
How to Use the Cuprizone Model to Study De- and Remyelination


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
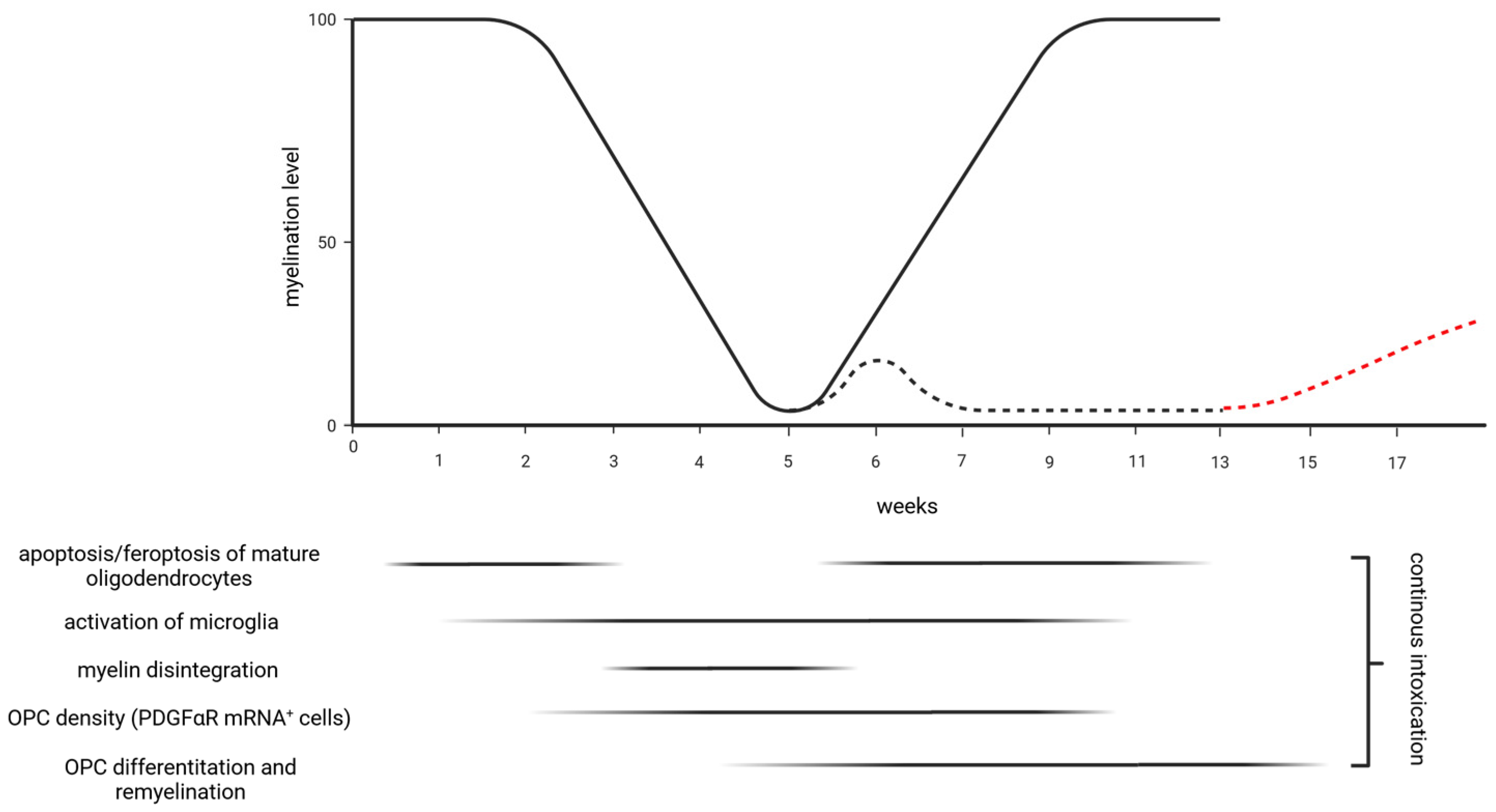
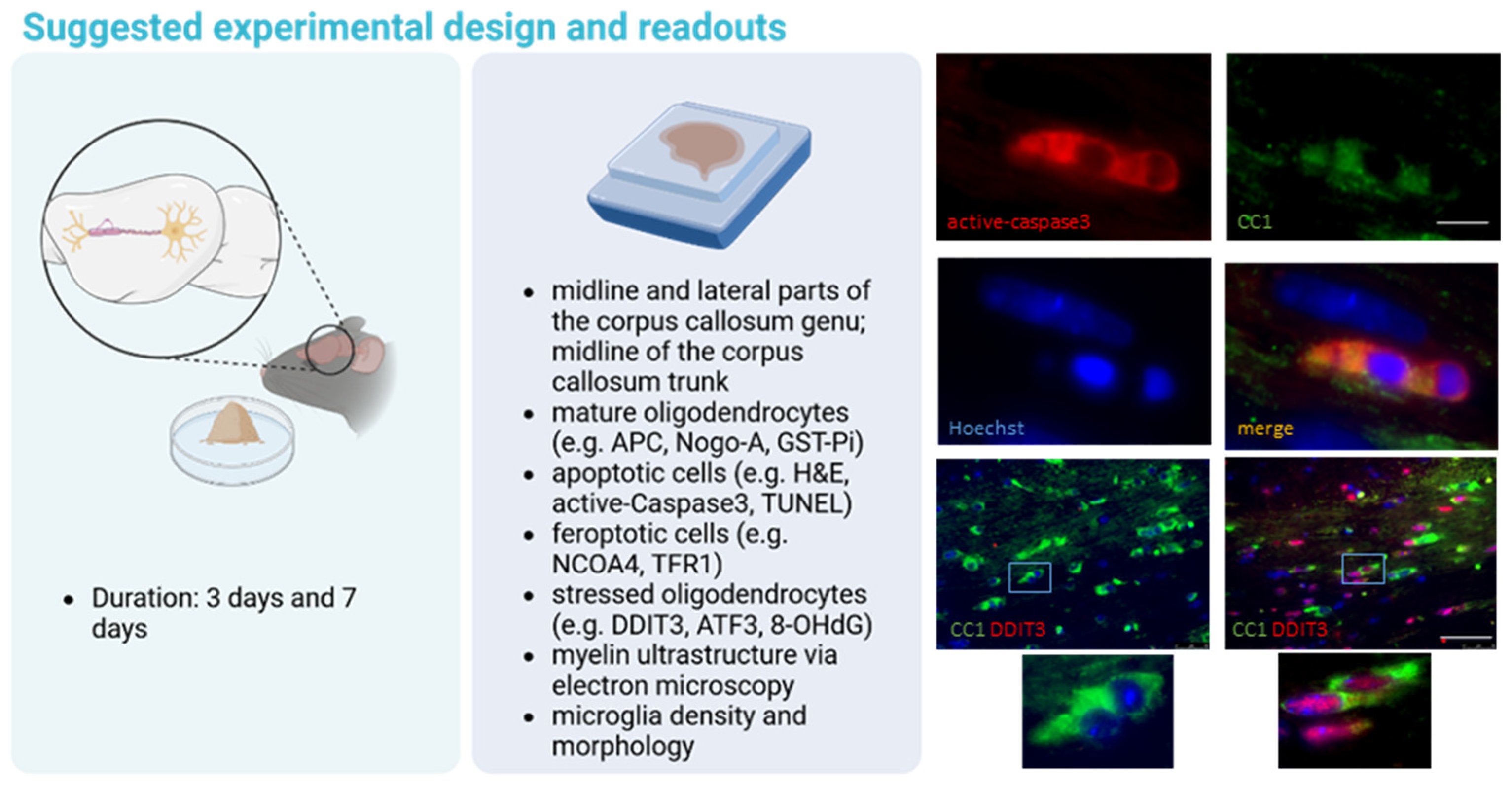
2. Oligodendrocyte Degeneration
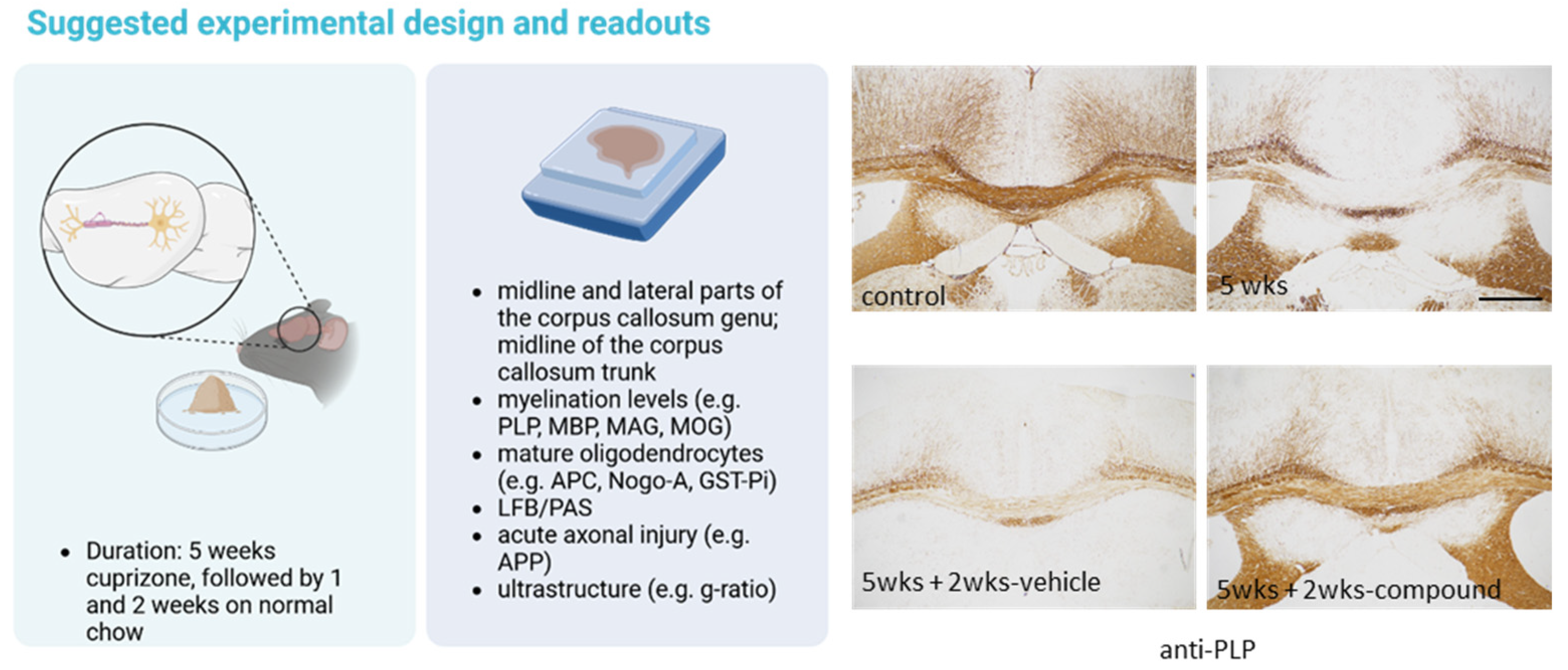
3. Demyelination
4. Remyelination
5. Conclusions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Hutchinson, N.A.; Koles, Z.J.; Smith, R.S. Conduction velocity in myelinated nerve fibres of Xenopus laevis. J. Physiol. 1970, 208, 279–289. [Google Scholar] [CrossRef]
- Farsi, Z.; Nicolella, A.; Simmons, S.K.; Aryal, S.; Shepard, N.; Brenner, K.; Lin, S.; Herzog, L.; Moran, S.P.; Stalnaker, K.J.; et al. Brain-region-specific changes in neurons and glia and dysregulation of dopamine signaling in Grin2a mutant mice. Neuron 2023, 111, 3378–3396.e9. [Google Scholar] [CrossRef]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Valori, C.F.; Sulmona, C.; Brambilla, L.; Rossi, D. Astrocytes: Dissecting Their Diverse Roles in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Cells 2023, 12, 1450. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Ortega, P.; Soria-Ortiz, M.B.; Rodríguez, V.M.; Vázquez-Martínez, E.O.; Díaz-Muñoz, M.; Reyes-Haro, D. Anorexia disrupts glutamate-glutamine homeostasis associated with astroglia in the prefrontal cortex of young female rats. Behav. Brain Res. 2022, 420, 113715. [Google Scholar] [CrossRef] [PubMed]
- Als, T.D.; Kurki, M.I.; Grove, J.; Voloudakis, G.; Therrien, K.; Tasanko, E.; Nielsen, T.T.; Naamanka, J.; Veerapen, K.; Levey, D.F.; et al. Depression pathophysiology, risk prediction of recurrence and comorbid psychiatric disorders using genome-wide analyses. Nat. Med. 2023, 29, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Klineova, S.; Lublin, F.D. Clinical Course of Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a028928. [Google Scholar] [CrossRef] [PubMed]
- Yarom, Y.; Naparstek, Y.; Lev-Ram, V.; Holoshitz, J.; Ben-Nun, A.; Cohen, I.R. Immunospecific inhibition of nerve conduction by T lymphocytes reactive to basic protein of myelin. Nature 1983, 303, 246–247. [Google Scholar] [CrossRef] [PubMed]
- Caprariello, A.V.; Rogers, J.A.; Morgan, M.L.; Hoghooghi, V.; Plemel, J.R.; Koebel, A.; Tsutsui, S.; Dunn, J.F.; Kotra, L.P.; Ousman, S.S.; et al. Biochemically altered myelin triggers autoimmune demyelination. Proc. Natl. Acad. Sci. USA 2018, 115, 5528–5533. [Google Scholar] [CrossRef] [PubMed]
- Scheld, M.; Rüther, B.J.; Große-Veldmann, R.; Ohl, K.; Tenbrock, K.; Dreymüller, D.; Fallier-Becker, P.; Zendedel, A.; Beyer, C.; Clarner, T.; et al. Neurodegeneration Triggers Peripheral Immune Cell Recruitment into the Forebrain. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 1410–1415. [Google Scholar] [CrossRef]
- Gudi, V.; Gingele, S.; Skripuletz, T.; Stangel, M. Glial response during cuprizone-induced de- and remyelination in the CNS: Lessons learned. Front. Cell. Neurosci. 2014, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Skripuletz, T.; Gudi, V.; Hackstette, D.; Stangel, M. De- and remyelination in the CNS white and grey matter induced by cuprizone: The old, the new, and the unexpected. Histol. Histopathol. 2011, 26, 1585–1597. [Google Scholar] [PubMed]
