
Nanoparticle-Based Immunotherapy for Reversing T-Cell Exhaustion

Abstract
:1. Introduction
2. T-Cell Exhaustion Is Common in Both Infectious Diseases and Cancer
3. Nanoparticle Classification and Application
3.1. Lipid-Based NPs
3.2. Polymeric NPs
3.3. Inorganic NPs
3.4. Other NPs
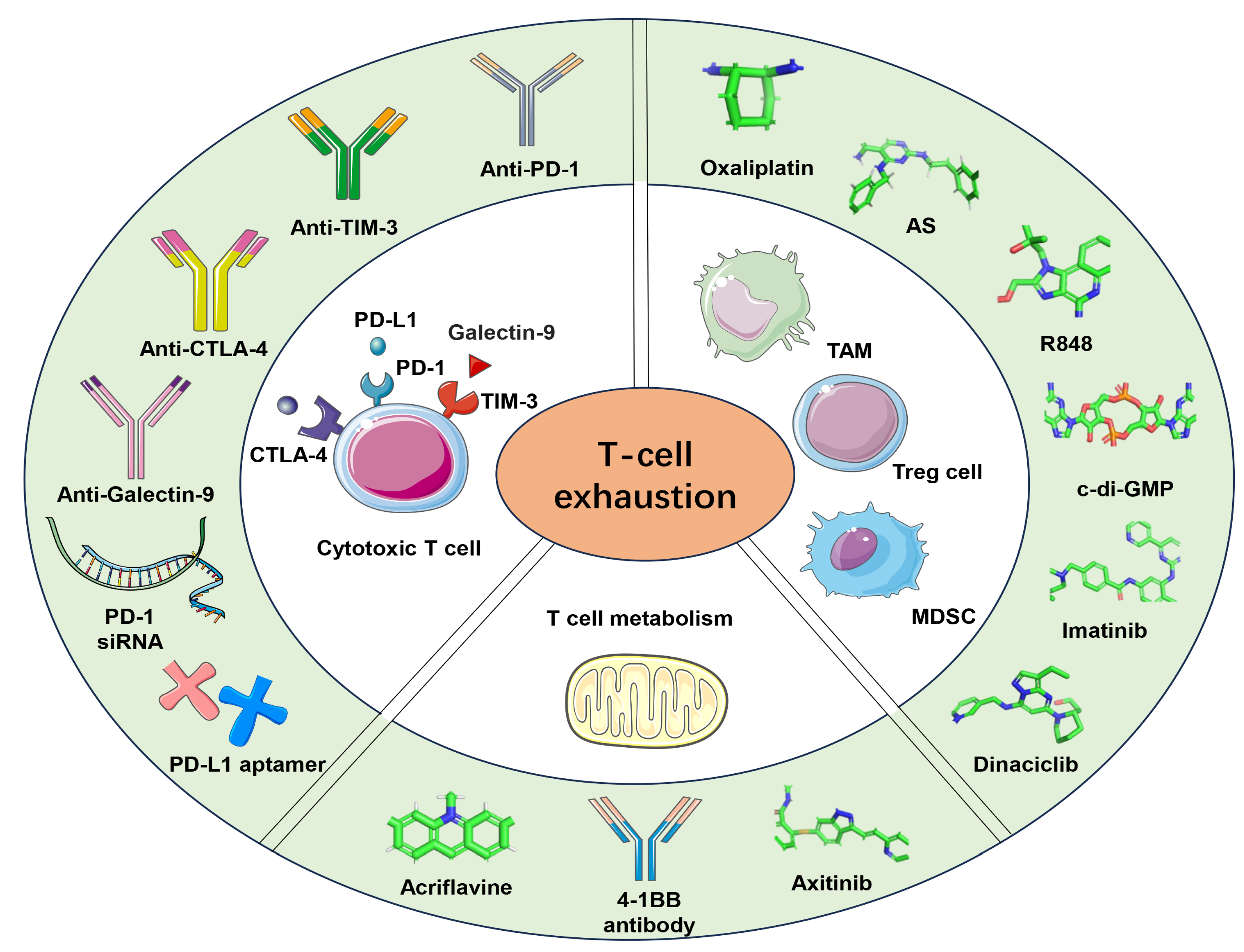
4. Nanoparticle-Based Immunotherapy in Reversing T-Cell Exhaustion
4.1. Targeting Checkpoint Blockade
4.2. Remodeling the Tumor Microenvironment
4.2.1. Targeting TAMs
- (1)
- Inducing TAMs repolarization
- (2)
- Inhibiting TAMs recruitment
- (3)
- TAMs depletion
4.2.2. Targeting Immunosuppressive Cells
4.3. Targeting T-Cell Metabolism

{kind=link}
| Strategy | Composition of NPs | Immunomodulators | Target Cells | Intervention Mechanism | Ref. |
|---|---|---|---|---|---|
| Targeting checkpoint blockade | PAMAM dendrimer-entrapped AuNPs | PD-1 siRNA; IDO inhibitor | T cells | Silencing PD-1 gene | [49] |
| lncRNA-edited tumor CM-camouflaged NPs | Anti-TIM-3 | DC cells and CD8+ T cells | Mediating antigen cross-presentation and dampening immunosuppression | [122] | |
| LMP2-mRNA LNPs | Anti-PD-1 | CD8+ T cells | Enhancing memory T-cell formation | [123] | |
| CaP-based nanoparticle vaccine | Anti-PD-L1; CpG; a virus-specific CD8+ T cell epitope | CD8+ T cell | Reactivating CD8+ T cell immunity | [127] | |
| T-cell-derived nanovesicles | PD-1; TGF-β receptor | Cancer cells | Blocking the PD-L1 pathway and eliminating TGF-β | [128] | |
| T-cell-membrane-coated NPs (TCMNPs) | LFA; Dacarbazine | Tumor cells | Blocking immune checkpoint interactions and inducing FasL-mediated apoptosis | [95] | |
| Gold nanorods (GNRs) | PD-L1 aptamer | Tumor cells | Activating CTLs and inhibiting Treg cells | [129] | |
| Remodeling the tumor microenvironment | MOFs coating with PM | Lactate oxidase; Oxaliplatin | TAMs | Promoting M2-to-M1 repolarization and decreasing Treg levels | [139] |
| PEG-decorated NPs | Anti-Galectin-9; AS | TAMs | Promoting M2-to-M1 repolarization and enhancing effector T-cell infiltration | [46] | |
| Cyclodextrin NPs | TLR7/8 agonist (R848); anti-PD-1 | TAMs | Promoting M2-to-M1 repolarization and inhibiting tumor growth | [142] | |
| Neutral cytidinyl lipid DNCA/cationic lipid CLD | c-di-GMP | Cancer cells | Triggering immunogenic cell death and increasing effector T-cell infiltration | [146] | |
| Cationic NPs | CCR2 siRNA | Monocytes | Inhibition of TAMs recruitment | [154] | |
| Liposome | Clodronate | TAMs | TAMs depletion | [160] | |
| Lipid NPs | CSF-1R siRNA; M2pep; a scavenger receptor targeting peptide | TAMs; scavenger receptor | TAMs depletion | [164] | |
| Hybrid NPs | tLyp1 peptide; Imatinib; Anti-CTLA-4 | Tregs | Downregulating Tregs suppression | [165] | |
| Lipid NPs | Dinaciclib; Anti-PD-L1 | MDSCs | Depleting MDSCs and attenuating their immunosuppressive functions | [166] | |
| Targeting T-cell metabolism | Tumor CM decorated vesicle | Axitinib; 4-1BB antibody; PF-06446846 | T cells | Promoting T-cell mitochondrial biogenesis and reducing hypoxia | [168] |
| MnO2 NPs | Acriflavine | Tumor cells | HIF-1α functional inhibition and subsequently activating tumor-specific immune responses | [171] |
5. Conclusions and Perspectives
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Pace, L. Temporal and epigenetic control of plasticity and fate decision during CD8+ T-cell memory differentiation. Cold Spring Harb. Perspect. Biol. 2021, 13, a037754. [Google Scholar] [CrossRef] [PubMed]
- Tanel, A.; Fonseca, S.G.; Yassine-Diab, B.; Bordi, R.; Zeidan, J.; Shi, Y.; Benne, C.; Sékaly, R.P. Cellular and molecular mechanisms of memory T-cell survival. Expert. Rev. Vaccines 2009, 8, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Gattinoni, L. Lineage relationship of effector and memory T cells. Curr. Opin. Immunol. 2013, 25, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Tan, J.T.; Wherry, E.J.; Konieczny, B.T.; Surh, C.D.; Ahmed, R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 2003, 4, 1191–1198. [Google Scholar] [CrossRef]
- Lugli, E.; Galletti, G.; Boi, S.K.; Youngblood, B.A. Stem, effector, and hybrid states of memory CD8+ T cells. Trends Immunol. 2020, 41, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Wherry, E.J. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity 2007, 27, 393–405. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef]
- Masopust, D.; Schenkel, J.M. The integration of T cell migration, differentiation and function. Nat. Rev. Immunol. 2013, 13, 309–320. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Dyck, L.; Mills, K.H.G. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur. J. Immunol. 2017, 47, 765–779. [Google Scholar] [CrossRef]
- Callahan, M.K.; Postow, M.A.; Wolchok, J.D. Targeting T cell co-receptors for cancer therapy. Immunity 2016, 44, 1069–1078. [Google Scholar] [CrossRef]
