
Functionalization of Bacterial Cellulose with the Antimicrobial Peptide KR-12 via Chimerical Cellulose-Binding Peptides



Abstract
1. Introduction
2. Results and Discussion
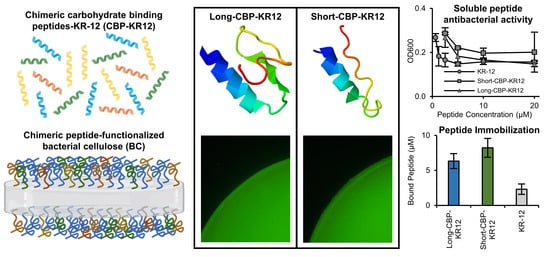
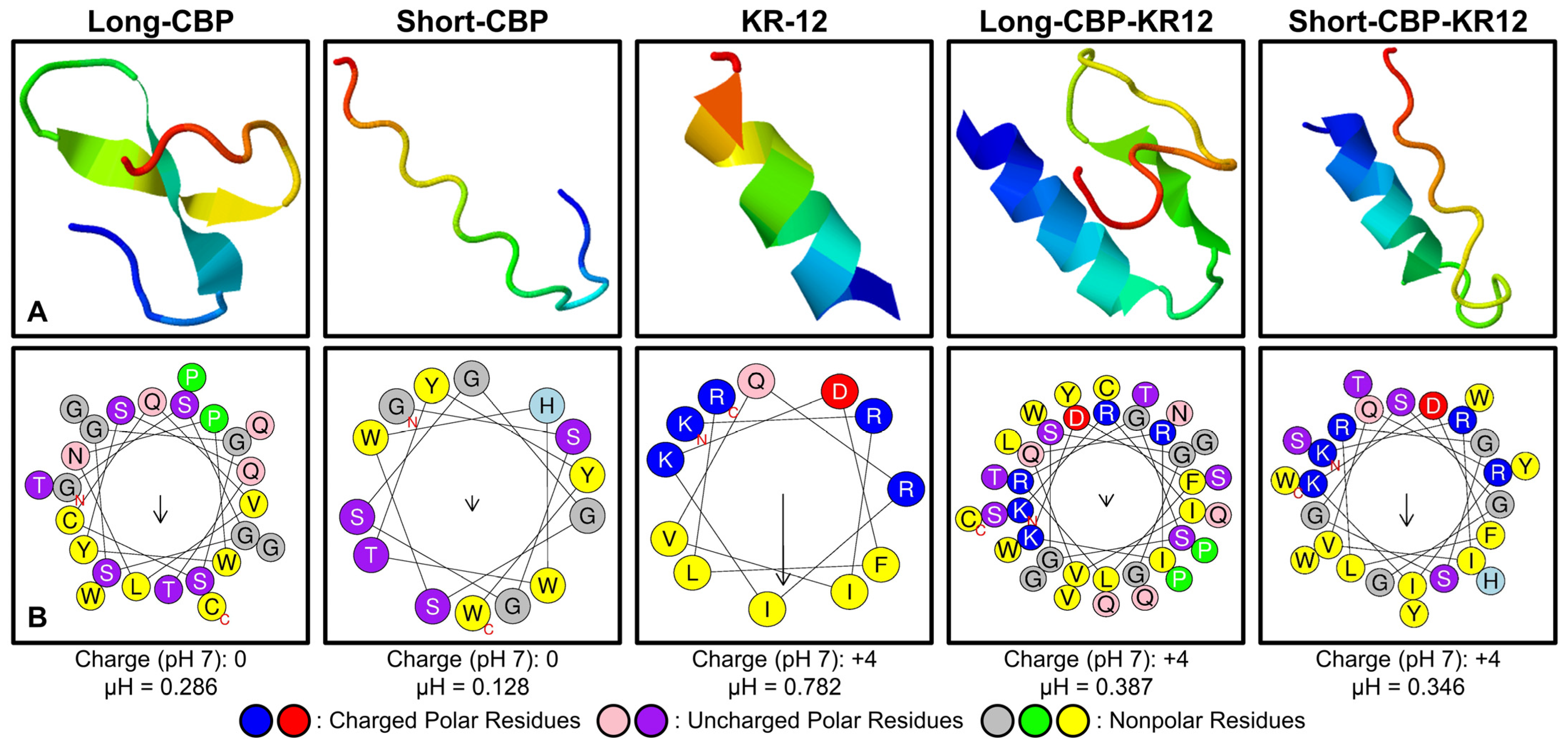
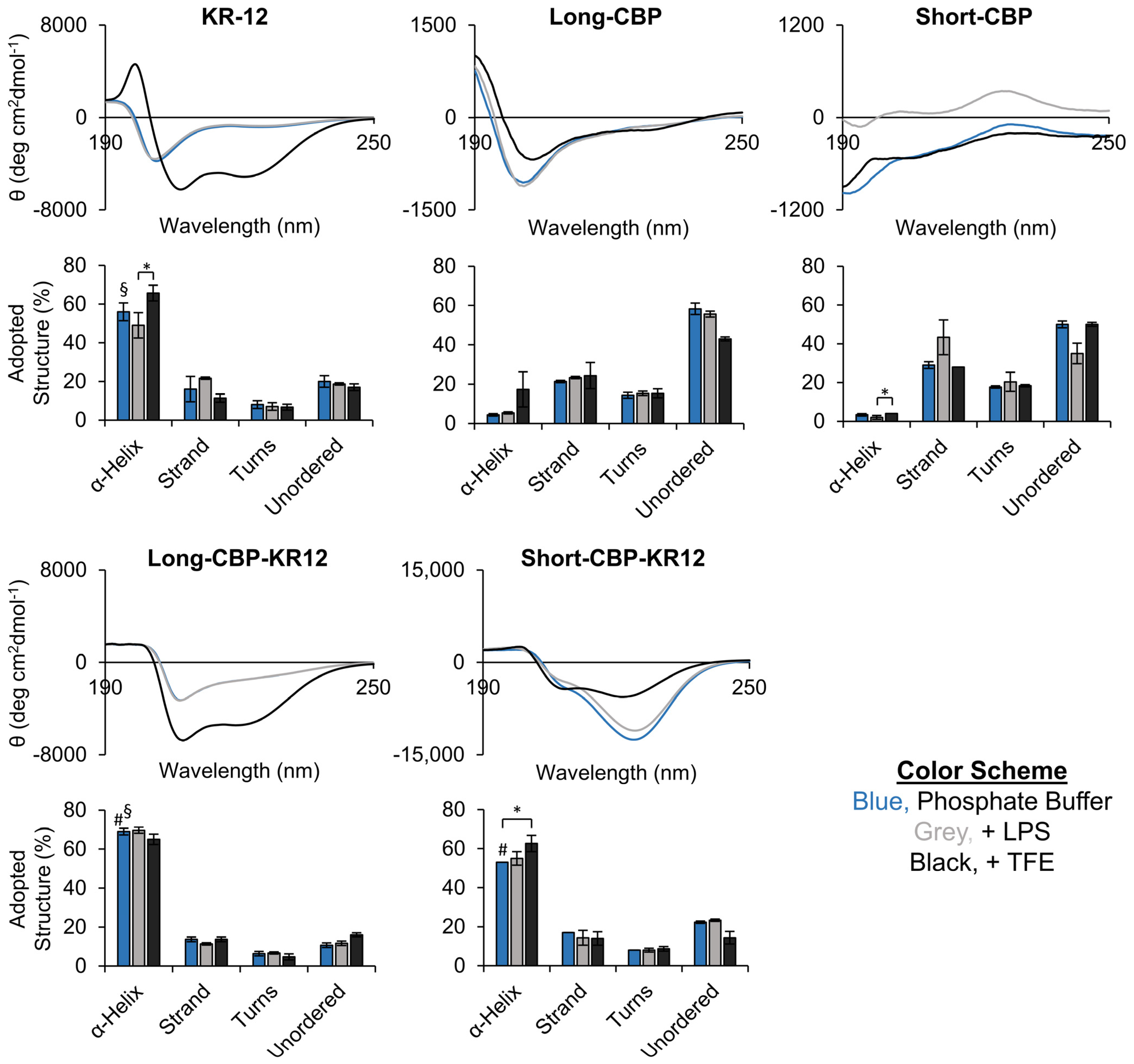
2.1. Peptide Structure and Helix Analysis
2.2. Free Peptide Cytotoxicity
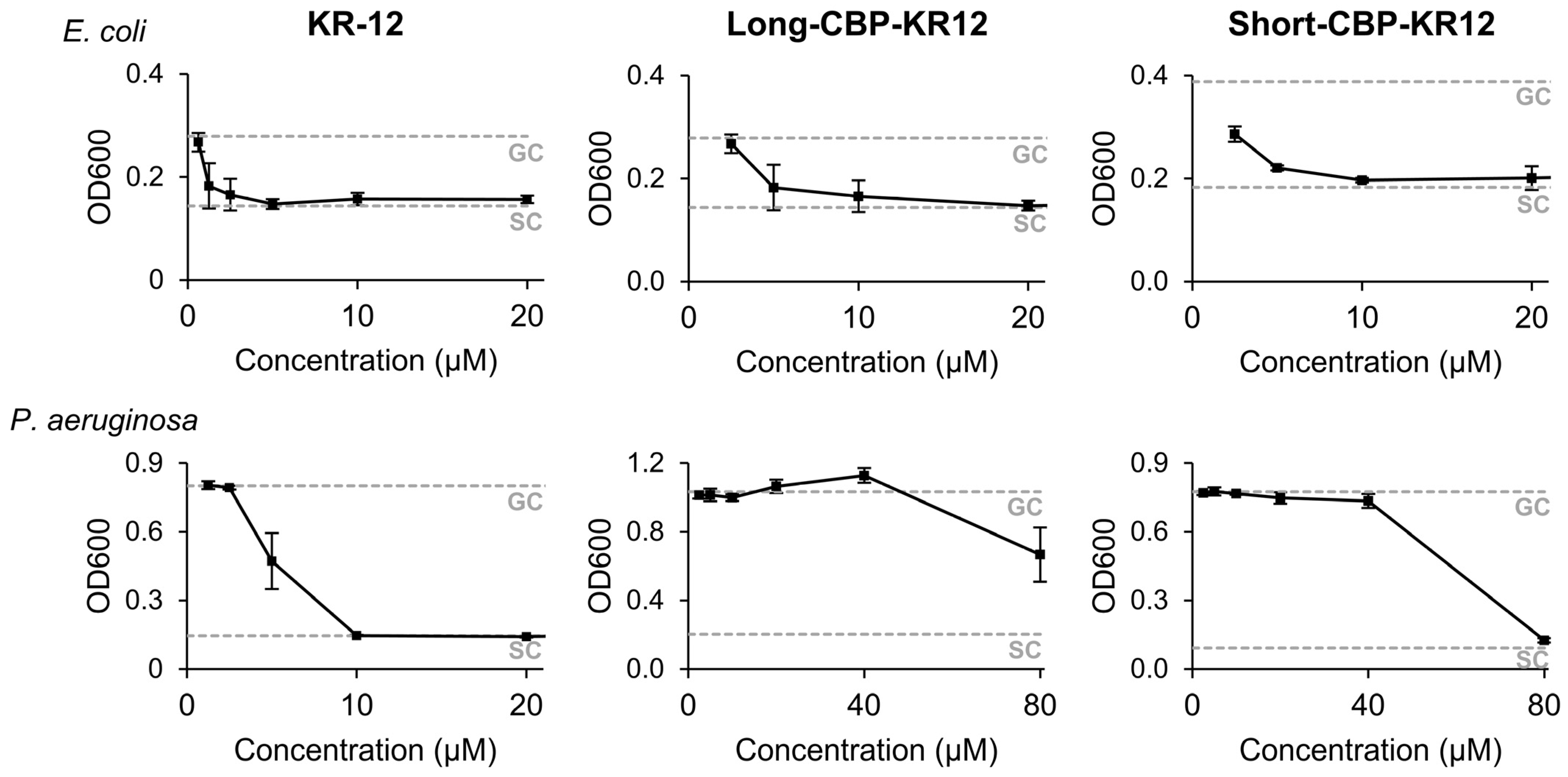
2.3. Peptide Minimum Inhibitory Concentration (MIC)
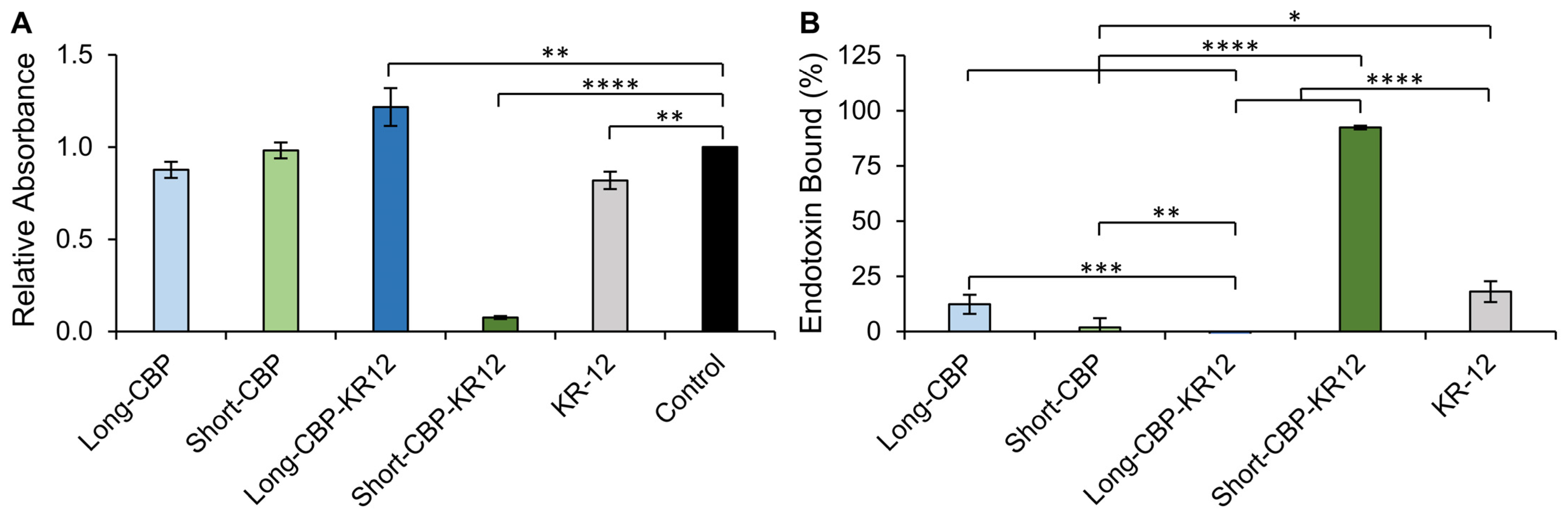
2.4. Endotoxin Binding
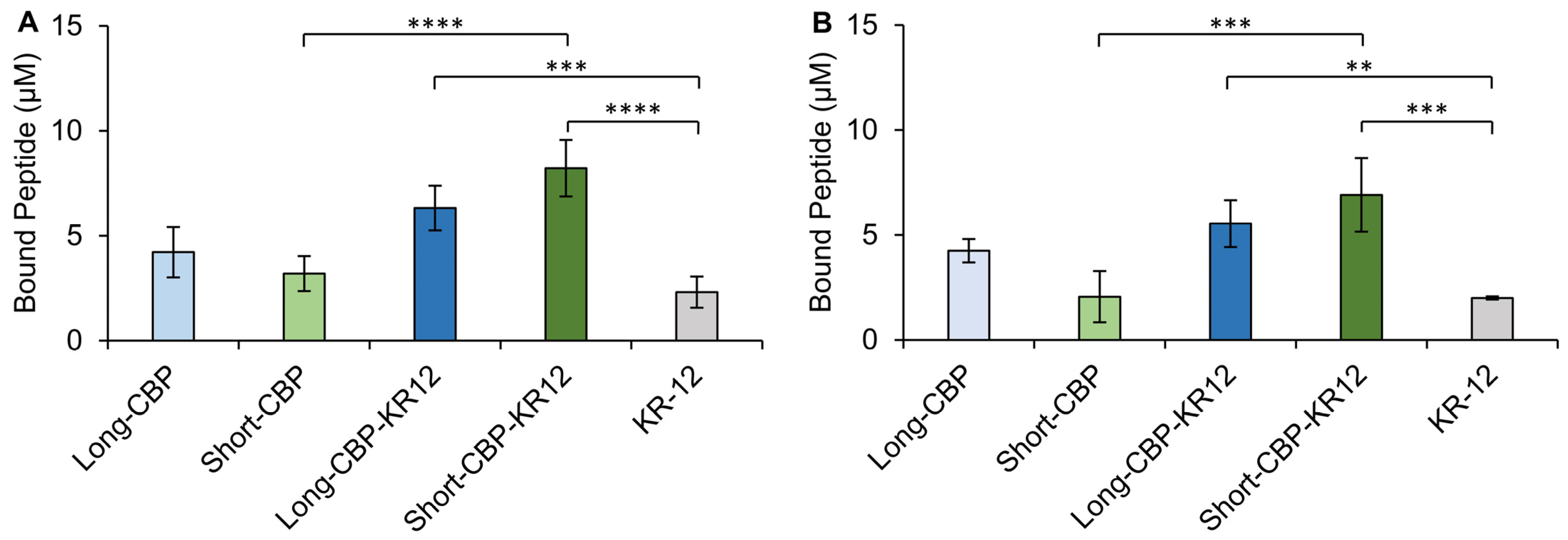
2.5. Cellulose-Binding Capacity of the CBPs and Chimeric Peptides
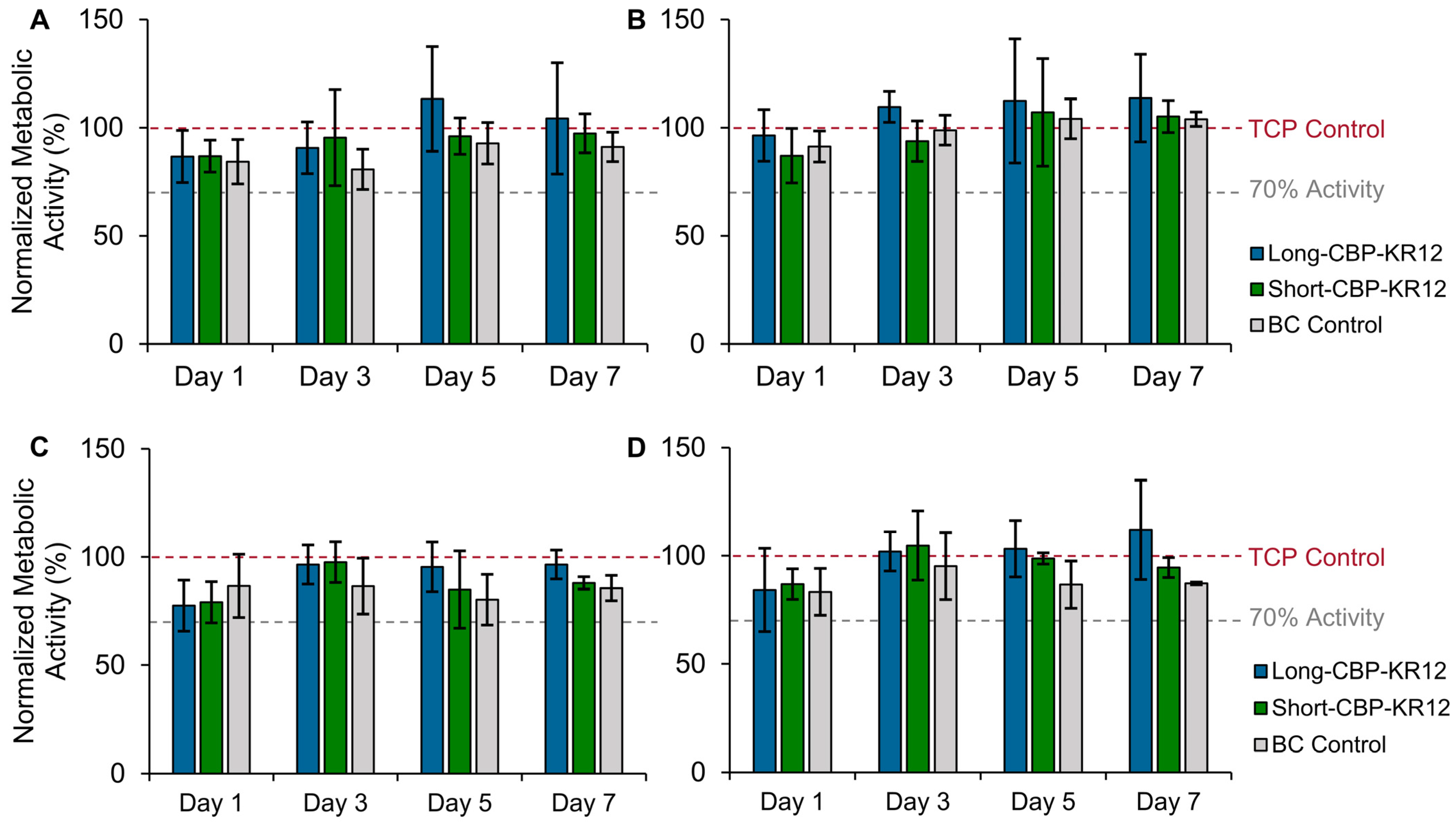
2.6. Effect of Peptide-Functionalized BC on In Vitro Cytotoxicity
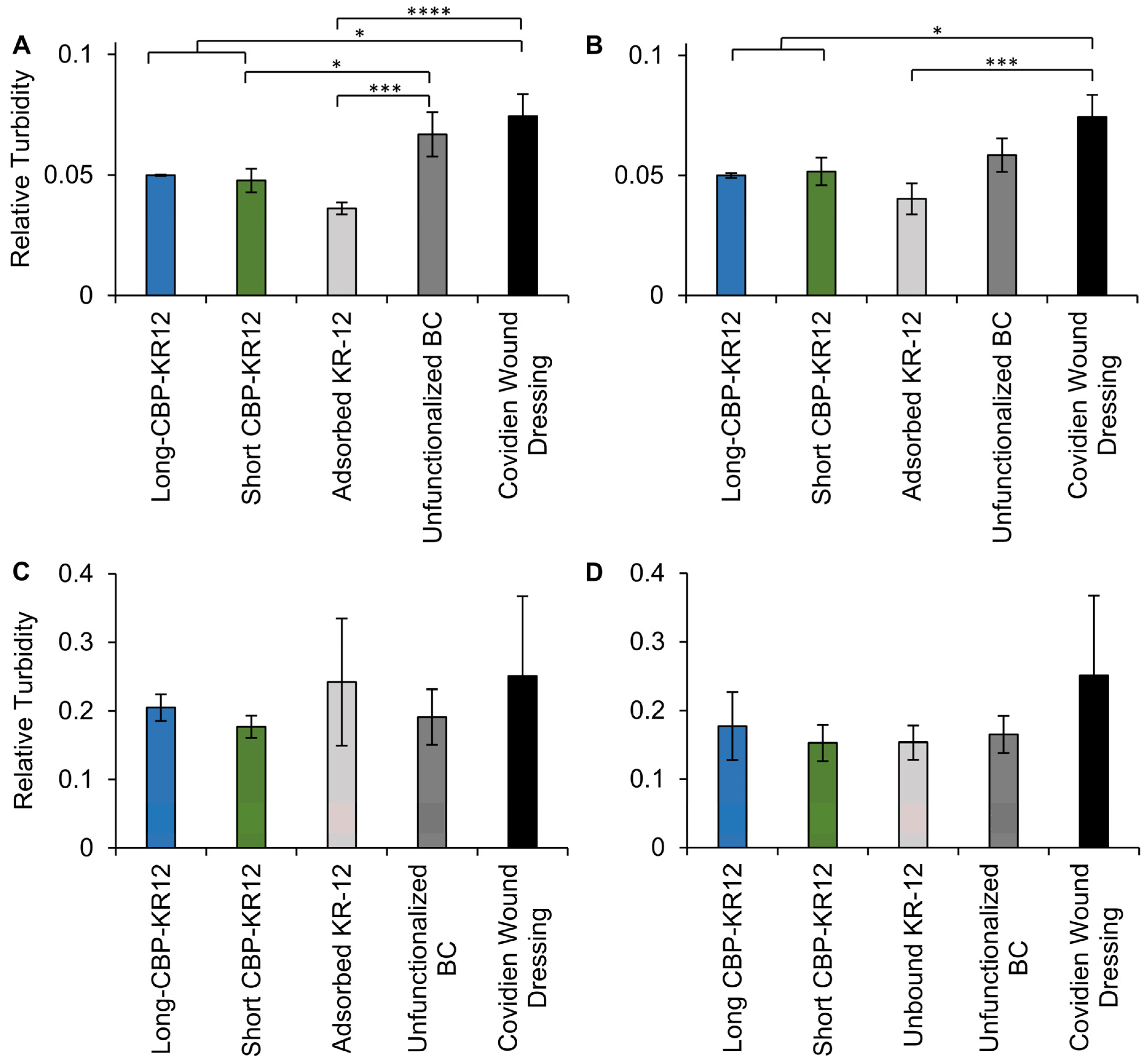
2.7. Surface Functionalized BC Antibacterial Activity
3. Materials and Methods
3.1. Materials
3.2. Peptide Design
3.3. Peptide Modeling and Helix Wheel
3.4. Circular Dichroism (CD) Spectroscopy
3.5. Mammalian Cell Culture Maintenance
3.6. Free Peptide Cytotoxicity
3.7. Peptide Minimum Inhibitory Concentration (MIC)
3.8. Endotoxin Binding
3.9. Cellulose Production
3.10. Peptide Binding to BC
3.11. Surface Functionalized BC Cytotoxicity
3.12. Surface Functionalized BC Bacterial Regrowth
3.13. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AI | Aliphatic index |
| ANOVA | Analysis of variance |
| ATCC | American Type Culture Collection |
| AMPs | Antimicrobial peptides |
| BC | Bacterial cellulose |
| CBD | Cellulose-binding domain |
| CBP | Cellulose-binding peptide |
| CD | Circular dichroism |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | Dimethyl sulfoxide |
| ECM | Extra cellular matrix |
| EU | Endotoxin units |
| FBS | Fetal bovine serum |
| GC | Growth control |
| H | Hydrophobicity |
| Hx | Helicity |
| HaCaT | Human epidermal keratinocytes |
| HS | Hestrin–Schramm |
| HSD | Honest significant difference |
| II | Instability index |
| I-TASSER | Iterative Threading ASSEmbly Refinement |
| LAL | Limulus amebocyte lysate |
| LPS | Lipopolysaccharide |
| MHA | Mueller–Hinton agar |
| MHB | Mueller–Hinton broth |
| Mw | Molecular weight |
| MIC | Minimum inhibitory concentration |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| NHDFs | Normal human dermal fibroblasts |
| PEG | Poly(ethylene glycol) |
| PI | Isoelectric point |
| q | Charge |
| ROS | Reactive oxygen species |
| SD | Standard deviation |
| SC | Sterile control |
| TBS | Tris-buffered saline |
| TCP | Tissue culture plastic |
| TFE | Trifluoroethanol |
| µH | Hydrophobic moment |
References
- Frykberg, R.G.; Banks, J. Challenges in the treatment of chronic wounds. Adv. Wound Care 2015, 4, 560–582. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, S.R.; Carter, M.J.; Fife, C.E.; DaVanzo, J.; Haught, R.; Nusgart, M.; Cartwright, D. An economic evaluation of the impact, cost, and medicare policy implications of chronic nonhealing wounds. Value Health 2018, 21, 27–32. [Google Scholar] [CrossRef]
- Guo, S.; DiPietro, L.A. Factors affecting wound healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef]
- Zeng, R.; Lin, C.; Lin, Z.; Chen, H.; Lu, W.; Lin, C.; Li, H. Approaches to cutaneous wound healing: Basics and future directions. Cell Tissue Res. 2018, 374, 217–232. [Google Scholar] [CrossRef]
