liu, K.H.; Jackling, F.C.; et al. Construction of developmental lineage relationships in the mouse mammary gland by single-cell RNA profiling. Nat. Commun. 2017, 8, 1627. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Zhu, S.; Gu, F.; Valencak, T.G.; Liu, J.X.; Sun, H.Z. Cross-tissue single-cell transcriptomic landscape reveals the key cell subtypes and their potential roles in the nutrient absorption and metabolism in dairy cattle. J. Adv. Res. 2022, 37, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.; Weikard, R.; Hadlich, F.; Kühn, C. Single-cell RNA sequencing of freshly isolated bovine milk cells and cultured primary mammary epithelial cells. Sci. Data 2021, 8, 177. [Google Scholar] [CrossRef]
- Fan, Y.; Jin, L.; He, Z.; Wei, T.; Luo, T.; Zhang, J.; Liu, C.; Dai, C.; A, C.; Liang, Y.; et al. A cell transcriptomic profile provides insights into adipocytes of porcine mammary gland across development. J. Anim. Sci. Biotechnol. 2023, 14, 126. [Google Scholar] [CrossRef]
- Gutierrez, G.; Sun, P.; Han, Y.; Dai, X. Defining mammary basal cell transcriptional states using single-cell RNA-sequencing. Sci. Rep. 2022, 12, 4893. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.; Nee, K.; Wei, R.; He, S.; Nguyen, Q.H.; Bai, S.; Blake, K.; Pein, M.; Gong, Y.; Sei, E.; et al. A spatially resolved single-cell genomic atlas of the adult human breast. Nature 2023, 620, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, E.; Deng, Q. Single-cell RNA sequencing: Technical advancements and biological applications. Mol. Asp. Med. 2018, 59, 36–46. [Google Scholar] [CrossRef] [PubMed]
- AlJanahi, A.A.; Danielsen, M.; Dunbar, C.E. An Introduction to the Analysis of Single-Cell RNA-Sequencing Data. Mol. Ther. Methods Clin. Dev. 2018, 10, 189–196. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.O.; Lee, K.M.; Lee, H.B.; Kim, K.T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef]
- Li, L.; Dong, J.; Yan, L.; Yong, J.; Liu, X.; Hu, Y.; Fan, X.; Wu, X.; Guo, H.; Wang, X.; et al. Single-Cell RNA-Seq Analysis Maps Development of Human Germline Cells and Gonadal Niche Interactions. Cell Stem Cell 2017, 20, 858–873.e854. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, L.H.; Liu, Y.; Li, Y.Q.; Chen, H.T.; Xu, J.H.; Peng, W.; Lin, G.W.; Wei, P.P.; Li, B.; et al. Single-cell transcriptome profiling of an adult human cell atlas of 15 major organs. Genome Biol. 2020, 21, 294. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Klughammer, J.; Farlik, M.; Penz, T.; Spittler, A.; Barbieux, C.; Berishvili, E.; Bock, C.; Kubicek, S. Single-cell transcriptomes reveal characteristic features of human pancreatic islet cell types. EMBO Rep. 2016, 17, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Guerrero-Juarez, C.F.; Chen, Z.; Tang, Y.; Ma, X.; Lv, C.; Bi, X.; Deng, M.; Bu, L.; Tian, Y.; et al. The Msi1-mTOR pathway drives the pathogenesis of mammary and extramammary Paget’s disease. Cell Res. 2020, 30, 854–872. [Google Scholar] [CrossRef]