- Praet, J.; Guglielmetti, C.; Berneman, Z.; Van der Linden, A.; Ponsaerts, P. Cellular and molecular neuropathology of the cuprizone mouse model: Clinical relevance for multiple sclerosis. Neurosci. Biobehav. Rev. 2014, 47, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M.; Nyamoya, S.; Hochstrasser, T.; Amor, S. Multiple sclerosis animal models: A clinical and histopathological perspective. Brain Pathol. 2017, 27, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Plemel, J.R.; Michaels, N.J.; Weishaupt, N.; Caprariello, A.V.; Keough, M.B.; Rogers, J.A.; Yukseloglu, A.; Lim, J.; Patel, V.V.; Rawji, K.S.; et al. Mechanisms of lysophosphatidylcholine-induced demyelination: A primary lipid disrupting myelinopathy. Glia 2018, 66, 327–347. [Google Scholar] [CrossRef] [PubMed]
- DePaula-Silva, A.B.; Hanak, T.J.; Libbey, J.E.; Fujinami, R.S. Theiler’s murine encephalomyelitis virus infection of SJL/J and C57BL/6J mice: Models for multiple sclerosis and epilepsy. J. Neuroimmunol. 2017, 308, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Jhelum, P.; Santos-Nogueira, E.; Teo, W.; Haumont, A.; Lenoël, I.; Stys, P.K.; David, S. Ferroptosis Mediates Cuprizone-Induced Loss of Oligodendrocytes and Demyelination. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 9327–9341. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, F.; Nedelcu, J.; Leopold, P.; Zhan, J.; Clarner, T.; Nellessen, L.; Beißel, C.; van Heuvel, Y.; Goswami, A.; Weis, J.; et al. Cuprizone-induced graded oligodendrocyte vulnerability is regulated by the transcription factor DNA damage-inducible transcript 3. Glia 2019, 67, 263–276. [Google Scholar] [CrossRef]
- Pasquini, L.A.; Calatayud, C.A.; Bertone Uña, A.L.; Millet, V.; Pasquini, J.M.; Soto, E.F. The neurotoxic effect of cuprizone on oligodendrocytes depends on the presence of pro-inflammatory cytokines secreted by microglia. Neurochem. Res. 2007, 32, 279–292. [Google Scholar] [CrossRef]
- Taraboletti, A.; Walker, T.; Avila, R.; Huang, H.; Caporoso, J.; Manandhar, E.; Leeper, T.C.; Modarelli, D.A.; Medicetty, S.; Shriver, L.P. Cuprizone Intoxication Induces Cell Intrinsic Alterations in Oligodendrocyte Metabolism Independent of Copper Chelation. Biochemistry 2017, 56, 1518–1528. [Google Scholar] [CrossRef]
- Morgan, M.L.; Teo, W.; Hernandez, Y.; Brideau, C.; Cummins, K.; Kuipers, H.F.; Stys, P.K. Cuprizone-induced Demyelination in Mouse Brain is not due to Depletion of Copper. ASN Neuro 2022, 14, 17590914221126367. [Google Scholar] [CrossRef]
- Buschmann, J.P.; Berger, K.; Awad, H.; Clarner, T.; Beyer, C.; Kipp, M. Inflammatory response and chemokine expression in the white matter corpus callosum and gray matter cortex region during cuprizone-induced demyelination. J. Mol. Neurosci. 2012, 48, 66–76. [Google Scholar] [CrossRef]
- Voss, E.V.; Škuljec, J.; Gudi, V.; Skripuletz, T.; Pul, R.; Trebst, C.; Stangel, M. Characterisation of microglia during de- and remyelination: Can they create a repair promoting environment? Neurobiol. Dis. 2012, 45, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, W.F. Observations on oligodendrocyte degeneration, the resolution of status spongiosus and remyelination in cuprizone intoxication in mice. J. Neurocytol. 1972, 1, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Buonvicino, D.; Ranieri, G.; Chiarugi, A. Cuprizone-Dependent De/Remyelination Responses and Functional Correlates in Mouse Strains Adopted to Model Relapsing, Chronic and Progressive Experimental Autoimmune Encephalomyelitis. Neurotox. Res. 2021, 39, 658–666. [Google Scholar] [CrossRef]
- Mason, J.L.; Langaman, C.; Morell, P.; Suzuki, K.; Matsushima, G.K. Episodic demyelination and subsequent remyelination within the murine central nervous system: Changes in axonal calibre. Neuropathol. Appl. Neurobiol. 2001, 27, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.C.; Le, T.Q.; Flint, N.C.; Vana, A.C.; Zhou, Y.X. Endogenous cell repair of chronic demyelination. J. Neuropathol. Exp. Neurol. 2006, 65, 245–256. [Google Scholar] [CrossRef]