- Walker, M.; Metcalf, D.; Parsons, D.; Bowler, P. A real-life clinical evaluation of a next-generation antimicrobial dressing on acute and chronic wounds. J. Wound Care 2015, 24, 11–22. [Google Scholar] [CrossRef]
- Jere, S.W.; Abrahamse, H.; Houreld, N.N. The JAK/STAT signaling pathway and photobiomodulation in chronic wound healing. Cytokine Growth Factor Rev. 2017, 38, 73–79. [Google Scholar] [CrossRef]
- Tandara, A.A.; Mustoe, T.A. Oxygen in wound healing—More than a nutrient. World J. Surg. 2004, 28, 294–300. [Google Scholar] [CrossRef]
- Rodriguez, P.G.; Felix, F.N.; Woodley, D.T.; Shim, E.K. The role of oxygen in wound healing: A review of the literature. Dermatol. Surg. 2008, 34, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Dhall, S.; Do, D.; Garcia, M.; Wijesinghe, D.S.; Brandon, A.; Kim, J.; Sanchez, A.; Lyubovitsky, J.; Gallagher, S.; Nothnagel, E.A. A novel model of chronic wounds: Importance of redox imbalance and biofilm-forming bacteria for establishment of chronicity. PLoS ONE 2014, 9, e109848. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Kim, J.H.; Saeed, S.; Martins-Green, M. Using systems biology approaches to identify signalling pathways activated during chronic wound initiation. Wound Repair Regen. 2021, 29, 881–898. [Google Scholar] [CrossRef]
- Ullah, H.; Santos, H.A.; Khan, T. Applications of bacterial cellulose in food, cosmetics and drug delivery. Cellulose 2016, 23, 2291–2314. [Google Scholar] [CrossRef]
- Thomas, G.W.; Rael, L.T.; Bar-Or, R.; Shimonkevitz, R.; Mains, C.W.; Slone, D.S.; Craun, M.L.; Bar-Or, D. Mechanisms of delayed wound healing by commonly used antiseptics. J. Trauma Acute Care Surg. 2009, 66, 82–91. [Google Scholar] [CrossRef]
- Brooks, S.E.; Walczak, M.A.; Hameed, R.; Coonan, P. Chlorhexidine resistance in antibiotic-resistant bacteria isolated from the surfaces of dispensers of soap containing chlorhexidine. Infect. Control Hosp. Epidemiol. 2002, 23, 692–695. [Google Scholar] [CrossRef]
- Koljalg, S.; Naaber, P.; Mikelsaar, M. Antibiotic resistance as an indicator of bacterial chlorhexidine susceptibility. J. Hosp. Infect. 2002, 51, 106–113. [Google Scholar] [CrossRef]
- George Broughton, I.; Janis, J.E.; Attinger, C.E. The basic science of wound healing. Plast. Reconstr. Surg. 2006, 117, 12S–34S. [Google Scholar] [CrossRef]
- Lozeau, L.D.; Grosha, J.; Smith, I.M.; Stewart, E.J.; Camesano, T.A.; Rolle, M.W. Alginate Affects Bioactivity of Chimeric Collagen Binding LL37 Antimicrobial Peptides Adsorbed to Collagen-Alginate Wound Dressings. ACS Biomater. Sci. Eng. 2020, 6, 3398–3410. [Google Scholar] [CrossRef]
- Hancock, R.E.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Izadpanah, A.; Gallo, R.L. Antimicrobial peptides. J. Am. Acad. Dermatol. 2005, 52, 381–390. [Google Scholar] [CrossRef]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Ciumac, D.; Gong, H.; Hu, X.; Lu, J.R. Membrane targeting cationic antimicrobial peptides. J. Colloid Interface Sci. 2019, 537, 163–185. [Google Scholar] [CrossRef]
- Talapko, J.; Meštrović, T.; Juzbašić, M.; Tomas, M.; Erić, S.; Horvat Aleksijević, L.; Bekić, S.; Schwarz, D.; Matić, S.; Neuberg, M.; et al. Antimicrobial Peptides-Mechanisms of Action, Antimicrobial Effects and Clinical Applications. Antibiotics 2022, 11, 1417. [Google Scholar] [CrossRef]