- Smith, D.M.; Simon, J.K.; Baker, J.R., Jr. Applications of nanotechnology for immunology. Nat. Rev. Immunol. 2013, 13, 592–605. [Google Scholar] [CrossRef]
- Cheng, R.; Santos, H.A. Smart nanoparticle-based platforms for regulating tumor microenvironment and cancer immunotherapy. Adv. Healthc. Mater. 2023, 12, e2202063. [Google Scholar] [CrossRef]
- Qi, J.; Jin, F.; Xu, X.; Du, Y. Combination cancer immunotherapy of nanoparticle-based immunogenic cell death inducers and immune checkpoint inhibitors. Int. J. Nanomed. 2021, 16, 1435–1456. [Google Scholar] [CrossRef]
- Jin, H.T.; Jeong, Y.H.; Park, H.J.; Ha, S.J. Mechanism of T cell exhaustion in a chronic environment. BMB Rep. 2011, 44, 217–231. [Google Scholar] [CrossRef]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef]
- Lombardi, A.; Villa, S.; Castelli, V.; Bandera, A.; Gori, A. T-cell exhaustion in mycobacterium tuberculosis and nontuberculous mycobacteria Infection: Pathophysiology and therapeutic perspectives. Microorganisms 2021, 9, 2460. [Google Scholar] [CrossRef]
- Scharping, N.E.; Rivadeneira, D.B.; Menk, A.V.; Vignali, P.D.A.; Ford, B.R.; Rittenhouse, N.L.; Peralta, R.; Wang, Y.; Wang, Y.; DePeaux, K.; et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 2021, 22, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-cell exhaustion: Characteristics, causes and conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef] [PubMed]
- Canale, F.P.; Ramello, M.C.; Núñez, N.; Araujo Furlan, C.L.; Bossio, S.N.; Gorosito Serrán, M.; Tosello Boari, J.; Del Castillo, A.; Ledesma, M.; Sedlik, C.; et al. CD39 expression defines cell exhaustion in tumor-infiltrating CD8+ T cells. Cancer Res. 2018, 78, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Usatorre, A.; Carmona, S.J.; Godfroid, C.; Yacoub Maroun, C.; Labiano, S.; Romero, P. Enhanced phenotype definition for precision isolation of precursor exhausted tumor-infiltrating CD8 T cells. Front. Immunol. 2020, 11, 340. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Shin, H.; Blackburn, S.D.; Intlekofer, A.M.; Kao, C.; Angelosanto, J.M.; Reiner, S.L.; Wherry, E.J. A role for the transcriptional repressor Blimp-1 in CD8+ T cell exhaustion during chronic viral infection. Immunity 2009, 31, 309–320. [Google Scholar] [CrossRef]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef]
- Kao, C.; Oestreich, K.J.; Paley, M.A.; Crawford, A.; Angelosanto, J.M.; Ali, M.A.; Intlekofer, A.M.; Boss, J.M.; Reiner, S.L.; Weinmann, A.S.; et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat. Immunol. 2011, 12, 663–671. [Google Scholar] [CrossRef]
- Hudson, W.H.; Gensheimer, J.; Hashimoto, M.; Wieland, A.; Valanparambil, R.M.; Li, P.; Lin, J.X.; Konieczny, B.T.; Im, S.J.; Freeman, G.J.; et al. Proliferating transitory T cells with an effector-like transcriptional signature emerge from PD-1+ stem-like CD8+ T cells during chronic infection. Immunity 2019, 51, 1043–1058.e4. [Google Scholar] [CrossRef]
- Kagoya, Y. Dissecting the heterogeneity of exhausted T cells at the molecular level. Int. Immunol. 2022, 34, 547–553. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ji, Z.; Ngiow, S.F.; Manne, S.; Cai, Z.; Huang, A.C.; Johnson, J.; Staupe, R.P.; Bengsch, B.; Xu, C.; et al. TCF-1-centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity 2019, 51, 840–855.e5. [Google Scholar] [CrossRef] [PubMed]
- Daniel, B.; Yost, K.E.; Hsiung, S.; Sandor, K.; Xia, Y.; Qi, Y.; Hiam-Galvez, K.J.; Black, M.; Raposo, C.J.; Shi, Q.; et al. Divergent clonal differentiation trajectories of T cell exhaustion. Nat. Immunol. 2022, 23, 1614–1627. [Google Scholar] [CrossRef] [PubMed]
- Adams, W.C.; Chen, Y.H.; Kratchmarov, R.; Yen, B.; Nish, S.A.; Lin, W.W.; Rothman, N.J.; Luchsinger, L.L.; Klein, U.; Busslinger, M.; et al. Anabolism-associated mitochondrial stasis driving lymphocyte differentiation over self-renewal. Cell Rep. 2016, 17, 3142–3152. [Google Scholar] [CrossRef] [PubMed]
- Schurich, A.; Pallett, L.J.; Jajbhay, D.; Wijngaarden, J.; Otano, I.; Gill, U.S.; Hansi, N.; Kennedy, P.T.; Nastouli, E.; Gilson, R.; et al. Distinct metabolic requirements of exhausted and functional virus-specific CD8 T cells in the same host. Cell Rep. 2016, 16, 1243–1252. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef]
- Siska, P.J.; Beckermann, K.E.; Mason, F.M.; Andrejeva, G.; Greenplate, A.R.; Sendor, A.B.; Chiang, Y.J.; Corona, A.L.; Gemta, L.F.; Vincent, B.G.; et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2017, 2, e93411. [Google Scholar] [CrossRef]
- McKinney, E.F.; Smith, K.G.C. Metabolic exhaustion in infection, cancer and autoimmunity. Nat. Immunol. 2018, 19, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Pallett, L.J.; Schmidt, N.; Schurich, A. T cell metabolism in chronic viral infection. Clin. Exp. Immunol. 2019, 197, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Vardhana, S.A.; Hwee, M.A.; Berisa, M.; Wells, D.K.; Yost, K.E.; King, B.; Smith, M.; Herrera, P.S.; Chang, H.Y.; Satpathy, A.T.; et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol. 2020, 21, 1022–1033. [Google Scholar] [CrossRef]