- Mason, J.L.; Toews, A.; Hostettler, J.D.; Morell, P.; Suzuki, K.; Goldman, J.E.; Matsushima, G.K. Oligodendrocytes and progenitors become progressively depleted within chronically demyelinated lesions. Am. J. Pathol. 2004, 164, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.; Fokuhl, J.; Linsmeier, F.; Trebst, C.; Stangel, M. Chronic toxic demyelination in the central nervous system leads to axonal damage despite remyelination. Neurosci. Lett. 2009, 453, 120–125. [Google Scholar] [CrossRef]
- Hussain, R.; Ghoumari, A.M.; Bielecki, B.; Steibel, J.; Boehm, N.; Liere, P.; Macklin, W.B.; Kumar, N.; Habert, R.; Mhaouty-Kodja, S.; et al. The neural androgen receptor: A therapeutic target for myelin repair in chronic demyelination. Brain 2013, 136 Pt 1, 132–146. [Google Scholar] [CrossRef]
- Gudi, V.; Moharregh-Khiabani, D.; Skripuletz, T.; Koutsoudaki, P.N.; Kotsiari, A.; Skuljec, J.; Trebst, C.; Stangel, M. Regional differences between grey and white matter in cuprizone induced demyelination. Brain Res. 2009, 1283, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Leo, H.; Kipp, M. Remyelination in Multiple Sclerosis: Findings in the Cuprizone Model. Int. J. Mol. Sci. 2022, 23, 16093. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, G.K.; Morell, P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001, 11, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537. [Google Scholar] [CrossRef]
- Doonan, F.; Cotter, T.G. Morphological assessment of apoptosis. Methods 2008, 44, 200–204. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Pati, S.; Singh Gautam, A.; Dey, M.; Tiwari, A.; Kumar Singh, R. Molecular and functional characteristics of receptor-interacting protein kinase 1 (RIPK1) and its therapeutic potential in Alzheimer’s disease. Drug Discov. Today 2023, 28, 103750. [Google Scholar] [CrossRef]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; Barros, P.L.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765. [Google Scholar] [CrossRef]
- Benarroch, E. What Is the Role of Ferroptosis in Neurodegeneration? Neurology 2023, 101, 312–319. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, J.; Zhao, Y.; Zhou, L.; Qiao, H.; Xu, Q.; Liu, Y. The role of ferroptosis in neurodegenerative diseases. Mol. Biol. Rep. 2023, 50, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Hesse, A.; Wagner, M.; Held, J.; Brück, W.; Salinas-Riester, G.; Hao, Z.; Waisman, A.; Kuhlmann, T. In toxic demyelination oligodendroglial cell death occurs early and is FAS independent. Neurobiol. Dis. 2010, 37, 362–369. [Google Scholar] [CrossRef]
- Briggs, D.T.; Martin, C.B.; Ingersoll, S.A.; Barnum, S.R.; Martin, B.K. Astrocyte-specific expression of a soluble form of the murine complement control protein Crry confers demyelination protection in the cuprizone model. Glia 2007, 55, 1405–1415. [Google Scholar] [CrossRef]
- Mojaverrostami, S.; Pasbakhsh, P.; Madadi, S.; Nekoonam, S.; Zarini, D.; Noori, L.; Shiri, E.; Salama, M.; Zibara, K.; Kashani, I.R. Calorie restriction promotes remyelination in a Cuprizone-Induced demyelination mouse model of multiple sclerosis. Metab. Brain Dis. 2020, 35, 1211–1224. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.; Daniel, M.; van Heuvel, Y.; Victor, M.; Beyer, C.; Clarner, T.; Kipp, M. Short-term cuprizone feeding induces selective amino acid deprivation with concomitant activation of an integrated stress response in oligodendrocytes. Cell. Mol. Neurobiol. 2013, 33, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Petković, F.; Campbell, I.L.; Gonzalez, B.; Castellano, B. Astrocyte-targeted production of interleukin-6 reduces astroglial and microglial activation in the cuprizone demyelination model: Implications for myelin clearance and oligodendrocyte maturation. Glia 2016, 64, 2104–2119. [Google Scholar] [CrossRef] [PubMed]