- Kang, J.; Dietz, M.J.; Li, B. Antimicrobial peptide LL-37 is bactericidal against Staphylococcus aureus biofilms. PLoS ONE 2019, 14, e0216676. [Google Scholar] [CrossRef]
- Lozeau, L.D.; Grosha, J.; Kole, D.; Prifti, F.; Dominko, T.; Camesano, T.A.; Rolle, M.W. Collagen tethering of synthetic human antimicrobial peptides cathelicidin LL37 and its effects on antimicrobial activity and cytotoxicity. Acta Biomater. 2017, 52, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Lozeau, L.D.; Youssefian, S.; Rahbar, N.; Camesano, T.A.; Rolle, M.W. Concentration-Dependent, Membrane-Selective Activity of Human LL37 Peptides Modified with Collagen Binding Domain Sequences. Biomacromolecules 2018, 19, 4513–4523. [Google Scholar] [CrossRef] [PubMed]
- Samuelsen, Ø.; Haukland, H.H.; Kahl, B.C.; von Eiff, C.; Proctor, R.A.; Ulvatne, H.; Sandvik, K.; Vorland, L.H. Staphylococcus aureus small colony variants are resistant to the antimicrobial peptide lactoferricin B. J. Antimicrob. Chemother. 2005, 56, 1126–1129. [Google Scholar] [CrossRef]
- Jacob, B.; Park, I.S.; Bang, J.K.; Shin, S.Y. Short KR-12 analogs designed from human cathelicidin LL-37 possessing both antimicrobial and antiendotoxic activities without mammalian cell toxicity. J. Pept. Sci. 2013, 19, 700–707. [Google Scholar] [CrossRef]
- Luo, Y.; McLean, D.T.; Linden, G.J.; McAuley, D.F.; McMullan, R.; Lundy, F.T. The naturally occurring host defense peptide, LL-37, and its truncated mimetics KE-18 and KR-12 have selected biocidal and antibiofilm activities against Candida albicans, Staphylococcus aureus, and Escherichia coli in vitro. Front. Microbiol. 2017, 8, 544. [Google Scholar] [CrossRef]
- Saporito, P.; Vang Mouritzen, M.; Løbner-Olesen, A.; Jenssen, H. LL-37 fragments have antimicrobial activity against Staphylococcus epidermidis biofilms and wound healing potential in HaCaT cell line. J. Pept. Sci. 2018, 24, e3080. [Google Scholar] [CrossRef]
- Song, D.W.; Kim, S.H.; Kim, H.H.; Lee, K.H.; Ki, C.S.; Park, Y.H. Multi-biofunction of antimicrobial peptide-immobilized silk fibroin nanofiber membrane: Implications for wound healing. Acta Biomater. 2016, 39, 146–155. [Google Scholar] [CrossRef]
- Blasi-Romero, A.; Ångström, M.; Franconetti, A.; Muhammad, T.; Jiménez-Barbero, J.; Göransson, U.; Palo-Nieto, C.; Ferraz, N. KR-12 Derivatives Endow Nanocellulose with Antibacterial and Anti-Inflammatory Properties: Role of Conjugation Chemistry. ACS Appl. Mater. Interfaces 2023, 15, 24186–24196. [Google Scholar] [CrossRef]
- Mookherjee, N.; Brown, K.L.; Bowdish, D.M.; Doria, S.; Falsafi, R.; Hokamp, K.; Roche, F.M.; Mu, R.; Doho, G.H.; Pistolic, J. Modulation of the TLR-mediated inflammatory response by the endogenous human host defense peptide LL-37. J. Immunol. 2006, 176, 2455–2464. [Google Scholar] [CrossRef] [PubMed]
- Jervis, E.J.; Haynes, C.A.; Kilburn, D.G. Surface diffusion of cellulases and their isolated binding domains on cellulose. J. Biol. Chem. 1997, 272, 24016–24023. [Google Scholar] [CrossRef]
- Khalid, A.; Khan, R.; Ul-Islam, M.; Khan, T.; Wahid, F. Bacterial cellulose-zinc oxide nanocomposites as a novel dressing system for burn wounds. Carbohydr. Polym. 2017, 164, 214–221. [Google Scholar] [CrossRef]
- Muangman, P.; Opasanon, S.; Suwanchot, S.; Thangthed, O. Efficiency of microbial cellulose dressing in partial-thickness burn wounds. J. Am. Coll. Certif. Wound Spec. 2011, 3, 16–19. [Google Scholar]