- Verdon, D.J.; Mulazzani, M.; Jenkins, M.R. Cellular and molecular mechanisms of CD8+ T cell differentiation, dysfunction and exhaustion. Int. J. Mol. Sci. 2020, 21, 7357. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Song, J.; Linghu, D.; Yang, R.; Liu, B.; Xue, Z.; Chen, Q.; Liu, C.; Zhong, D.; Hung, M.C.; et al. Galectin-9 blockade synergizes with ATM inhibition to induce potent anti-tumor immunity. Int. J. Biol. Sci. 2023, 19, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Zhang, Q.; Yi, J.; Wang, R.; Zhang, Z.; Luo, P.; Zeng, R.; Wang, Y.; Tu, M. PEG-sheddable nanodrug remodels tumor microenvironment to promote effector T cell infiltration and revise their exhaustion for breast cancer immunotherapy. Small 2023, 19, e2301749. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hakeem, M.S.; Manne, S.; Beltra, J.C.; Stelekati, E.; Chen, Z.; Nzingha, K.; Ali, M.A.; Johnson, J.L.; Giles, J.R.; Mathew, D.; et al. Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat. Immunol. 2021, 22, 1008–1019. [Google Scholar] [CrossRef]
- Tabana, Y.; Moon, T.C.; Siraki, A.; Elahi, S.; Barakat, K. Reversing T-cell exhaustion in immunotherapy: A review on current approaches and limitations. Expert Opin. Ther. Targets 2021, 25, 347–363. [Google Scholar] [CrossRef]
- Gao, Y.; Ouyang, Z.; Yang, C.; Song, C.; Jiang, C.; Song, S.; Shen, M.; Shi, X. Overcoming T cell exhaustion via immune checkpoint modulation with a dendrimer-based hybrid nanocomplex. Adv. Healthc. Mater. 2021, 10, e2100833. [Google Scholar] [CrossRef]
- Wu, P.; Han, J.; Gong, Y.; Liu, C.; Yu, H.; Xie, N. Nanoparticle-based drug delivery systems targeting tumor microenvironment for cancer immunotherapy resistance: Current advances and applications. Pharmaceutics 2022, 14, 1990. [Google Scholar] [CrossRef]
- Xu, C.; Lei, C.; Yu, C. Mesoporous silica nanoparticles for protein protection and delivery. Front. Chem. 2019, 7, 290. [Google Scholar] [CrossRef]
- Chong, G.; Zang, J.; Han, Y.; Su, R.; Weeranoppanant, N.; Dong, H.; Li, Y. Bioengineering of nano metal-organic frameworks for cancer immunotherapy. Nano Res. 2021, 14, 1244–1259. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, M.D.; Liang, L.; Xing, H.; Zhang, C.W.; Shen, F.; Huang, D.S.; Yang, T. Nanotechnology for hepatocellular carcinoma: From surveillance, diagnosis to management. Small 2021, 17, e2005236. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y. Nanotechnology for next-generation cancer immunotherapy: State of the art and future perspectives. J. Control. Release 2023, 356, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Li, S.R.; Huo, F.Y.; Wang, H.Q.; Wang, J.; Xu, C.; Liu, B.; Bu, L.L. Recent advances in porous nanomaterials-based drug delivery systems for cancer immunotherapy. J. Nanobiotechnol. 2022, 20, 277. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Dai, H.; Fan, Q.; Wang, C. Recent applications of immunomodulatory biomaterials for disease immunotherapy. Exploration 2022, 2, 20210157. [Google Scholar] [CrossRef]
- Liu, R.; Luo, C.; Pang, Z.Q.; Zhang, J.M.; Ruan, S.B.; Wu, M.Y.; Wang, L.; Sun, T.; Li, N.; Han, L.; et al. Advances of nanoparticles as drug delivery systems for disease diagnosis and treatment. Chin. Chem. Lett. 2023, 34, 107518. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef] [PubMed]
- Fenton, O.S.; Olafson, K.N.; Pillai, P.S.; Mitchell, M.J.; Langer, R. Advances in biomaterials for drug delivery. Adv. Mater. 2018, 30, e1705328. [Google Scholar] [CrossRef] [PubMed]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Multifunctional nanocarriers. Adv. Drug Deliv. Rev. 2006, 58, 1532–1555. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Santos, B.; Gremião, M.P.; Chorilli, M. Nanotechnology-based drug delivery systems for the treatment of Alzheimer’s disease. Int. J. Nanomed. 2015, 10, 4981–5003. [Google Scholar] [CrossRef] [PubMed]
- Hoshyar, N.; Gray, S.; Han, H.; Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 2016, 11, 673–692. [Google Scholar] [CrossRef]
- Kim, M.; Jeong, M.; Hur, S.; Cho, Y.; Park, J.; Jung, H.; Seo, Y.; Woo, H.A.; Nam, K.T.; Lee, K.; et al. Engineered ionizable lipid nanoparticles for targeted delivery of RNA therapeutics into different types of cells in the liver. Sci. Adv. 2021, 7, eabf4398. [Google Scholar] [CrossRef]
- Han, J.; Lim, J.; Wang, C.J.; Han, J.H.; Shin, H.E.; Kim, S.N.; Jeong, D.; Lee, S.H.; Chun, B.H.; Park, C.G.; et al. Lipid nanoparticle-based mRNA delivery systems for cancer immunotherapy. Nano Converg. 2023, 10, 36. [Google Scholar] [CrossRef]