- Venkatakrishnan, A.J.; Puranik, A.; Anand, A.; Zemmour, D.; Yao, X.; Wu, X.; Chilaka, R.; Murakowski, D.K.; Standish, K.; Raghunathan, B.; et al. Knowledge synthesis of 100 million biomedical documents augments the deep expression profiling of coronavirus receptors. Elife 2020, 9, e58040. [Google Scholar] [CrossRef] [PubMed]
- Nugteren, S.; Samsom, J.N. Secretory Leukocyte Protease Inhibitor (SLPI) in mucosal tissues: Protects against inflammation, but promotes cancer. Cytokine Growth Factor Rev. 2021, 59, 22–35. [Google Scholar] [CrossRef]
- Farmer, D.T.; Nathan, S.; Finley, J.K.; Shengyang Yu, K.; Emmerson, E.; Byrnes, L.E.; Sneddon, J.B.; McManus, M.T.; Tward, A.D.; Knox, S.M. Defining epithelial cell dynamics and lineage relationships in the developing lacrimal gland. Development 2017, 144, 2517–2528. [Google Scholar] [CrossRef] [PubMed]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, H.; Ma, Q.; Chen, C.; Yue, J.; Li, B.; Zhang, X. Time-course single-cell RNA sequencing reveals transcriptional dynamics and heterogeneity of limbal stem cells derived from human pluripotent stem cells. Cell Biosci. 2021, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Van den Eynde, C.; De Clercq, K.; Van Bree, R.; Luyten, K.; Annibali, D.; Amant, F.; Han, S.; Van Nieuwenhuysen, E.; Baert, T.; Peeraer, K.; et al. TRP channel expression correlates with the epithelial-mesenchymal transition and high-risk endometrial carcinoma. Cell Mol. Life Sci. 2021, 79, 26. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, R.J.; Lloyd, C.M. Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol. 2021, 21, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Kester, L.; Seinstra, D.; van Rossum, A.G.J.; Vennin, C.; Hoogstraat, M.; van der Velden, D.; Opdam, M.; van Werkhoven, E.; Hahn, K.; Nederlof, I.; et al. Differential Survival and Therapy Benefit of Patients with Breast Cancer Are Characterized by Distinct Epithelial and Immune Cell Microenvironments. Clin. Cancer Res. 2022, 28, 960–971. [Google Scholar] [CrossRef]
- Pal, B.; Chen, Y.; Vaillant, F.; Capaldo, B.D.; Joyce, R.; Song, X.; Bryant, V.L.; Penington, J.S.; Di Stefano, L.; Tubau Ribera, N.; et al. A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. Embo J. 2021, 40, e107333. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.J.; Ferdinand, J.R.; Young, M.D.; Mitchell, T.J.; Loudon, K.W.; Riding, A.M.; Richoz, N.; Frazer, G.L.; Staniforth, J.U.L.; Vieira Braga, F.A.; et al. Spatiotemporal immune zonation of the human kidney. Science 2019, 365, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef]
- Bischoff, P.; Trinks, A.; Obermayer, B.; Pett, J.P.; Wiederspahn, J.; Uhlitz, F.; Liang, X.; Lehmann, A.; Jurmeister, P.; Elsner, A.; et al. Single-cell RNA sequencing reveals distinct tumor microenvironmental patterns in lung adenocarcinoma. Oncogene 2021, 40, 6748–6758. [Google Scholar] [CrossRef] [PubMed]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstråhle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 1661–1662. [Google Scholar] [CrossRef]
- Wu, G.; Lee, Y.Y.; Gulla, E.M.; Potter, A.; Kitzmiller, J.; Ruben, M.D.; Salomonis, N.; Whitsett, J.A.; Francey, L.J.; Hogenesch, J.B.; et al. Short-term exposure to intermittent hypoxia leads to changes in gene expression seen in chronic pulmonary disease. Elife 2021, 10, e63003. [Google Scholar] [CrossRef] [PubMed]