- van Zyl, E.M.; Coburn, J.M. Hierarchical structure of bacterial-derived cellulose and its impact on biomedical applications. Curr. Opin. Chem. Eng. 2019, 24, 122–130. [Google Scholar] [CrossRef]
- van Zyl, E.M.; Kennedy, M.A.; Nason, W.; Fenlon, S.J.; Young, E.M.; Smith, L.J.; Bhatia, S.R.; Coburn, J.M. Structural properties of optically clear bacterial cellulose produced by Komagataeibacter hansenii using arabitol. Biomater. Adv. 2023, 148, 213345. [Google Scholar] [CrossRef]
- Weishaupt, R.; Zünd, J.N.; Heuberger, L.; Zuber, F.; Faccio, G.; Robotti, F.; Ferrari, A.; Fortunato, G.; Ren, Q.; Maniura-Weber, K. Antibacterial, Cytocompatible, Sustainably Sourced: Cellulose Membranes with Bifunctional Peptides for Advanced Wound Dressings. Adv. Healthc. Mater. 2020, 9, 1901850. [Google Scholar] [CrossRef]
- Khazanov, N.; Iline-Vul, T.; Noy, E.; Goobes, G.; Senderowitz, H. Design of Compact Biomimetic Cellulose Binding Peptides as Carriers for Cellulose Catalytic Degradation. J. Phys. Chem. B 2016, 120, 309–319. [Google Scholar] [CrossRef]
- Kim, E.Y.; Rajasekaran, G.; Shin, S.Y. LL-37-derived short antimicrobial peptide KR-12-a5 and its d-amino acid substituted analogs with cell selectivity, anti-biofilm activity, synergistic effect with conventional antibiotics, and anti-inflammatory activity. Eur. J. Med. Chem. 2017, 136, 428–441. [Google Scholar] [CrossRef]
- Gullo, M.; La China, S.; Falcone, P.M.; Giudici, P. Biotechnological production of cellulose by acetic acid bacteria: Current state and perspectives. Appl. Microbiol. Biotechnol. 2018, 102, 6885–6898. [Google Scholar] [CrossRef]
- Hodel, K.V.S.; Fonseca, L.M.D.S.; Santos, I.M.D.S.; Cerqueira, J.C.; Santos-Júnior, R.E.D.; Nunes, S.B.; Barbosa, J.D.V.; Machado, B.A.S. Evaluation of Different Methods for Cultivating Gluconacetobacter hansenii for Bacterial Cellulose and Montmorillonite Biocomposite Production: Wound-Dressing Applications. Polymers 2020, 12, 267. [Google Scholar] [CrossRef]
- Barbosa, M.; Simões, H.; Pinto, S.N.; Macedo, A.S.; Fonte, P.; Prazeres, D.M.F. Fusions of a carbohydrate binding module with the small cationic hexapeptide RWRWRW confer antimicrobial properties to cellulose-based materials. Acta Biomater. 2022, 143, 216–232. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Catchmark, J.M.; Mohamed, M.N.A.; Benesi, A.J.; Tien, M.; Kao, T.-H.; Watts, H.D.; Kubicki, J.D. Identification and characterization of a cellulose binding heptapeptide revealed by phage display. Biomacromolecules 2013, 14, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Diosa, J.; Guzman, F.; Bernal, C.; Mesa, M. Formation mechanisms of chitosan-silica hybrid materials and its performance as solid support for KR-12 peptide adsorption: Impact on KR-12 antimicrobial activity and proteolytic stability. J. Mater. Res. Technol. 2020, 9, 890–901. [Google Scholar] [CrossRef]
- Liu, M.; Liu, T.; Zhang, X.; Jian, Z.; Xia, H.; Yang, J.; Hu, X.; Xing, M.; Luo, G.; Wu, J. Fabrication of KR-12 peptide-containing hyaluronic acid immobilized fibrous eggshell membrane effectively kills multi-drug-resistant bacteria, promotes angiogenesis and accelerates re-epithelialization. Int. J. Nanomed. 2019, 14, 3345. [Google Scholar] [CrossRef]