- Leung, A.K.; Tam, Y.Y.; Chen, S.; Hafez, I.M.; Cullis, P.R. Microfluidic mixing: A general method for encapsulating macromolecules in lipid nanoparticle systems. J. Phys. Chem. B 2015, 119, 8698–8706. [Google Scholar] [CrossRef]
- Berraondo, P.; Martini, P.G.V.; Avila, M.A.; Fontanellas, A. Messenger RNA therapy for rare genetic metabolic diseases. Gut 2019, 68, 1323–1330. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef]
- Zhang, L.; Beatty, A.; Lu, L.; Abdalrahman, A.; Makris, T.M.; Wang, G.; Wang, Q. Microfluidic-assisted polymer-protein assembly to fabricate homogeneous functionalnanoparticles. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 111, 110768. [Google Scholar] [CrossRef]
- Xu, Y.; Ren, X.; Yu, M.; Weng, W.; Liu, Y.; Song, B.; Niu, J.; Qiao, Z.; Lin, Y.; Cao, Y.; et al. Nanotechnology-based drug delivery strategies for cancer therapy. Chin. Sci. Bull. 2023, 68, 4346–4372. (In Chinese) [Google Scholar] [CrossRef]
- Xu, Q.; Wang, Y.; Zheng, Y.; Zhu, Y.; Li, Z.; Liu, Y.; Ding, M. Polymersomes in drug delivery—From experiment to computational modeling. Biomacromolecules 2023. [Google Scholar] [CrossRef]
- Leong, J.; Teo, J.Y.; Aakalu, V.K.; Yang, Y.Y.; Kong, H. Engineering polymersomes for diagnostics and therapy. Adv. Healthc. Mater. 2018, 7, e1701276. [Google Scholar] [CrossRef]
- Rideau, E.; Dimova, R.; Schwille, P.; Wurm, F.R.; Landfester, K. Liposomes and polymersomes: A comparative review towards cell mimicking. Chem. Soc. Rev. 2018, 47, 8572–8610. [Google Scholar] [CrossRef]
- Zelmer, C.; Zweifel, L.P.; Kapinos, L.E.; Craciun, I.; Güven, Z.P.; Palivan, C.G.; Lim, R.Y.H. Organelle-specific targeting of polymersomes into the cell nucleus. Proc. Natl. Acad. Sci. USA 2020, 117, 2770–2778. [Google Scholar] [CrossRef]
- Gautam, M.; Jozic, A.; Su, G.L.; Herrera-Barrera, M.; Curtis, A.; Arrizabalaga, S.; Tschetter, W.; Ryals, R.C.; Sahay, G. Lipid nanoparticles with PEG-variant surface modifications mediate genome editing in the mouse retina. Nat. Commun. 2023, 14, 6468. [Google Scholar] [CrossRef]
- Ramezanpour, S.; Tavatoni, P.; Akrami, M.; Navaei-Nigjeh, M.; Shiri, P. Potential wound healing of PLGA nanoparticles containing a novel L-Carnitine–GHK peptide conjugate. J. Nanomater. 2022, 2022, 6165759. [Google Scholar] [CrossRef]
- Liu, K.; Hou, G.; Mao, J.; Xu, Z.; Yan, P.; Li, H.; Guo, X.; Bai, S.; Zhang, Z.C. Genesis of electron deficient Pt1(0) in PDMS-PEG aggregates. Nat. Commun. 2019, 10, 996. [Google Scholar] [CrossRef]
- Fishburn, C.S. The pharmacology of PEGylation: Balancing PD with PK to generate novel therapeutics. J. Pharm. Sci. 2008, 97, 4167–4183. [Google Scholar] [CrossRef]
- Swider, E.; Koshkina, O.; Tel, J.; Cruz, L.J.; de Vries, I.J.M.; Srinivas, M. Customizing poly(lactic-co-glycolic acid) particles for biomedical applications. Acta Biomater. 2018, 73, 38–51. [Google Scholar] [CrossRef]
- Liu, X.; Li, C.; Lv, J.; Huang, F.; An, Y.; Shi, L.; Ma, R. Glucose and H2O2 dual-responsive polymeric micelles for the self-regulated release of insulin. ACS Appl. Bio. Mater. 2020, 3, 1598–1606. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, Y.M.; Cho, C.H.; Kim, Y.T.; Kim, S.M.; Hur, S.Y.; Kim, J.H.; Kim, B.G.; Kim, S.C.; Ryu, H.S.; et al. An open-label, randomized, parallel, phase II trial to evaluate the efficacy and safety of a cremophor-free polymeric micelle formulation of paclitaxel as first-line treatment for ovarian cancer: A korean gynecologic oncology group study (KGOG-3021). Cancer Res. Treat. 2018, 50, 195–203. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, H.; Wu, Y. Dendrimer advances for the central nervous system delivery of therapeutics. ACS Chem. Neurosci. 2014, 5, 2–13. [Google Scholar] [CrossRef]
- Yokoyama, M. Drug targeting with nano-sized carrier systems. J. Artif. Organs. 2005, 8, 77–84. [Google Scholar] [CrossRef]
- Lee, K.X.; Shameli, K.; Yew, Y.P.; Teow, S.Y.; Jahangirian, H.; Rafiee-Moghaddam, R.; Webster, T.J. Recent developments in the facile bio-synthesis of gold nanoparticles (AuNPs) and their biomedical applications. Int. J. Nanomed. 2020, 15, 275–300. [Google Scholar] [CrossRef]
- Arias, L.S.; Pessan, J.P.; Vieira, A.P.M.; Lima, T.M.T.; Delbem, A.C.B.; Monteiro, D.R. Iron oxide nanoparticles for biomedical applications: A perspective on synthesis, drugs, antimicrobial activity, and toxicity. Antibiotics 2018, 7, 46. [Google Scholar] [CrossRef]
- Djayanti, K.; Maharjan, P.; Cho, K.H.; Jeong, S.; Kim, M.S.; Shin, M.C.; Min, K.A. Mesoporous silica nanoparticles as a potential nanoplatform: Therapeutic applications and considerations. Int. J. Mol. Sci. 2023, 24, 6349. [Google Scholar] [CrossRef]
- Huang, Y.; Ruan, Y.; Ma, Y.; Chen, D.; Zhang, T.; Fan, S.; Lin, W.; Huang, Y.; Lu, H.; Xu, J.F.; et al. Immunomodulatory activity of manganese dioxide nanoparticles: Promising for novel vaccines and immunotherapeutics. Front. Immunol. 2023, 14, 1128840. [Google Scholar] [CrossRef]