- Cherry, C.; Maestas, D.R.; Han, J.; Andorko, J.I.; Cahan, P.; Fertig, E.J.; Garmire, L.X.; Elisseeff, J.H. Computational reconstruction of the signalling networks surrounding implanted biomaterials from single-cell transcriptomics. Nat. Biomed. Eng. 2021, 5, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Gillich, A.; Zhang, F.; Farmer, C.G.; Travaglini, K.J.; Tan, S.Y.; Gu, M.; Zhou, B.; Feinstein, J.A.; Krasnow, M.A.; Metzger, R.J. Capillary cell-type specialization in the alveolus. Nature 2020, 586, 785–789. [Google Scholar] [CrossRef]
- Schelker, M.; Feau, S.; Du, J.; Ranu, N.; Klipp, E.; MacBeath, G.; Schoeberl, B.; Raue, A. Estimation of immune cell content in tumour tissue using single-cell RNA-seq data. Nat. Commun. 2017, 8, 2032. [Google Scholar] [CrossRef]
- Theocharidis, G.; Thomas, B.E.; Sarkar, D.; Mumme, H.L.; Pilcher, W.J.R.; Dwivedi, B.; Sandoval-Schaefer, T.; Sîrbulescu, R.F.; Kafanas, A.; Mezghani, I.; et al. Single cell transcriptomic landscape of diabetic foot ulcers. Nat. Commun. 2022, 13, 181. [Google Scholar] [CrossRef] [PubMed]
- Räsänen, M.; Sultan, I.; Paech, J.; Hemanthakumar, K.A.; Yu, W.; He, L.; Tang, J.; Sun, Y.; Hlushchuk, R.; Huan, X.; et al. VEGF-B Promotes Endocardium-Derived Coronary Vessel Development and Cardiac Regeneration. Circulation 2021, 143, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, K.; Das, A. TWIST1-Reprogrammed Endothelial Cell Transplantation Potentiates Neovascularization-Mediated Diabetic Wound Tissue Regeneration. Diabetes 2020, 69, 1232–1247. [Google Scholar] [CrossRef] [PubMed]
- Brosseau, J.P.; Sathe, A.A.; Wang, Y.; Nguyen, T.; Glass, D.A., 2nd; Xing, C.; Le, L.Q. Human cutaneous neurofibroma matrisome revealed by single-cell RNA sequencing. Acta Neuropathol. Commun. 2021, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Richer, A.L.; Riemondy, K.A.; Hardie, L.; Hesselberth, J.R. Simultaneous measurement of biochemical phenotypes and gene expression in single cells. Nucleic Acids Res. 2020, 48, e59. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Mao, A.; Zheng, C.B.; Kan, H.; Zhang, K.; Zhang, Z.; Feng, L.; Ma, X. Aortic heterogeneity across segments and under high fat/salt/glucose conditions at the single-cell level. Natl. Sci. Rev. 2020, 7, 881–896. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Mitchell, T.J.; Vieira Braga, F.A.; Tran, M.G.B.; Stewart, B.J.; Ferdinand, J.R.; Collord, G.; Botting, R.A.; Popescu, D.M.; Loudon, K.W.; et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 2018, 361, 594–599. [Google Scholar] [CrossRef]
- Ren, X.; Wen, W.; Fan, X.; Hou, W.; Su, B.; Cai, P.; Li, J.; Liu, Y.; Tang, F.; Zhang, F.; et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell 2021, 184, 1895–1913.e1819. [Google Scholar] [CrossRef] [PubMed]
- Govaere, O.; Cockell, S.; Tiniakos, D.; Queen, R.; Younes, R.; Vacca, M.; Alexander, L.; Ravaioli, F.; Palmer, J.; Petta, S.; et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 2020, 12, eaba4448. [Google Scholar] [CrossRef]