- Voskuhl, R.R.; Itoh, N.; Tassoni, A.; Matsukawa, M.A.; Ren, E.; Tse, V.; Jang, E.; Suen, T.T.; Itoh, Y. Gene expression in oligodendrocytes during remyelination reveals cholesterol homeostasis as a therapeutic target in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 10130–10139. [Google Scholar] [CrossRef]
- Lee, H.N.; Jeon, G.S.; Kim, D.W.; Cho, I.H.; Cho, S.S. Expression of adenomatous polyposis coli protein in reactive astrocytes in hippocampus of kainic acid-induced rat. Neurochem. Res. 2010, 35, 114–121. [Google Scholar] [CrossRef]
- Luo, M.; Deng, M.; Yu, Z.; Zhang, Y.; Xu, S.; Hu, S.; Xu, H. Differential Susceptibility and Vulnerability of Brain Cells in C57BL/6 Mouse to Mitochondrial Dysfunction Induced by Short-Term Cuprizone Exposure. Front. Neuroanat. 2020, 14, 30. [Google Scholar] [CrossRef]
- Tezuka, T.; Tamura, M.; Kondo, M.A.; Sakaue, M.; Okada, K.; Takemoto, K.; Fukunari, A.; Miwa, K.; Ohzeki, H.; Kano, S.; et al. Cuprizone short-term exposure: Astrocytic IL-6 activation and behavioral changes relevant to psychosis. Neurobiol. Dis. 2013, 59, 63–68. [Google Scholar] [CrossRef]
- Joost, S.; Schweiger, F.; Pfeiffer, F.; Ertl, C.; Keiler, J.; Frank, M.; Kipp, M. Cuprizone Intoxication Results in Myelin Vacuole Formation. Front. Cell. Neurosci. 2022, 16, 709596. [Google Scholar] [CrossRef] [PubMed]
- Wittekindt, M.; Kaddatz, H.; Joost, S.; Staffeld, A.; Bitar, Y.; Kipp, M.; Frintrop, L. Different Methods for Evaluating Microglial Activation Using Anti-Ionized Calcium-Binding Adaptor Protein-1 Immunohistochemistry in the Cuprizone Model. Cells 2022, 11, 1723. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Popko, B. Endoplasmic reticulum stress in disorders of myelinating cells. Nat. Neurosci. 2009, 12, 379–385. [Google Scholar] [CrossRef]
- Coelho, D.S.; Domingos, P.M. Physiological roles of regulated Ire1 dependent decay. Front. Genet. 2014, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 2008, 118, 3378–3389. [Google Scholar] [CrossRef]
- Ji, C.; Mehrian-Shai, R.; Chan, C.; Hsu, Y.H.; Kaplowitz, N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol. Clin. Exp. Res. 2005, 29, 1496–1503. [Google Scholar] [CrossRef]
- Namba, T.; Tanaka, K.; Ito, Y.; Ishihara, T.; Hoshino, T.; Gotoh, T.; Endo, M.; Sato, K.; Mizushima, T. Positive role of CCAAT/enhancer-binding protein homologous protein, a transcription factor involved in the endoplasmic reticulum stress response in the development of colitis. Am. J. Pathol. 2009, 174, 1786–1798. [Google Scholar] [CrossRef]
- Thorp, E.; Li, G.; Seimon, T.A.; Kuriakose, G.; Ron, D.; Tabas, I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metab. 2009, 9, 474–481. [Google Scholar] [CrossRef]
- Fu, H.Y.; Okada, K.; Liao, Y.; Tsukamoto, O.; Isomura, T.; Asai, M.; Sawada, T.; Okuda, K.; Asano, Y.; Sanada, S.; et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 2010, 122, 361–369. [Google Scholar] [CrossRef]