- Cassin, M.E.; Ford, A.J.; Orbach, S.M.; Saverot, S.E.; Rajagopalan, P. The design of antimicrobial LL37-modified collagen-hyaluronic acid detachable multilayers. Acta Biomater. 2016, 40, 119–129. [Google Scholar] [CrossRef]
- Reddy Chichili, V.P.; Kumar, V.; Sivaraman, J. Linkers in the structural biology of protein–protein interactions. Protein Sci. 2013, 22, 153–167. [Google Scholar] [CrossRef]
- Nagase, H. Substrate specificity of MMPs. In Matrix Metalloproteinase Inhibitors in Cancer Therapy; Springer: Berlin/Heidelberg, Germany, 2001; pp. 39–66. [Google Scholar]
- Mishra, B.; Wang, G. Titanium surfaces immobilized with the major antimicrobial fragment FK-16 of human cathelicidin LL-37 are potent against multiple antibiotic-resistant bacteria. Biofouling 2017, 33, 544–555. [Google Scholar] [CrossRef]
- Monné, M.; Hermansson, M.; von Heijne, G. A turn propensity scale for transmembrane helices. J. Mol. Biol. 1999, 288, 141–145. [Google Scholar] [CrossRef]
- Monné, M.; Nilsson, I.; Elofsson, A.; von Heijne, G. Turns in transmembrane helices: Determination of the minimal length of a “helical hairpin” and derivation of a fine-grained turn propensity scale. J. Mol. Biol. 1999, 293, 807–814. [Google Scholar] [CrossRef]
- Ajish, C.; Yang, S.; Kumar, S.D.; Kim, E.Y.; Min, H.J.; Lee, C.W.; Shin, S.-H.; Shin, S.Y. A novel hybrid peptide composed of LfcinB6 and KR-12-a4 with enhanced antimicrobial, anti-inflammatory and anti-biofilm activities. Sci. Rep. 2022, 12, 4365. [Google Scholar] [CrossRef] [PubMed]
- Pathak, N.; Salas-Auvert, R.; Ruche, G.; Janna, M.H.; McCarthy, D.; Harrison, R.G. Comparison of the effects of hydrophobicity, amphiphilicity, and α-helicity on the activities of antimicrobial peptides. Proteins Struct. Funct. Bioinform. 1995, 22, 182–186. [Google Scholar] [CrossRef]
- Piotto, S.P.; Sessa, L.; Concilio, S.; Iannelli, P. YADAMP: Yet another database of antimicrobial peptides. Int. J. Antimicrob. Agents 2012, 39, 346–351. [Google Scholar] [CrossRef]
- Omardien, S.; Drijfhout, J.W.; Zaat, S.A.; Brul, S. Cationic amphipathic antimicrobial peptides perturb the inner membrane of germinated spores thus inhibiting their outgrowth. Front. Microbiol. 2018, 9, 2277. [Google Scholar] [CrossRef]
- Lohner, K.; Blondelle, S. Molecular mechanisms of membrane perturbation by antimicrobial peptides and the use of biophysical studies in the design of novel peptide antibiotics. Comb. Chem. High Throughput Screen. 2005, 8, 241–256. [Google Scholar] [CrossRef]
- Zsila, F.; Kohut, G.; Beke-Somfai, T. Disorder-to-helix conformational conversion of the human immunomodulatory peptide LL-37 induced by antiinflammatory drugs, food dyes and some metabolites. Int. J. Biol. Macromol. 2019, 129, 50–60. [Google Scholar] [CrossRef]
- Osorio, D.; Rondón-Villarreal, P.; Torres, R. Peptides: A package for data mining of antimicrobial peptides. Small 2015, 12, 44–444. [Google Scholar] [CrossRef]
- Wilson, I.; Henry, M.; RD, Q.; PJ, B. The pH of Varicose Ulcer Surfaces and Its Relationship to Healing. VASA Z. Fur. Gefasskrankh. 1979, 8, 339–342. [Google Scholar]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.E.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. HELIQUEST: A web server to screen sequences with specific α-helical properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef]