- Manshian, B.B.; Jiménez, J.; Himmelreich, U.; Soenen, S.J. Personalized medicine and follow-up of therapeutic delivery through exploitation of quantum dot toxicity. Biomaterials 2017, 127, 1–12. [Google Scholar] [CrossRef]
- Chugh, V.; Vijaya Krishna, K.; Pandit, A. Cell Membrane-coated mimics: A methodological approach for fabrication, characterization for therapeutic applications, and challenges for clinical translation. ACS Nano 2021, 15, 17080–17123. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Wang, J.; Lang, T.; Kong, Y.; Rong, R.; Cai, Y.; Ran, W.; Xiong, F.; Zheng, C.; Wang, Y.; et al. T lymphocyte membrane-decorated epigenetic nanoinducer of interferons for cancer immunotherapy. Nat. Nanotechnol. 2021, 16, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.Z.; Li, Z.H.; Bai, X.F.; Liu, C.J.; Zhang, X.Z. Hybrid vesicles based on autologous tumor cell membrane and bacterial outer membrane to enhance innate immune response and personalized tumor immunotherapy. Nano Lett. 2021, 21, 8609–8618. [Google Scholar] [CrossRef] [PubMed]
- Parodi, A.; Quattrocchi, N.; van de Ven, A.L.; Chiappini, C.; Evangelopoulos, M.; Martinez, J.O.; Brown, B.S.; Khaled, S.Z.; Yazdi, I.K.; Enzo, M.V.; et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat. Nanotechnol. 2013, 8, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Hong, J.; Jung, M.; Kwon, S.P.; Song, S.Y.; Kim, H.Y.; Lee, J.R.; Kang, S.; Han, J.; Koo, J.H.; et al. T-cell-mimicking nanoparticles for cancer immunotherapy. Adv. Mater. 2020, 32, e2003368. [Google Scholar] [CrossRef] [PubMed]
- Karami, Z.; Mehrzad, J.; Akrami, M.; Hosseinkhani, S. Anti-inflammation-based treatment of atherosclerosis using Gliclazide-loaded biomimetic nanoghosts. Sci. Rep. 2023, 13, 13880. [Google Scholar] [CrossRef] [PubMed]
- Karami, Z.; Akrami, M.; Mehrzad, J.; Esfandyari-Manesh, M.; Haririan, I.; Nateghi, S. An anti-inflammatory Glyburide-loaded nanoghost for atherosclerosis therapy: A red blood cell based bio-mimetic strategy. Giant 2023, 16, 100206. [Google Scholar] [CrossRef]
- Hong, J.; Jung, M.; Kim, C.; Kang, M.; Go, S.; Sohn, H.; Moon, S.; Kwon, S.; Song, S.Y.; Kim, B.S. Senescent cancer cell-derived nanovesicle as a personalized therapeutic cancer vaccine. Exp. Mol. Med. 2023, 55, 541–554. [Google Scholar] [CrossRef]
- Rao, L.; Wu, L.; Liu, Z.; Tian, R.; Yu, G.; Zhou, Z.; Yang, K.; Xiong, H.G.; Zhang, A.; Yu, G.T.; et al. Hybrid cellular membrane nanovesicles amplify macrophage immune responses against cancer recurrence and metastasis. Nat. Commun. 2020, 11, 4909. [Google Scholar] [CrossRef]
- Jiang, X.C.; Zhang, T.; Gao, J.Q. The in vivo fate and targeting engineering of crossover vesicle-based gene delivery system. Adv. Drug Deliv. Rev. 2022, 187, 114324. [Google Scholar] [CrossRef]
- Li, Y.J.; Wu, J.Y.; Liu, J.; Xu, W.; Qiu, X.; Huang, S.; Hu, X.B.; Xiang, D.X. Artificial exosomes for translational nanomedicine. J. Nanobiotechnol. 2021, 19, 242. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Miao, Y.; Wang, Y.; He, S.; Guo, L.; Mao, J.; Chen, M.; Yang, Y.; Zhang, X.; Gan, Y. Tumour-derived extracellular vesicle membrane hybrid lipid nanovesicles enhance siRNA delivery by tumour-homing and intracellular freeway transportation. J. Extracell Vesicles 2022, 11, e12198. [Google Scholar] [CrossRef] [PubMed]
- Jo, W.; Kim, J.; Yoon, J.; Jeong, D.; Cho, S.; Jeong, H.; Yoon, Y.J.; Kim, S.C.; Gho, Y.S.; Park, J. Large-scale generation of cell-derived nanovesicles. Nanoscale 2014, 6, 12056–12064. [Google Scholar] [CrossRef] [PubMed]
- Sayyed, A.A.; Gondaliya, P.; Yan, I.K.; Carrington, J.; Driscoll, J.; Moirangthem, A.; Patel, T. Engineering cell-derived nanovesicles for targeted immunomodulation. Nanomaterials 2023, 13, 2751. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.R.; Kyung, J.W.; Kumar, H.; Kwon, S.P.; Song, S.Y.; Han, I.B.; Kim, B.S. Targeted delivery of mesenchymal stem cell-derived nanovesicles for spinal cord injury treatment. Int. J. Mol. Sci. 2020, 21, 4185. [Google Scholar] [CrossRef]
- Harada, T.; Tsuboi, I.; Hino, H.; Yuda, M.; Hirabayashi, Y.; Hirai, S.; Aizawa, S. Age-related exacerbation of hematopoietic organ damage induced by systemic hyper-inflammation in senescence-accelerated mice. Sci. Rep. 2021, 11, 23250. [Google Scholar] [CrossRef]
- Lian, X.; Huang, Y.; Zhu, Y.; Fang, Y.; Zhao, R.; Joseph, E.; Li, J.; Pellois, J.P.; Zhou, H.C. Enzyme-MOF nanoreactor activates nontoxic paracetamol for cancer therapy. Angew. Chem. Int. Ed. Engl. 2018, 57, 5725–5730. [Google Scholar] [CrossRef]
- Gkaniatsou, E.; Sicard, C.; Ricoux, R.; Benahmed, L.; Bourdreux, F.; Zhang, Q.; Serre, C.; Mahy, J.P.; Steunou, N. Enzyme encapsulation in mesoporous metal-organic frameworks for selective biodegradation of harmful dye molecules. Angew. Chem. Int. Ed. Engl. 2018, 57, 16141–16146. [Google Scholar] [CrossRef]