- Silva, R.M.; Ries, V.; Oo, T.F.; Yarygina, O.; Jackson-Lewis, V.; Ryu, E.J.; Lu, P.D.; Marciniak, S.J.; Ron, D.; Przedborski, S.; et al. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J. Neurochem. 2005, 95, 974–986. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Ostrowski, R.P.; Sun, X.; Ma, Q.; Huang, B.; Zhan, Y.; Zhang, J.H. CHOP silencing reduces acute brain injury in the rat model of subarachnoid hemorrhage. Stroke 2012, 43, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Prasanthi, J.R.; Larson, T.; Schommer, J.; Ghribi, O. Silencing GADD153/CHOP gene expression protects against Alzheimer’s disease-like pathology induced by 27-hydroxycholesterol in rabbit hippocampus. PLoS ONE 2011, 6, e26420. [Google Scholar] [CrossRef] [PubMed]
- Makioka, K.; Yamazaki, T.; Fujita, Y.; Takatama, M.; Nakazato, Y.; Okamoto, K. Involvement of endoplasmic reticulum stress defined by activated unfolded protein response in multiple system atrophy. J. Neurol. Sci. 2010, 297, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Ohri, S.S.; Maddie, M.A.; Zhao, Y.; Qiu, M.S.; Hetman, M.; Whittemore, S.R. Attenuating the endoplasmic reticulum stress response improves functional recovery after spinal cord injury. Glia 2011, 59, 1489–1502. [Google Scholar] [CrossRef]
- Cunnea, P.; Mhaille, A.N.; McQuaid, S.; Farrell, M.; McMahon, J.; FitzGerald, U. Expression profiles of endoplasmic reticulum stress-related molecules in demyelinating lesions and multiple sclerosis. Mult. Scler. 2011, 17, 808–818. [Google Scholar] [CrossRef]
- Mhaille, A.N.; McQuaid, S.; Windebank, A.; Cunnea, P.; McMahon, J.; Samali, A.; FitzGerald, U. Increased expression of endoplasmic reticulum stress-related signaling pathway molecules in multiple sclerosis lesions. J. Neuropathol. Exp. Neurol. 2008, 67, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Bailey, S.L.; Ho, H.; Harding, H.P.; Ron, D.; Miller, S.D.; Popko, B. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J. Clin. Investig. 2007, 117, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Kamarehei, M.; Kabudanian Ardestani, S.; Firouzi, M.; Zahednasab, H.; Keyvani, H.; Harirchian, M.H. Increased expression of endoplasmic reticulum stress-related caspase-12 and CHOP in the hippocampus of EAE mice. Brain Res. Bull. 2019, 147, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Tanaka-Arakawa, M.M.; Matsui, M.; Tanaka, C.; Uematsu, A.; Uda, S.; Miura, K.; Sakai, T.; Noguchi, K. Developmental changes in the corpus callosum from infancy to early adulthood: A structural magnetic resonance imaging study. PLoS ONE 2015, 10, e0118760. [Google Scholar] [CrossRef]
- Tomasch, J. Size, distribution, and number of fibres in the human corpus callosum. Anat. Rec. 1954, 119, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.; Clarner, T.; Beyer, C.; Kipp, M. Anatomical Distribution of Cuprizone-Induced Lesions in C57BL6 Mice. J. Mol. Neurosci. MN 2015, 57, 166–175. [Google Scholar] [CrossRef]
- Available online: https://www.hms.harvard.edu/research/brain/atlas.html (accessed on 18 January 2024).
- Schmidt, T.; Awad, H.; Slowik, A.; Beyer, C.; Kipp, M.; Clarner, T. Regional heterogeneity of cuprizone-induced demyelination: Topographical aspects of the midline of the corpus callosum. J. Mol. Neurosci. MN 2013, 49, 80–88. [Google Scholar] [CrossRef]
- Rüther, B.J.; Scheld, M.; Dreymueller, D.; Clarner, T.; Kress, E.; Brandenburg, L.O.; Swartenbroekx, T.; Hoornaert, C.; Ponsaerts, P.; Fallier-Becker, P.; et al. Combination of cuprizone and experimental autoimmune encephalomyelitis to study inflammatory brain lesion formation and progression. Glia 2017, 65, 1900–1913. [Google Scholar] [CrossRef]
- Behrangi, N.; Heinig, L.; Frintrop, L.; Santrau, E.; Kurth, J.; Krause, B.; Atanasova, D.; Clarner, T.; Fragoulis, A.; Joksch, M.; et al. Siponimod ameliorates metabolic oligodendrocyte injury via the sphingosine-1 phosphate receptor 5. Proc. Natl. Acad. Sci. USA 2022, 119, e2204509119. [Google Scholar] [CrossRef]