- Miles, A.J.; Ramalli, S.G.; Wallace, B. DichroWeb, a website for calculating protein secondary structure from circular dichroism spectroscopic data. Protein Sci. 2022, 31, 37–46. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32 (Suppl. S2), W668–W673. [Google Scholar] [CrossRef]
- Zeng, Q.; Han, Y.; Li, H.; Chang, J. Design of a thermosensitive bioglass/agarose–alginate composite hydrogel for chronic wound healing. J. Mat. Chem. B 2015, 3, 8856–8864. [Google Scholar] [CrossRef]
- ISO 10993-5:2009; Biological Evaluation of Medical Devices—Part 5: Tests for In Vitro Cytotoxicity. International Organization for Standardization: Geneva, Switzerland, 2009.
- Wang, G.; Watson, K.M.; Buckheit, R.W., Jr. Anti-human immunodeficiency virus type 1 activities of antimicrobial peptides derived from human and bovine cathelicidins. Antimicrob. Agents Chemother. 2008, 52, 3438–3440. [Google Scholar] [CrossRef]
- Gunasekera, S.; Muhammad, T.; Strömstedt, A.A.; Rosengren, K.J.; Göransson, U. Alanine and lysine scans of the LL-37-derived peptide fragment KR-12 reveal key residues for antimicrobial activity. ChemBioChem 2018, 19, 931–939. [Google Scholar] [CrossRef]
- Kai-Larsen, Y.; Agerberth, B. The role of the multifunctional peptide LL-37 in host defense. Front. Biosci. Landmark 2008, 13, 3760–3767. [Google Scholar] [CrossRef]
- Fricks-Lima, J.; Hendrickson, C.; Allgaier, M.; Zhuo, H.; Wiener-Kronish, J.; Lynch, S.; Yang, K. Differences in biofilm formation and antimicrobial resistance of Pseudomonas aeruginosa isolated from airways of mechanically ventilated patients and cystic fibrosis patients. Int. J. Antimicrob. Agents 2011, 37, 309–315. [Google Scholar] [CrossRef]
- Torres, M.D.; Pedron, C.N.; Higashikuni, Y.; Kramer, R.M.; Cardoso, M.H.; Oshiro, K.G.; Franco, O.L.; Silva Junior, P.I.; Silva, F.D.; Oliveira Junior, V.X. Structure-function-guided exploration of the antimicrobial peptide polybia-CP identifies activity determinants and generates synthetic therapeutic candidates. Commun. Biol. 2018, 1, 221. [Google Scholar] [CrossRef]
- Scott, A.; Weldon, S.; Buchanan, P.J.; Schock, B.; Ernst, R.K.; McAuley, D.F.; Tunney, M.M.; Irwin, C.R.; Elborn, J.S.; Taggart, C.C. Evaluation of the ability of LL-37 to neutralise LPS in vitro and ex vivo. PLoS ONE 2011, 6, e26525. [Google Scholar] [CrossRef]
- Finger, S.; Kerth, A.; Dathe, M.; Blume, A. The efficacy of trivalent cyclic hexapeptides to induce lipid clustering in PG/PE membranes correlates with their antimicrobial activity. Biochim. Biophys. Acta (BBA)-Biomembr. 2015, 1848, 2998–3006. [Google Scholar] [CrossRef]
- Wadhwani, P.; Epand, R.; Heidenreich, N.; Bürck, J.; Ulrich, A.; Epand, R. Membrane-active peptides and the clustering of anionic lipids. Biophys. J. 2012, 103, 265–274. [Google Scholar] [CrossRef]
- Bhattacharjya, S. De novo designed lipopolysaccharide binding peptides: Structure based development of antiendotoxic and antimicrobial drugs. Curr. Med. Chem. 2010, 17, 3080–3093. [Google Scholar] [CrossRef]