- Wang, Y.; Chaffee, T.S.; LaRue, R.S.; Huggins, D.N.; Witschen, P.M.; Ibrahim, A.M.; Nelson, A.C.; Machado, H.L.; Schwertfeger, K.L. Tissue-resident macrophages promote extracellular matrix homeostasis in the mammary gland stroma of nulliparous mice. Elife 2020, 9, e57438. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Gao, Y.; Miao, J.; Chen, S.; Li, J.; Li, Z.; Yin, C.; Yue, W. Single-cell RNA-seq highlights a specific carcinoembryonic cluster in ovarian cancer. Cell Death Dis. 2021, 12, 1082. [Google Scholar] [CrossRef]
- Marinari, E.; Allard, M.; Gustave, R.; Widmer, V.; Philippin, G.; Merkler, D.; Tsantoulis, P.; Dutoit, V.; Dietrich, P.Y. Inflammation and lymphocyte infiltration are associated with shorter survival in patients with high-grade glioma. Oncoimmunology 2020, 9, 1779990. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, S.; Liu, Y.; He, X.; Qu, M.; Xu, G.; Wang, H.; Huang, M.; Pan, J.; Liu, Z.; et al. Single-cell RNA sequencing reveals the heterogeneity of liver-resident immune cells in human. Cell Discov. 2020, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- O’Flanagan, C.H.; Campbell, K.R.; Zhang, A.W.; Kabeer, F.; Lim, J.L.P.; Biele, J.; Eirew, P.; Lai, D.; McPherson, A.; Kong, E.; et al. Dissociation of solid tumor tissues with cold active protease for single-cell RNA-seq minimizes conserved collagenase-associated stress responses. Genome Biol. 2019, 20, 210. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.T.; Dolgalev, I.; Evensen, N.A.; Ma, C.; Chambers, T.; Roberts, K.G.; Sreeram, S.; Dai, Y.; Tikhonova, A.N.; Lasry, A.; et al. Extensive Remodeling of the Immune Microenvironment in B Cell Acute Lymphoblastic Leukemia. Cancer Cell 2020, 37, 867–882.e812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Moorlag, S.J.; Dominguez-Andres, J.; Bulut, Ö.; Kilic, G.; Liu, Z.; van Crevel, R.; Xu, C.J.; Joosten, L.A.; Netea, M.G.; et al. Single-cell RNA sequencing reveals induction of distinct trained-immunity programs in human monocytes. J. Clin. Investig. 2022, 132, 1–17. [Google Scholar] [CrossRef]
- Dussiau, C.; Boussaroque, A.; Gaillard, M.; Bravetti, C.; Zaroili, L.; Knosp, C.; Friedrich, C.; Asquier, P.; Willems, L.; Quint, L.; et al. Hematopoietic differentiation is characterized by a transient peak of entropy at a single-cell level. BMC Biol. 2022, 20, 60. [Google Scholar] [CrossRef]
- Tinholt, M.; Stavik, B.; Tekpli, X.; Garred, Ø.; Borgen, E.; Kristensen, V.; Sahlberg, K.K.; Sandset, P.M.; Iversen, N. Coagulation factor V is a marker of tumor-infiltrating immune cells in breast cancer. Oncoimmunology 2020, 9, 1824644. [Google Scholar] [CrossRef] [PubMed]
- Egelston, C.A.; Guo, W.; Tan, J.; Avalos, C.; Simons, D.L.; Lim, M.H.; Huang, Y.J.; Nelson, M.S.; Chowdhury, A.; Schmolze, D.B.; et al. Tumor-infiltrating exhausted CD8+ T cells dictate reduced survival in premenopausal estrogen receptor-positive breast cancer. JCI Insight 2022, 7, e153963. [Google Scholar] [CrossRef] [PubMed]
- Jaitin, D.A.; Weiner, A.; Yofe, I.; Lara-Astiaso, D.; Keren-Shaul, H.; David, E.; Salame, T.M.; Tanay, A.; van Oudenaarden, A.; Amit, I. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 2016, 167, 1883–1896.e1815. [Google Scholar] [CrossRef]