- Wan, X.; Song, L.; Pan, W.; Zhong, H.; Li, N.; Tang, B. Tumor-targeted cascade nanoreactor based on metal-organic frameworks for synergistic ferroptosis-starvation anticancer therapy. ACS Nano 2020, 14, 11017–11028. [Google Scholar] [CrossRef]
- Wan, X.; Zhong, H.; Pan, W.; Li, Y.; Chen, Y.; Li, N.; Tang, B. Programmed release of dihydroartemisinin for synergistic cancer therapy using a CaCO3 mineralized metal-organic framework. Angew. Chem. Int. Ed. Engl. 2019, 58, 14134–14139. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, F.; Liu, C.; Wang, Z.; Kang, L.; Huang, Y.; Dong, K.; Ren, J.; Qu, X. Nanozyme decorated metal-organic frameworks for enhanced photodynamic therapy. ACS Nano 2018, 12, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Diercks, C.S.; Yaghi, O.M. The atom, the molecule, and the covalent organic framework. Science 2017, 355, eaal1585. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Liu, Y.; Lau, J.; Fan, W.; Li, Q.; Zhang, C.; Huang, P.; Chen, X. Recent progress in nanoscale metal-organic frameworks for drug release and cancer therapy. Nanomedicine 2019, 14, 1343–1365. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Kang, H.; Lee, H.H.; Kim, C.W. Programmed cell death 1 (PD-1) and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) in viral hepatitis. Int. J. Mol. Sci. 2017, 18, 1517. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef] [PubMed]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef]
- Marshall, H.T.; Djamgoz, M.B.A. Immuno-oncology: Emerging targets and combination therapies. Front. Oncol. 2018, 8, 315. [Google Scholar] [CrossRef]
- Urbanavicius, D.; Alvarez, T.; Such, G.K.; Johnston, A.P.R.; Mintern, J.D. The potential of nanoparticle vaccines as a treatment for cancer. Mol. Immunol. 2018, 98, 2–7. [Google Scholar] [CrossRef]
- Ahmad, A.; Khan, F.; Mishra, R.K.; Khan, R. Precision cancer nanotherapy: Evolving Role of multifunctional nanoparticles for cancer active targeting. J. Med. Chem. 2019, 62, 10475–10496. [Google Scholar] [CrossRef]
- Wu, Y.; Gu, W.; Li, L.; Chen, C.; Xu, Z.P. Enhancing PD-1 gene silence in T lymphocytes by comparing the delivery performance of two inorganic nanoparticle platforms. Nanomaterials 2019, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, F.; Tan, L.; Li, X.; Dai, Z.; Cheng, Q.; Liu, J.; Wang, Y.; Huang, L.; Wang, L.; et al. LncRNA-edited biomimetic nanovaccines combined with anti-TIM-3 for augmented immune checkpoint blockade immunotherapy. J. Control. Release 2023, 361, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Tian, M.; Huang, J.; Li, Y.; Li, G.; Li, X.; Jiang, Z.; Song, X.; Ma, X. LMP2-mRNA lipid nanoparticle sensitizes EBV-related tumors to anti-PD-1 therapy by reversing T cell exhaustion. J. Nanobiotechnol. 2023, 21, 324. [Google Scholar] [CrossRef] [PubMed]
- Knuschke, T.; Bayer, W.; Rotan, O.; Sokolova, V.; Wadwa, M.; Kirschning, C.J.; Hansen, W.; Dittmer, U.; Epple, M.; Buer, J.; et al. Prophylactic and therapeutic vaccination with a nanoparticle-based peptide vaccine induces efficient protective immunity during acute and chronic retroviral infection. Nanomedicine 2014, 10, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Knuschke, T.; Rotan, O.; Bayer, W.; Kollenda, S.; Dickow, J.; Sutter, K.; Hansen, W.; Dittmer, U.; Lang, K.S.; Epple, M.; et al. Induction of type I interferons by therapeutic nanoparticle-based vaccination is indispensable to reinforce cytotoxic CD8+ T cell responses during chronic retroviral infection. Front. Immunol. 2018, 9, 614. [Google Scholar] [CrossRef]
- Heße, C.; Kollenda, S.; Rotan, O.; Pastille, E.; Adamczyk, A.; Wenzek, C.; Hansen, W.; Epple, M.; Buer, J.; Westendorf, A.M.; et al. A tumor-peptide-based nanoparticle vaccine elicits efficient tumor growth control in antitumor immunotherapy. Mol. Cancer Ther. 2019, 18, 1069–1080. [Google Scholar] [CrossRef]
- Knuschke, T.; Kollenda, S.; Wenzek, C.; Zelinskyy, G.; Steinbach, P.; Dittmer, U.; Buer, J.; Epple, M.; Westendorf, A.M. A combination of anti-PD-L1 treatment and therapeutic vaccination facilitates improved retroviral clearance via reactivation of highly exhausted T cells. mBio 2021, 12, 10–1128. [Google Scholar] [CrossRef]
- Hong, J.; Kang, M.; Jung, M.; Lee, Y.Y.; Cho, Y.; Kim, C.; Song, S.Y.; Park, C.G.; Doh, J.; Kim, B.S. T-cell-derived nanovesicles for cancer immunotherapy. Adv. Mater. 2021, 33, e2101110. [Google Scholar] [CrossRef]