- Schmued, L.C. A rapid, sensitive histochemical stain for myelin in frozen brain sections. J. Histochem. Cytochem. 1990, 38, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Salthouse, T.N. Luxol fast blue g as a myelin stain. Stain Technol. 1964, 39, 123. [Google Scholar] [PubMed]
- Margolis, G.; Pickett, J.P. New applications of the Luxol fast blue myelin stain. Lab. Investig. A J. Tech. Methods Pathol. 1956, 5, 459–474. [Google Scholar]
- Scholtz, C.L. Quantitative histochemistry of myelin using Luxol Fast Blue MBS. Histochem. J. 1977, 9, 759–765. [Google Scholar] [CrossRef]
- Morgan, M.L.; Brideau, C.; Teo, W.; Caprariello, A.V.; Stys, P.K. Label-free assessment of myelin status using birefringence microscopy. J. Neurosci. Methods 2021, 360, 109226. [Google Scholar] [CrossRef]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Brück, W.; Lucchinetti, C.; Lassmann, H. Remyelination is extensive in a subset of multiple sclerosis patients. Brain A J. Neurol. 2006, 129 Pt 12, 3165–3172. [Google Scholar] [CrossRef]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Brück, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Honmou, O.; Felts, P.A.; Waxman, S.G.; Kocsis, J.D. Restoration of normal conduction properties in demyelinated spinal cord axons in the adult rat by transplantation of exogenous Schwann cells. J. Neurosci. 1996, 16, 3199–3208. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, R.; Osanai, Y.; Kouki, T.; Huang, J.K.; Ohno, N. Pharmacological treatment promoting remyelination enhances motor function after internal capsule demyelination in mice. Neurochem. Int. 2023, 164, 105505. [Google Scholar] [CrossRef] [PubMed]
- Nave, K.A. Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 2010, 11, 275–283. [Google Scholar] [CrossRef]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef]
- Menn, B.; Garcia-Verdugo, J.M.; Yaschine, C.; Gonzalez-Perez, O.; Rowitch, D.; Alvarez-Buylla, A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J. Neurosci. 2006, 26, 7907–7918. [Google Scholar] [CrossRef]
- Nait-Oumesmar, B.; Picard-Riera, N.; Kerninon, C.; Decker, L.; Seilhean, D.; Höglinger, G.U.; Hirsch, E.C.; Reynolds, R.; Baron-Van Evercooren, A. Activation of the subventricular zone in multiple sclerosis: Evidence for early glial progenitors. Proc. Natl. Acad. Sci. USA 2007, 104, 4694–4699. [Google Scholar] [CrossRef]
- Serwanski, D.R.; Rasmussen, A.L.; Brunquell, C.B.; Perkins, S.S.; Nishiyama, A. Sequential Contribution of Parenchymal and Neural Stem Cell-Derived Oligodendrocyte Precursor Cells toward Remyelination. Neuroglia 2018, 1, 8. [Google Scholar] [CrossRef]
- Butti, E.; Bacigaluppi, M.; Chaabane, L.; Ruffini, F.; Brambilla, E.; Berera, G.; Montonati, C.; Quattrini, A.; Martino, G. Neural Stem Cells of the Subventricular Zone Contribute to Neuroprotection of the Corpus Callosum after Cuprizone-Induced Demyelination. J. Neurosci. 2019, 39, 5481–5492. [Google Scholar] [CrossRef]
- Gruchot, J.; Weyers, V.; Göttle, P.; Förster, M.; Hartung, H.P.; Küry, P.; Kremer, D. The Molecular Basis for Remyelination Failure in Multiple Sclerosis. Cells 2019, 8, 825. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.R.; Polito, A.; Levine, J.M.; Reynolds, R. NG2-expressing glial progenitor cells: An abundant and widespread population of cycling cells in the adult rat CNS. Mol. Cell. Neurosci. 2003, 24, 476–488. [Google Scholar] [CrossRef]
- Terai, K.; Soga, T.; Takahashi, M.; Kamohara, M.; Ohno, K.; Yatsugi, S.; Okada, M.; Yamaguchi, T. Edg-8 receptors are preferentially expressed in oligodendrocyte lineage cells of the rat CNS. Neuroscience 2003, 116, 1053–1062. [Google Scholar] [CrossRef]