- Rosenfeld, Y.; Papo, N.; Shai, Y. Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides: Peptide properties and plausible modes of action. J. Biol. Chem. 2006, 281, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, Y.; Lev, N.; Shai, Y. Effect of the hydrophobicity to net positive charge ratio on antibacterial and anti-endotoxin activities of structurally similar antimicrobial peptides. Biochemistry 2010, 49, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Lin, Z.; Ren, J.; Yao, P.; Wang, X.; Wang, Z.; Zhang, Q. The non-specific binding of fluorescent-labeled MiRNAs on cell surface by hydrophobic interaction. PLoS ONE 2016, 11, e0149751. [Google Scholar] [CrossRef] [PubMed]
- Tjabringa, G.S.; Aarbiou, J.; Ninaber, D.K.; Drijfhout, J.W.; Sørensen, O.E.; Borregaard, N.; Rabe, K.F.; Hiemstra, P.S. The antimicrobial peptide LL-37 activates innate immunity at the airway epithelial surface by transactivation of the epidermal growth factor receptor. J. Immunol. 2003, 171, 6690–6696. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Costa, F.; Carvalho, I.F.; Montelaro, R.C.; Gomes, P.; Martins, M.C.L. Covalent immobilization of antimicrobial peptides (AMPs) onto biomaterial surfaces. Acta Biomater. 2011, 7, 1431–1440. [Google Scholar] [CrossRef]
- Haynie, S.L.; Crum, G.A.; Doele, B.A. Antimicrobial activities of amphiphilic peptides covalently bonded to a water-insoluble resin. Antimicrob. Agents Chemother. 1995, 39, 301–307. [Google Scholar] [CrossRef]
- Cho, W.-M.; Joshi, B.P.; Cho, H.; Lee, K.-H. Design and synthesis of novel antibacterial peptide-resin conjugates. Bioorg. Med. Chem. Lett. 2007, 17, 5772–5776. [Google Scholar] [CrossRef]
- Long, T.; Li, H.; Wang, X.; Yue, B. A comparative analysis of antibacterial properties and inflammatory responses for the KR-12 peptide on titanium and PEGylated titanium surfaces. RSC Adv. 2017, 7, 34321–34330. [Google Scholar]
- Gabriel, M.; Nazmi, K.; Veerman, E.C.; Nieuw Amerongen, A.V.; Zentner, A. Preparation of LL-37-grafted titanium surfaces with bactericidal activity. Bioconj. Chem. 2006, 17, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, M.; Beyermann, M.; Dathe, M. Immobilization reduces the activity of surface-bound cationic antimicrobial peptides with no influence upon the activity spectrum. Antimicrob. Agents Chemother. 2009, 53, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.M.; Maia, S.R.; Gomes, P.A.; Martins, M.C.L. Dhvar5 antimicrobial peptide (AMP) chemoselective covalent immobilization results on higher antiadherence effect than simple physical adsorption. Biomaterials 2015, 52, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, T.; Strömstedt, A.A.; Gunasekera, S.; Göransson, U. Transforming Cross-Linked Cyclic Dimers of KR-12 into Stable and Potent Antimicrobial Drug Leads. Biomedicines 2023, 11, 504. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Separovic, F.; O’Brien-Simpson, N.M.; Wade, J.D. Chemically modified and conjugated antimicrobial peptides against superbugs. Chem. Soc. Rev. 2021, 50, 4932–4973. [Google Scholar] [CrossRef] [PubMed]
- Ong, Z.Y.; Wiradharma, N.; Yang, Y.Y. Strategies employed in the design and optimization of synthetic antimicrobial peptide amphiphiles with enhanced therapeutic potentials. Adv. Drug Del. Rev. 2014, 78, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Pinto, I.B.; dos Santos Machado, L.; Meneguetti, B.T.; Nogueira, M.L.; Carvalho, C.M.E.; Roel, A.R.; Franco, O.L. Utilization of antimicrobial peptides, analogues and mimics in creating antimicrobial surfaces and bio-materials. Biochem. Eng. J. 2019, 150, 107237. [Google Scholar] [CrossRef]