- Chang, R.; Li, T.; Fu, Y.; Chen, Z.; He, Y.; Sun, X.; Deng, Y.; Zhong, Y.; Xie, Z.; Yang, Y.; et al. A PD-L1 targeting nanotheranostic for effective photoacoustic imaging guided photothermal-immunotherapy of tumor. J. Mater. Chem. B 2023, 11, 8492–8505. [Google Scholar] [CrossRef]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab. 2019, 30, 143–156.e5. [Google Scholar] [CrossRef]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Wang, W.; Wang, S.; Yang, T.; Zhang, G.; Wang, D.; Ju, R.; Lu, Y.; Wang, H.; Wang, L. Tumor microenvironment remodeling and tumor therapy based on M2-like tumor associated macrophage-targeting nano-complexes. Theranostics 2021, 11, 2892–2916. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Kitamura, T. Macrophage targeting: Opening new possibilities for cancer immunotherapy. Immunology 2018, 155, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Yrigoyen, M.; Cassetta, L.; Pollard, J.W. Macrophage targeting in cancer. Ann. N. Y. Acad. Sci. 2021, 1499, 18–41. [Google Scholar] [CrossRef]
- Li, R.; Serrano, J.C.; Xing, H.; Lee, T.A.; Azizgolshani, H.; Zaman, M.; Kamm, R.D. Interstitial flow promotes macrophage polarization toward an M2 phenotype. Mol. Biol. Cell 2018, 29, 1927–1940. [Google Scholar] [CrossRef]
- Bai, R.; Li, Y.; Jian, L.; Yang, Y.; Zhao, L.; Wei, M. The hypoxia-driven crosstalk between tumor and tumor-associated macrophages: Mechanisms and clinical treatment strategies. Mol. Cancer 2022, 21, 177. [Google Scholar] [CrossRef]
- Wang, H.; Wu, C.; Tong, X.; Chen, S. A biomimetic metal-organic framework nanosystem modulates immunosuppressive tumor microenvironment metabolism to amplify immunotherapy. J. Control. Release 2023, 353, 727–737. [Google Scholar] [CrossRef]
- Leone, R.D.; Powell, J.D. Metabolism of immune cells in cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Saeed, M.; Gao, J.; Shi, Y.; Lammers, T.; Yu, H. Engineering nanoparticles to reprogram the tumor immune microenvironment for improved cancer immunotherapy. Theranostics 2019, 9, 7981–8000. [Google Scholar] [CrossRef] [PubMed]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Khong, H.; Dai, Z.; Huang, X.F.; Wargo, J.A.; Cooper, Z.A.; Vasilakos, J.P.; Hwu, P.; Overwijk, W.W. Effective innate and adaptive antimelanoma immunity through localized TLR7/8 activation. J. Immunol. 2014, 193, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, J.; Alu, A.; Han, X.; Wei, Y.; Wei, X. cGAS-STING pathway in cancer biotherapy. Mol. Cancer 2020, 19, 136. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yu, J.; Dai, H.; Deng, C.; Sun, X.; Long, S.; Jiang, Z.; Jin, H.; Guan, Z.; Yang, Z. Novel formulation of c-di-GMP with cytidinyl/cationic lipid reverses T cell exhaustion and activates stronger anti-tumor immunity. Theranostics 2022, 12, 6723–6739. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef]
- Argyle, D.; Kitamura, T. Targeting macrophage-recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Front. Immunol. 2018, 9, 2629. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Choi, S.H. Tumor-associated macrophages in cancer: Recent advancements in cancer nanoimmunotherapies. J. Exp. Clin. Cancer Res. 2022, 41, 68. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Zhang, Y.; Chen, K.G.; Luo, Y.L.; Wang, J. Cationic polymeric nanoparticle delivering CCR2 siRNA to inflammatory monocytes for tumor microenvironment modification and cancer therapy. Mol. Pharm. 2018, 15, 3642–3653. [Google Scholar] [CrossRef] [PubMed]
- Schmall, A.; Al-Tamari, H.M.; Herold, S.; Kampschulte, M.; Weigert, A.; Wietelmann, A.; Vipotnik, N.; Grimminger, F.; Seeger, W.; Pullamsetti, S.S.; et al. Macrophage and cancer cell cross-talk via CCR2 and CX3CR1 is a fundamental mechanism driving lung cancer. Am. J. Respir. Crit. Care Med. 2015, 191, 437–447. [Google Scholar] [CrossRef]
- Han, R.; Gu, S.; Zhang, Y.; Luo, A.; Jing, X.; Zhao, L.; Zhao, X.; Zhang, L. Estrogen promotes progression of hormone-dependent breast cancer through CCL2-CCR2 axis by upregulation of Twist via PI3K/AKT/NF-κB signaling. Sci. Rep. 2018, 8, 9575. [Google Scholar] [CrossRef] [PubMed]
- Tu, M.M.; Abdel-Hafiz, H.A.; Jones, R.T.; Jean, A.; Hoff, K.J.; Duex, J.E.; Chauca-Diaz, A.; Costello, J.C.; Dancik, G.M.; Tamburini, B.A.J.; et al. Inhibition of the CCL2 receptor, CCR2, enhances tumor response to immune checkpoint therapy. Commun. Biol. 2020, 3, 720. [Google Scholar] [CrossRef]
- Fritz, J.M.; Tennis, M.A.; Orlicky, D.J.; Lin, H.; Ju, C.; Redente, E.F.; Choo, K.S.; Staab, T.A.; Bouchard, R.J.; Merrick, D.T.; et al. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front. Immunol. 2014, 5, 587. [Google Scholar] [CrossRef]
- Kersten, K.; Hu, K.H.; Combes, A.J.; Samad, B.; Harwin, T.; Ray, A.; Rao, A.A.; Cai, E.; Marchuk, K.; Artichoker, J.; et al. Spatiotemporal co-dependency between macrophages and exhausted CD8+ T cells in cancer. Cancer Cell 2022, 40, 624–638.e9. [Google Scholar] [CrossRef]
- Hiraoka, K.; Zenmyo, M.; Watari, K.; Iguchi, H.; Fotovati, A.; Kimura, Y.N.; Hosoi, F.; Shoda, T.; Nagata, K.; Osada, H.; et al. Inhibition of bone and muscle metastases of lung cancer cells by a decrease in the number of monocytes/macrophages. Cancer Sci. 2008, 99, 1595–1602. [Google Scholar] [CrossRef]