- Sun, L.O.; Mulinyawe, S.B.; Collins, H.Y.; Ibrahim, A.; Li, Q.; Simon, D.J.; Tessier-Lavigne, M.; Barres, B.A. Spatiotemporal Control of CNS Myelination by Oligodendrocyte Programmed Cell Death through the TFEB-PUMA Axis. Cell 2018, 175, 1811–1826.e21. [Google Scholar] [CrossRef] [PubMed]
- Tomassy, G.S.; Berger, D.R.; Chen, H.H.; Kasthuri, N.; Hayworth, K.J.; Vercelli, A.; Seung, H.S.; Lichtman, J.W.; Arlotta, P. Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science 2014, 344, 319–324. [Google Scholar] [CrossRef]
- Watanabe, M.; Toyama, Y.; Nishiyama, A. Differentiation of proliferated NG2-positive glial progenitor cells in a remyelinating lesion. J. Neurosci. Res. 2002, 69, 826–836. [Google Scholar] [CrossRef]
- Duncan, I.D.; Radcliff, A.B.; Heidari, M.; Kidd, G.; August, B.K.; Wierenga, L.A. The adult oligodendrocyte can participate in remyelination. Proc. Natl. Acad. Sci. USA 2018, 115, E11807–E11816. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.S.Y.; Djelloul, M.; Steiner, E.; Bernard, S.; Salehpour, M.; Possnert, G.; Brundin, L.; Frisén, J. Dynamics of oligodendrocyte generation in multiple sclerosis. Nature 2019, 566, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Zilkha-Falb, R.; Kaushansky, N.; Ben-Nun, A. The Median Eminence, A New Oligodendrogenic Niche in the Adult Mouse Brain. Stem Cell Rep. 2020, 14, 1076–1092. [Google Scholar] [CrossRef] [PubMed]
- Palhol, J.S.C.; Balia, M.; Sánchez-Román Terán, F.; Labarchède, M.; Gontier, E.; Battefeld, A. Direct association with the vascular basement membrane is a frequent feature of myelinating oligodendrocytes in the neocortex. Fluids Barriers CNS 2023, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, E.N.; Albrecht, S.; Groll, K.; Thomas, C.; Wallhorn, L.; Herold, M.; Hucke, S.; Klotz, L.; Kuhlmann, T. Influx of T cells into corpus callosum increases axonal injury, but does not change the course of remyelination in toxic demyelination. Glia 2023, 71, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Marenna, S.; Huang, S.C.; Dalla Costa, G.; d’Isa, R.; Castoldi, V.; Rossi, E.; Comi, G.; Leocani, L. Visual Evoked Potentials to Monitor Myelin Cuprizone-Induced Functional Changes. Front. Neurosci. 2022, 16, 820155. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Guo, Y.; Chen, X.; Du, S.; Han, Y.; Yan, Z.; Zhu, Q.; Li, Y. Demyelination and remyelination detected in an alternative cuprizone mouse model of multiple sclerosis with 7.0 T multiparameter magnetic resonance imaging. Sci. Rep. 2021, 11, 11060. [Google Scholar] [CrossRef] [PubMed]
- Höflich, K.M.; Beyer, C.; Clarner, T.; Schmitz, C.; Nyamoya, S.; Kipp, M.; Hochstrasser, T. Acute axonal damage in three different murine models of multiple sclerosis: A comparative approach. Brain Res. 2016, 1650, 125–133. [Google Scholar] [CrossRef]
- Nack, A.; Brendel, M.; Nedelcu, J.; Daerr, M.; Nyamoya, S.; Beyer, C.; Focke, C.; Deussing, M.; Hoornaert, C.; Ponsaerts, P.; et al. Expression of Translocator Protein and [18F]-GE180 Ligand Uptake in Multiple Sclerosis Animal Models. Cells 2019, 8, 94. [Google Scholar] [CrossRef]
- Licht-Mayer, S.; Campbell, G.R.; Mehta, A.R.; McGill, K.; Symonds, A.; Al-Azki, S.; Pryce, G.; Zandee, S.; Zhao, C.; Kipp, M.; et al. Axonal response of mitochondria to demyelination and complex IV activity within demyelinated axons in experimental models of multiple sclerosis. Neuropathol. Appl. Neurobiol. 2023, 49, e12851. [Google Scholar] [CrossRef]