- Xu, K.; Wang, R.; Xie, H.; Hu, L.; Wang, C.; Xu, J.; Zhu, C.; Liu, Y.; Gao, F.; Li, X.; et al. Single-cell RNA sequencing reveals cell heterogeneity and transcriptome profile of breast cancer lymph node metastasis. Oncogenesis 2021, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Joseph, D.B.; Henry, G.H.; Malewska, A.; Reese, J.C.; Mauck, R.J.; Gahan, J.C.; Hutchinson, R.C.; Malladi, V.S.; Roehrborn, C.G.; Vezina, C.M.; et al. Single-cell analysis of mouse and human prostate reveals novel fibroblasts with specialized distribution and microenvironment interactions. J. Pathol. 2021, 255, 141–154. [Google Scholar] [CrossRef]
- Ge, G.; Han, Y.; Zhang, J.; Li, X.; Liu, X.; Gong, Y.; Lei, Z.; Wang, J.; Zhu, W.; Xu, Y.; et al. Single-Cell RNA-seq Reveals a Developmental Hierarchy Super-Imposed Over Subclonal Evolution in the Cellular Ecosystem of Prostate Cancer. Adv. Sci. 2022, 9, e2105530. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Zhao, Q.; Nguyen, T.; Pjanic, M.; Cheng, P.; Wirka, R.; Travisano, S.; Nagao, M.; Kundu, R.; Quertermous, T. Environment-Sensing Aryl Hydrocarbon Receptor Inhibits the Chondrogenic Fate of Modulated Smooth Muscle Cells in Atherosclerotic Lesions. Circulation 2020, 142, 575–590. [Google Scholar] [CrossRef]
- Milich, L.M.; Choi, J.S.; Ryan, C.; Cerqueira, S.R.; Benavides, S.; Yahn, S.L.; Tsoulfas, P.; Lee, J.K. Single-cell analysis of the cellular heterogeneity and interactions in the injured mouse spinal cord. J. Exp. Med. 2021, 218, e20210040. [Google Scholar] [CrossRef]
- Park, J.; Shrestha, R.; Qiu, C.; Kondo, A.; Huang, S.; Werth, M.; Li, M.; Barasch, J.; Suszták, K. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 2018, 360, 758–763. [Google Scholar] [CrossRef]
- Shami, A.N.; Zheng, X.; Munyoki, S.K.; Ma, Q.; Manske, G.L.; Green, C.D.; Sukhwani, M.; Orwig, K.E.; Li, J.Z.; Hammoud, S.S. Single-Cell RNA Sequencing of Human, Macaque, and Mouse Testes Uncovers Conserved and Divergent Features of Mammalian Spermatogenesis. Dev. Cell 2020, 54, 529–547.e512. [Google Scholar] [CrossRef] [PubMed]
- Li, C.M.; Shapiro, H.; Tsiobikas, C.; Selfors, L.M.; Chen, H.; Rosenbluth, J.; Moore, K.; Gupta, K.P.; Gray, G.K.; Oren, Y.; et al. Aging-Associated Alterations in Mammary Epithelia and Stroma Revealed by Single-Cell RNA Sequencing. Cell Rep. 2020, 33, 108566. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Mora, F.; Salomon, R.; Gloss, B.S.; Law, A.M.K.; Venhuizen, J.; Castillo, L.; Murphy, K.J.; Magenau, A.; Papanicolaou, M.; Rodriguez de la Fuente, L.; et al. Single-cell transcriptomics reveals involution mimicry during the specification of the basal breast cancer subtype. Cell Rep. 2021, 35, 108945. [Google Scholar] [CrossRef] [PubMed]
- Fawkner-Corbett, D.; Antanaviciute, A.; Parikh, K.; Jagielowicz, M.; Gerós, A.S.; Gupta, T.; Ashley, N.; Khamis, D.; Fowler, D.; Morrissey, E.; et al. Spatiotemporal analysis of human intestinal development at single-cell resolution. Cell 2021, 184, 810–826.e823. [Google Scholar] [CrossRef] [PubMed]