- Cieslewicz, M.; Tang, J.; Yu, J.L.; Cao, H.; Zavaljevski, M.; Motoyama, K.; Lieber, A.; Raines, E.W.; Pun, S.H. Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc. Natl. Acad. Sci. USA 2013, 110, 15919–15924. [Google Scholar] [CrossRef] [PubMed]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Kowal, J.; Akkari, L.; Schuhmacher, A.J.; Huse, J.T.; West, B.L.; Joyce, J.A. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 2017, 36, 6049–6058. [Google Scholar] [CrossRef]
- Qian, Y.; Qiao, S.; Dai, Y.; Xu, G.; Dai, B.; Lu, L.; Yu, X.; Luo, Q.; Zhang, Z. Molecular-targeted immunotherapeutic strategy for melanoma via dual-targeting nanoparticles delivering small interfering RNA to tumor-associated macrophages. ACS Nano 2017, 11, 9536–9549. [Google Scholar] [CrossRef]
- Ou, W.; Thapa, R.K.; Jiang, L.; Soe, Z.C.; Gautam, M.; Chang, J.H.; Jeong, J.H.; Ku, S.K.; Choi, H.G.; Yong, C.S.; et al. Regulatory T cell-targeted hybrid nanoparticles combined with immuno-checkpoint blockage for cancer immunotherapy. J. Control Release 2018, 281, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Miska, J.; Lee-Chang, C.; Rashidi, A.; Panek, W.K.; An, S.; Zannikou, M.; Lopez-Rosas, A.; Han, Y.; Xiao, T.; et al. Therapeutic targeting of tumor-associated myeloid cells synergizes with radiation therapy for glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 23714–23723. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, Q.; Chen, X.; Nie, X.; Xue, F.; Xu, W.; Luan, Y. An injectable hydrogel to modulate T cells for cancer immunotherapy. Small 2022, 18, e2202663. [Google Scholar] [CrossRef]
- Wu, H.; Zhao, X.; Hochrein, S.M.; Eckstein, M.; Gubert, G.F.; Knöpper, K.; Mansilla, A.M.; Öner, A.; Doucet-Ladevèze, R.; Schmitz, W.; et al. Mitochondrial dysfunction promotes the transition of precursor to terminally exhausted T cells through HIF-1α-mediated glycolytic reprogramming. Nat. Commun. 2023, 14, 6858. [Google Scholar] [CrossRef]
- Shurin, M.R.; Umansky, V. Cross-talk between HIF and PD-1/PD-L1 pathways in carcinogenesis and therapy. J. Clin. Investg. 2022, 132, e159473. [Google Scholar] [CrossRef]
- Meng, L.; Cheng, Y.; Tong, X.; Gan, S.; Ding, Y.; Zhang, Y.; Wang, C.; Xu, L.; Zhu, Y.; Wu, J.; et al. Tumor oxygenation and hypoxia inducible factor-1 functional inhibition via a reactive oxygen species responsive nanoplatform for enhancing radiation therapy and abscopal effects. ACS Nano 2018, 12, 8308–8322. [Google Scholar] [CrossRef] [PubMed]
- Neetika; Sharma, M.; Thakur, P.; Gaur, P.; Rani, G.M.; Rustagi, S.; Talreja, R.K.; Chaudhary, V. Cancer treatment and toxicity outlook of nanoparticles. Environ. Res. 2023, 237, 116870. [Google Scholar]
- Labouta, H.I.; Asgarian, N.; Rinker, K.; Cramb, D.T. Meta-analysis of nanoparticle cytotoxicity via data-mining the literature. ACS Nano 2019, 13, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; Aggarwal, P.; Hall, J.B.; McNeil, S.E. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol. Pharm. 2008, 5, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Elias, D.R.; Poloukhtine, A.; Popik, V.; Tsourkas, A. Effect of ligand density, receptor density, and nanoparticle size on cell targeting. Nanomedicine 2013, 9, 194–201. [Google Scholar] [CrossRef]
- Grimaldi, A.M.; Incoronato, M.; Salvatore, M.; Soricelli, A. Nanoparticle-based strategies for cancer immunotherapy and immunodiagnostics. Nanomedicine 2017, 12, 2349–2365. [Google Scholar] [CrossRef]
- Getts, D.R.; Shea, L.D.; Miller, S.D.; King, N.J. Harnessing nanoparticles for immune modulation. Trends Immunol. 2015, 36, 419–427. [Google Scholar] [CrossRef]
- Getts, D.R.; Martin, A.J.; McCarthy, D.P.; Terry, R.L.; Hunter, Z.N.; Yap, W.T.; Getts, M.T.; Pleiss, M.; Luo, X.; King, N.J.; et al. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat. Biotechnol. 2012, 30, 1217–1224. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Wang, Y.; Chen, D.; Du, Y. Nanoparticle-Based Immunotherapy for Reversing T-Cell Exhaustion. Int. J. Mol. Sci. 2024, 25, 1396. https://doi.org/10.3390/ijms25031396
Li F, Wang Y, Chen D, Du Y. Nanoparticle-Based Immunotherapy for Reversing T-Cell Exhaustion. International Journal of Molecular Sciences. 2024; 25(3):1396. https://doi.org/10.3390/ijms25031396
Chicago/Turabian StyleLi, Fei, Yahong Wang, Dandan Chen, and Yunjie Du. 2024. "Nanoparticle-Based Immunotherapy for Reversing T-Cell Exhaustion" International Journal of Molecular Sciences 25, no. 3: 1396. https://doi.org/10.3390/ijms25031396
APA StyleLi, F., Wang, Y., Chen, D., & Du, Y. (2024). Nanoparticle-Based Immunotherapy for Reversing T-Cell Exhaustion. International Journal of Molecular Sciences, 25(3), 1396. https://doi.org/10.3390/ijms25031396