- Zhou, X.; Zheng, W.; Li, Y.; Pearce, R.; Zhang, C.; Bell, E.W.; Zhang, G.; Zhang, Y. I-TASSER-MTD: A deep-learning-based platform for multi-domain protein structure and function prediction. Nat. Protoc. 2022, 17, 2326–2353. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolym. Orig. Res. Biomol. 2008, 89, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Compton, L.A.; Johnson, W.C., Jr. Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal. Biochem. 1986, 155, 155–167. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48 (Suppl. S1), 5–16. [Google Scholar] [CrossRef]
- Wang, X.; Mishra, B.; Lushnikova, T.; Narayana, J.L.; Wang, G. Amino acid composition determines peptide activity spectrum and hot-spot-based design of merecidin. Adv. Biosyst. 2018, 2, 1700259. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Label | Sequence/Predicted Secondary Structure |
|---|---|
| Long-CBP | GSGSGGSCQVLNPWYSQTTPGWGQC CCCCCCCCSSSCCHHCCCCCCCCCC |
| Short-CBP | GSGSGGSWHWTYYW CCCCCCCSSSSSSC |
| KR-12 | KRIVQRIKDFLR CHHHHHHHHHHC |
| Long-CBP-KR12 | KRIVQRIKDFLRGSGSGGSCQVLNPWYSQTTPGWGQC CHHHHHHHHHHCCCCCCCCCSSCCHHHHCCCCCCCCC |
| Short-CBP-KR12 | KRIVQRIKDFLRGSGSGGSWHWTYYW CHHHHHHHHHHCCCCCCCCSSSSSSC |
| Peptide Label | Mw (Da) a | pI a | q b | AI a | Hx (%) c | µH b | H b | II a |
|---|---|---|---|---|---|---|---|---|
| Long-CBP | 2557.75 | 5.51 | 0 | 27.2 | 4.3 | 0.286 | 0.480 | 24.72 |
| Short-CBP | 1630.70 | 6.74 | 0 | 0 | 3.3 | 0.128 | 0.639 | 23.63 |
| KR-12 | 1571.93 | 11.72 | +4 | 121.67 | 56.0 | 0.782 | 0.193 | 31.95 |
| Long-CBP-KR12 | 4111.67 | 9.84 | +4 | 57.84 | 69.0 | 0.387 | 0.387 | 25.04 |
| Short-CBP-KR12 | 3184.61 | 10.43 | +4 | 56.15 | 53.0 | 0.346 | 0.435 | 24.59 |
| Peptide Label | Peptide Sequence |
|---|---|
| Short-CBP | WHWTYYW-NH2 |
| Long-CBP | CQVLNPWYSQTTPGWGQC-NH2 |
| KR-12 | KRIVQRIKDFLR-NH2 |
| Short-CBP-KR12 | KRIVQRIKDFLR-GSGSGGS-WHWTYYW-NH2 |
| Long-CBP-KR12 | KRIVQRIKDFLR-GSGSGGS- CQVLNPWYSQTTPGWGQC-NH2 |
| Short-CBP-FAM | WHWTYYW-(mini-PEG)-K(5-FAM)-NH2 |
| Long-CBP-FAM | CQVLNPWYSQTTPGWGQC-(mini-PEG)-K(5-FAM)-NH2 |
| KR-12-FAM | KRIVQRIKDFLR-(min-iPEG)-K(5-FAM)-NH2 |
| Short-CBP-KR12-FAM | KRIVQRIKDFLR-GSGSGGS-WHWTYYW-NH2 |
| Long-CBP-KR12-FAM | KRIVQRIKDFLR-GSGSGGS-CQVLNPWYSQTTPGWGQC-(mini-PEG)-K(5-FAM)-NH2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Zyl, E.M.; Coburn, J.M. Functionalization of Bacterial Cellulose with the Antimicrobial Peptide KR-12 via Chimerical Cellulose-Binding Peptides. Int. J. Mol. Sci. 2024, 25, 1462. https://doi.org/10.3390/ijms25031462
van Zyl EM, Coburn JM. Functionalization of Bacterial Cellulose with the Antimicrobial Peptide KR-12 via Chimerical Cellulose-Binding Peptides. International Journal of Molecular Sciences. 2024; 25(3):1462. https://doi.org/10.3390/ijms25031462
Chicago/Turabian Stylevan Zyl, Elizabeth M., and Jeannine M. Coburn. 2024. "Functionalization of Bacterial Cellulose with the Antimicrobial Peptide KR-12 via Chimerical Cellulose-Binding Peptides" International Journal of Molecular Sciences 25, no. 3: 1462. https://doi.org/10.3390/ijms25031462
APA Stylevan Zyl, E. M., & Coburn, J. M. (2024). Functionalization of Bacterial Cellulose with the Antimicrobial Peptide KR-12 via Chimerical Cellulose-Binding Peptides. International Journal of Molecular Sciences, 25(3), 1462. https://doi.org/10.3390/ijms25031462