- Plum, T.; Wang, X.; Rettel, M.; Krijgsveld, J.; Feyerabend, T.B.; Rodewald, H.R. Human Mast Cell Proteome Reveals Unique Lineage, Putative Functions, and Structural Basis for Cell Ablation. Immunity 2020, 52, 404–416.e405. [Google Scholar] [CrossRef]
- Valent, P.; Orazi, A.; Savona, M.R.; Patnaik, M.M.; Onida, F.; van de Loosdrecht, A.A.; Haase, D.; Haferlach, T.; Elena, C.; Pleyer, L.; et al. Proposed diagnostic criteria for classical chronic myelomonocytic leukemia (CMML), CMML variants and pre-CMML conditions. Haematologica 2019, 104, 1935–1949. [Google Scholar] [CrossRef]
- Zheng, S.; Zou, Y.; Xie, X.; Liang, J.Y.; Yang, A.; Yu, K.; Wang, J.; Tang, H.; Xie, X. Development and validation of a stromal immune phenotype classifier for predicting immune activity and prognosis in triple-negative breast cancer. Int. J. Cancer 2020, 147, 542–553. [Google Scholar] [CrossRef]
- Theocharidis, G.; Baltzis, D.; Roustit, M.; Tellechea, A.; Dangwal, S.; Khetani, R.S.; Shu, B.; Zhao, W.; Fu, J.; Bhasin, S.; et al. Integrated Skin Transcriptomics and Serum Multiplex Assays Reveal Novel Mechanisms of Wound Healing in Diabetic Foot Ulcers. Diabetes 2020, 69, 2157–2169. [Google Scholar] [CrossRef]
- Mitterberger, M.C.; Lechner, S.; Mattesich, M.; Kaiser, A.; Probst, D.; Wenger, N.; Pierer, G.; Zwerschke, W. DLK1(PREF1) is a negative regulator of adipogenesis in CD105+/CD90+/CD34+/CD31−/FABP4− adipose-derived stromal cells from subcutaneous abdominal fat pats of adult women. Stem Cell Res. 2012, 9, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Denisenko, E.; Guo, B.B.; Jones, M.; Hou, R.; de Kock, L.; Lassmann, T.; Poppe, D.; Clément, O.; Simmons, R.K.; Lister, R.; et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 2020, 21, 130. [Google Scholar] [CrossRef]
- Qiu, Y.; Wang, J.; Lei, J.; Roeder, K. Identification of cell-type-specific marker genes from co-expression patterns in tissue samples. Bioinformatics 2021, 37, 3228–3234. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yang, G.; Zhang, Y.; Wang, Y. Spatial transcriptomics in development and disease. Mol. Biomed. 2023, 4, 32. [Google Scholar] [CrossRef]
- Baran, Y.; Doğan, B. scMAGS: Marker gene selection from scRNA-seq data for spatial transcriptomics studies. Comput. Biol. Med. 2023, 155, 106634. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.Y.; You, L.; Hardillo, J.A.U.; Chien, M.P. Spatial Transcriptomic Technologies. Cells 2023, 12, 2042. [Google Scholar] [CrossRef]
- Wang, R.; Peng, G.; Tam, P.P.L.; Jing, N. Integration of Computational Analysis and Spatial Transcriptomics in Single-cell Studies. Genom. Proteom. Bioinform. 2023, 21, 13–23. [Google Scholar] [CrossRef]
- Yue, L.; Liu, F.; Hu, J.; Yang, P.; Wang, Y.; Dong, J.; Shu, W.; Huang, X.; Wang, S. A guidebook of spatial transcriptomic technologies, data resources and analysis approaches. Comput. Struct. Biotechnol. J. 2023, 21, 940–955. [Google Scholar] [CrossRef] [PubMed]
- Robles-Remacho, A.; Sanchez-Martin, R.M.; Diaz-Mochon, J.J. Spatial Transcriptomics: Emerging Technologies in Tissue Gene Expression Profiling. Anal. Chem. 2023, 95, 15450–15460. [Google Scholar] [CrossRef] [PubMed]