- Gudi, V.; Gai, L.; Herder, V.; Tejedor, L.S.; Kipp, M.; Amor, S.; Sühs, K.W.; Hansmann, F.; Beineke, A.; Baumgärtner, W.; et al. Synaptophysin Is a Reliable Marker for Axonal Damage. J. Neuropathol. Exp. Neurol. 2017, 76, 109–125. [Google Scholar] [CrossRef]
- Mason, J.L.; Jones, J.J.; Taniike, M.; Morell, P.; Suzuki, K.; Matsushima, G.K. Mature oligodendrocyte apoptosis precedes IGF-1 production and oligodendrocyte progenitor accumulation and differentiation during demyelination/remyelination. J. Neurosci. Res. 2000, 61, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Baxi, E.G.; DeBruin, J.; Jin, J.; Strasburger, H.J.; Smith, M.D.; Orthmann-Murphy, J.L.; Schott, J.T.; Fairchild, A.N.; Bergles, D.E.; Calabresi, P.A. Lineage tracing reveals dynamic changes in oligodendrocyte precursor cells following cuprizone-induced demyelination. Glia 2017, 65, 2087–2098. [Google Scholar] [CrossRef]
- Ludwin, S.K. An autoradiographic study of cellular proliferation in remyelination of the central nervous system. Am. J. Pathol. 1979, 95, 683–696. [Google Scholar] [PubMed]
- Xing, Y.L.; Röth, P.T.; Stratton, J.A.; Chuang, B.H.; Danne, J.; Ellis, S.L.; Ng, S.W.; Kilpatrick, T.J.; Merson, T.D. Adult neural precursor cells from the subventricular zone contribute significantly to oligodendrocyte regeneration and remyelination. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 14128–14146. [Google Scholar] [CrossRef] [PubMed]
- Samanta, J.; Grund, E.M.; Silva, H.M.; Lafaille, J.J.; Fishell, G.; Salzer, J.L. Inhibition of Gli1 mobilizes endogenous neural stem cells for remyelination. Nature 2015, 526, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Lubrich, C.; Giesler, P.; Kipp, M. Motor Behavioral Deficits in the Cuprizone Model: Validity of the Rotarod Test Paradigm. Int. J. Mol. Sci. 2022, 23, 11342. [Google Scholar] [CrossRef]
- Yamate-Morgan, H.; Lauderdale, K.; Horeczko, J.; Merchant, U.; Tiwari-Woodruff, S.K. Functional Effects of Cuprizone-Induced Demyelination in the Presence of the mTOR-Inhibitor Rapamycin. Neuroscience 2019, 406, 667–683. [Google Scholar] [CrossRef]
- Crawford, D.K.; Mangiardi, M.; Xia, X.; López-Valdés, H.E.; Tiwari-Woodruff, S.K. Functional recovery of callosal axons following demyelination: A critical window. Neuroscience 2009, 164, 1407–1421. [Google Scholar] [CrossRef]
- Land, R.; Sentis, S.C.; Kral, A. Topographical EEG Recordings of Visual Evoked Potentials in Mice using Multichannel Thin-film Electrodes. J. Vis. Exp. JoVE 2022, e64034. [Google Scholar] [CrossRef]
- Almuslehi, M.S.M.; Sen, M.K.; Shortland, P.J.; Mahns, D.A.; Coorssen, J.R. Histological and Top-Down Proteomic Analyses of the Visual Pathway in the Cuprizone Demyelination Model. J. Mol. Neurosci. MN 2022, 72, 1374–1401. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Zhou, F. Cuproptosis: A new form of programmed cell death. Cell. Mol. Immunol. 2022, 19, 867–868. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kipp, M. How to Use the Cuprizone Model to Study De- and Remyelination. Int. J. Mol. Sci. 2024, 25, 1445. https://doi.org/10.3390/ijms25031445
Kipp M. How to Use the Cuprizone Model to Study De- and Remyelination. International Journal of Molecular Sciences. 2024; 25(3):1445. https://doi.org/10.3390/ijms25031445
Chicago/Turabian StyleKipp, Markus. 2024. "How to Use the Cuprizone Model to Study De- and Remyelination" International Journal of Molecular Sciences 25, no. 3: 1445. https://doi.org/10.3390/ijms25031445
APA StyleKipp, M. (2024). How to Use the Cuprizone Model to Study De- and Remyelination. International Journal of Molecular Sciences, 25(3), 1445. https://doi.org/10.3390/ijms25031445