- Antico, F.; Gai, M.; Arigoni, M. Tissue RNA Integrity in Visium Spatial Protocol (Fresh Frozen Samples). Methods Mol. Biol. 2023, 2584, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.M.; Ge, J.Y.; Chen, Y.F.; Liu, T.; Chen, L.; Liu, C.C.; Ma, D.; Chen, Y.Y.; Cai, Y.W.; Xu, Y.Y.; et al. Combined Single-Cell and Spatial Transcriptomics Reveal the Metabolic Evolvement of Breast Cancer during Early Dissemination. Adv. Sci. 2023, 10, e2205395. [Google Scholar] [CrossRef]
- Yoshitake, R.; Mori, H.; Ha, D.; Wu, X.; Wang, J.; Wang, X.; Saeki, K.; Chang, G.; Shim, H.J.; Chan, Y.; et al. Molecular features of luminal breast cancer defined through spatial and single-cell transcriptomics. Clin. Transl. Med. 2024, 14, e1548. [Google Scholar] [CrossRef] [PubMed]
- Gray, G.K.; Li, C.M.; Rosenbluth, J.M.; Selfors, L.M.; Girnius, N.; Lin, J.R.; Schackmann, R.C.J.; Goh, W.L.; Moore, K.; Shapiro, H.K.; et al. A human breast atlas integrating single-cell proteomics and transcriptomics. Dev. Cell 2022, 57, 1400–1420.e1407. [Google Scholar] [CrossRef] [PubMed]
- Bhat-Nakshatri, P.; Gao, H.; Khatpe, A.S.; Adebayo, A.K.; McGuire, P.C.; Erdogan, C.; Chen, D.; Jiang, G.; New, F.; German, R.; et al. Single-nucleus chromatin accessibility and transcriptomic map of breast tissues of women of diverse genetic ancestry. Nat. Med. 2024, 30, 3482–3494. [Google Scholar] [CrossRef]
- Iglesia, M.D.; Jayasinghe, R.G.; Chen, S.; Terekhanova, N.V.; Herndon, J.M.; Storrs, E.; Karpova, A.; Zhou, D.C.; Naser Al Deen, N.; Shinkle, A.T.; et al. Differential chromatin accessibility and transcriptional dynamics define breast cancer subtypes and their lineages. Nat. Cancer 2024, 5, 1713–1736. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, X.; Bi, J.; Jiang, X.; Zhang, L.; Li, Z.; Shao, M. Integrating single-cell and spatial transcriptomes reveals COL4A1/2 facilitates the spatial organisation of stromal cells differentiation in breast phyllodes tumours. Clin. Transl. Med. 2024, 14, e1611. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Ma, X.; La, Y.; Guo, X.; Chu, M.; Bao, P.; Yan, P.; Wu, X.; Liang, C. Advancements in the Application of scRNA-Seq in Breast Research: A Review. Int. J. Mol. Sci. 2024, 25, 13706. https://doi.org/10.3390/ijms252413706
Zhang Z, Ma X, La Y, Guo X, Chu M, Bao P, Yan P, Wu X, Liang C. Advancements in the Application of scRNA-Seq in Breast Research: A Review. International Journal of Molecular Sciences. 2024; 25(24):13706. https://doi.org/10.3390/ijms252413706
Chicago/Turabian StyleZhang, Zhenyu, Xiaoming Ma, Yongfu La, Xian Guo, Min Chu, Pengjia Bao, Ping Yan, Xiaoyun Wu, and Chunnian Liang. 2024. "Advancements in the Application of scRNA-Seq in Breast Research: A Review" International Journal of Molecular Sciences 25, no. 24: 13706. https://doi.org/10.3390/ijms252413706
APA StyleZhang, Z., Ma, X., La, Y., Guo, X., Chu, M., Bao, P., Yan, P., Wu, X., & Liang, C. (2024). Advancements in the Application of scRNA-Seq in Breast Research: A Review. International Journal of Molecular Sciences, 25(24), 13706. https://doi.org/10.3390/ijms252413706