

Exploring the Molecular Aspects of Myeloproliferative Neoplasms Associated with Unusual Site Vein Thrombosis: Review of the Literature and Latest Insights
 , , ,
, , ,
Abstract
:1. Introduction
1.1. MPNs and Thrombotic Risk
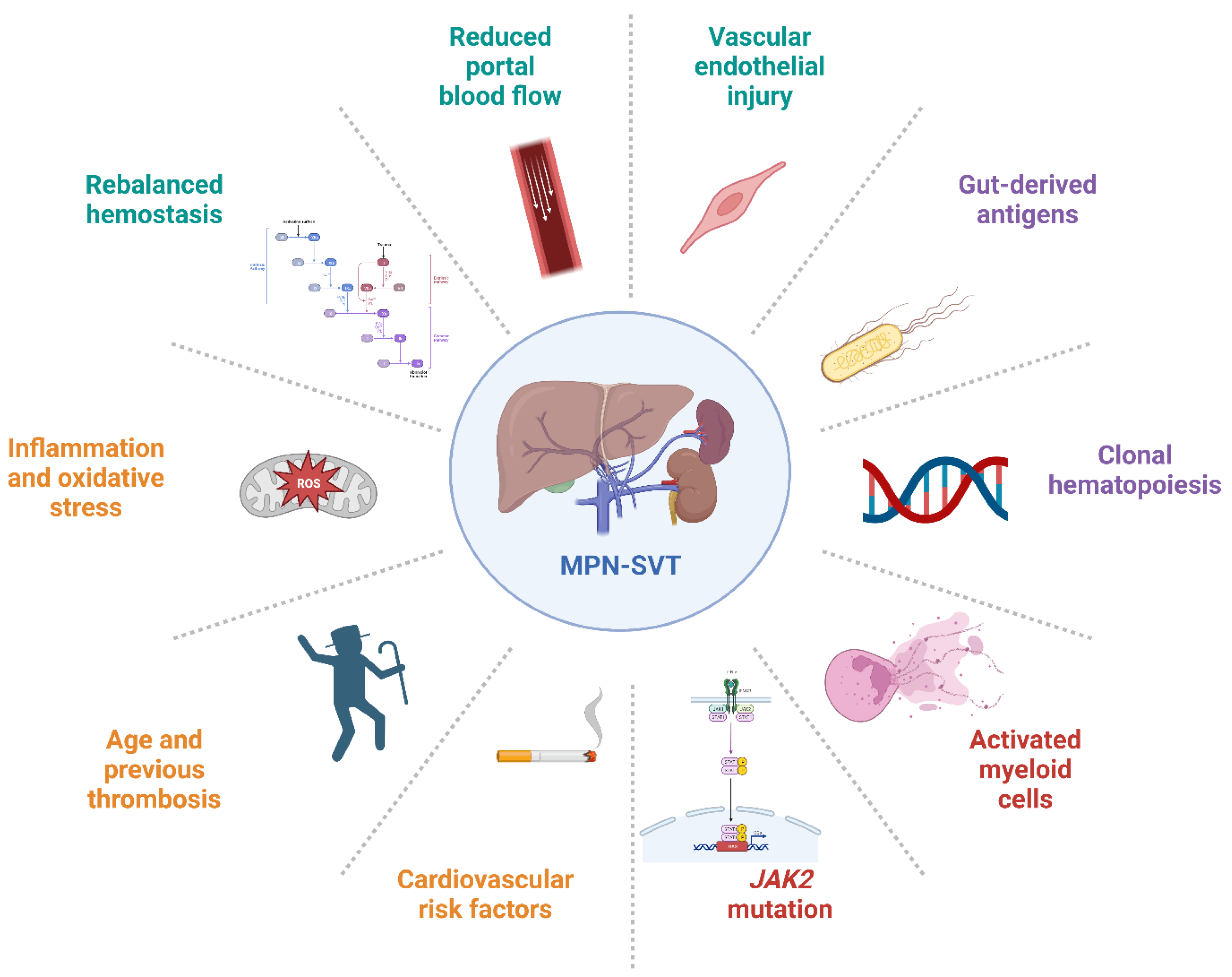
1.2. MPN and Unusual Site Thrombosis
1.3. MPNs: Driver and Additional Mutations
2. Myeloproliferative Neoplasms Associated with Unusual Site Thrombosis
2.1. Peculiar Clinical Presentation
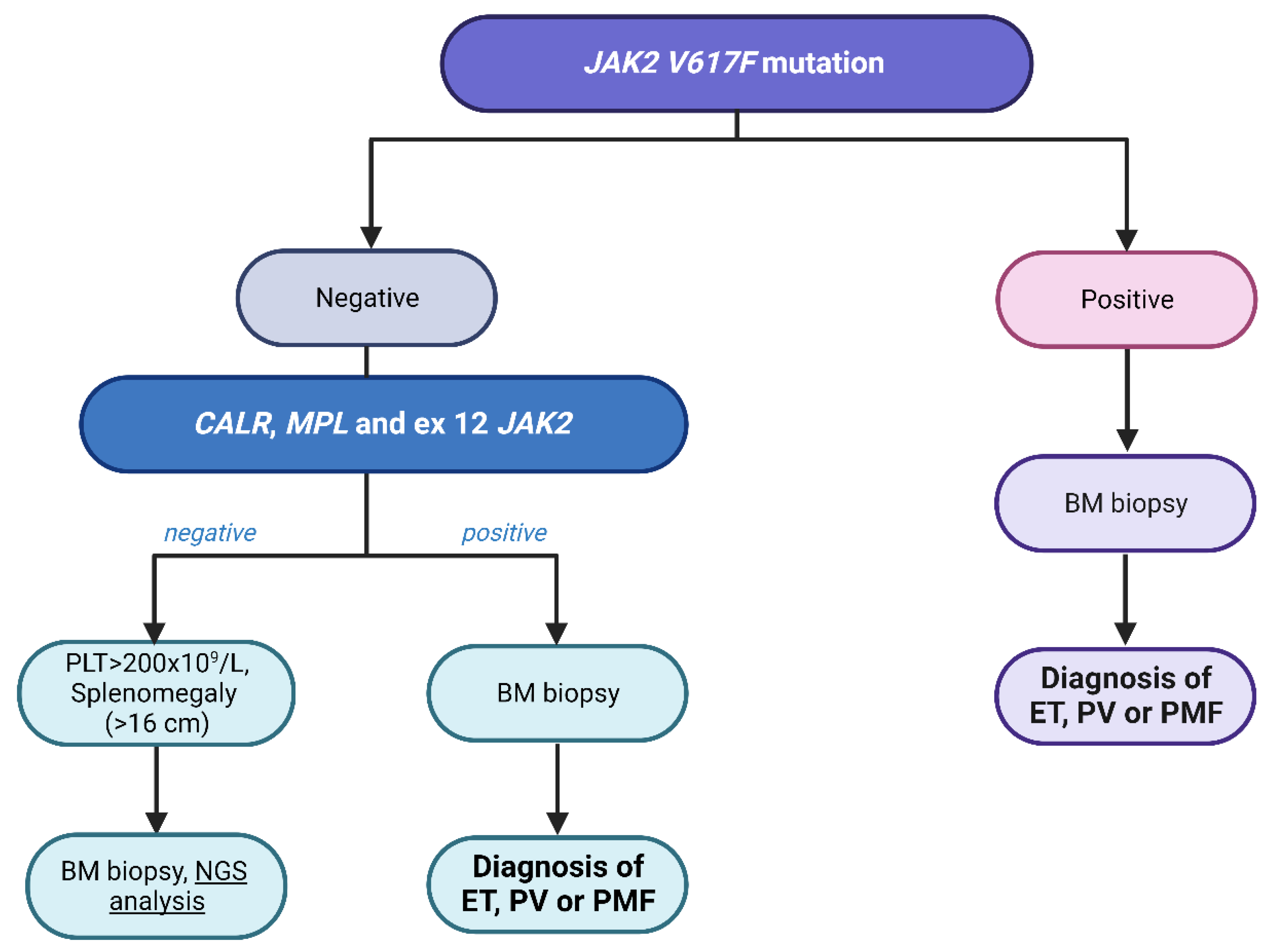
2.2. Role of Driver Mutation in Diagnosis and Prognosis
2.3. Comprehensive Genomic Profiling of Thrombosis Risk
2.3.1. Other Molecular Drivers of MPN-Associated SVT
2.3.2. Next-Generation Sequencing in the Diagnosis of Non-Cirrhotic Splanchnic Vein Thrombosis
3. Discussion
4. Conclusions and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. Special Report International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Elvers, M.; Schafer, A.I. The pathobiology of thrombosis, microvascular disease, and hemorrhage in the myeloproliferative neoplasms. Blood 2021, 137, 2152–2160. [Google Scholar] [CrossRef]
- Marchioli, R.; Finazzi, G.; Specchia, G.; Cacciola, R.; Cavazzina, R.; Cilloni, D.; De Stefano, V.; Elli, E.; Iurlo, A.; Latagliata, R.; et al. Cardiovascular events and intensity of treatment in polycythemia vera. N. Engl. J. Med. 2013, 368, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, G. A prospective analysis of thrombotic events in the European collaboration study on low-dose aspirin in polycythemia (ECLAP). Pathol. Biol. 2004, 52, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Hultcrantz, M.; Björkholm, M.; Dickman, P.W.; Landgren, O.; Derolf, R.; Kristinsson, S.Y.; Andersson, T.M.; Moliterno, A.R.; Ratchford, E.V.; Zhao, J.V.; et al. Risk for Arterial and Venous Thrombosis in Patients With Myeloproliferative Neoplasms: A Population-Based Cohort Study. Ann. Intern. Med. 2018, 168, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; Vannucchi, A.M.; Antonioli, E.; et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: An international study of 891 patients. Blood 2011, 117, 5857–5859. [Google Scholar] [CrossRef] [PubMed]
- Saliba, W.; Mishchenko, E.; Cohen, S.; Rennert, G.; Preis, M. Association between myelofibrosis and thromboembolism: A population-based retrospective cohort study. J. Thromb. Haemost. 2020, 18, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Reeves, B.N.; Moliterno, A.R. Thrombosis in myeloproliferative neoplasms: Update in pathophysiology. Curr. Opin. Hematol. 2021, 28, 285–291. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, V.; Martinelli, I. Splanchnic vein thrombosis: Clinical presentation, risk factors and treatment. Intern. Emerg. Med. 2010, 5, 487–494. [Google Scholar] [CrossRef]
- Dentali, F.; Galli, M.; Gianni, M.; Ageno, W. Inherited thrombophilic abnormalities and risk of portal vein thrombosis. a meta-analysis. Thromb. Haemost. 2008, 99, 675–682. [Google Scholar] [CrossRef]
- Sant’Antonio, E.; Guglielmelli, P.; Pieri, L.; Primignani, M.; Randi, M.L.; Santarossa, C.; Rumi, E.; Cervantes, F.; Delaini, F.; Carobbio, A.; et al. Splanchnic vein thromboses associated with myeloproliferative neoplasms: An international, retrospective study on 518 cases. Am. J. Hematol. 2020, 95, 156–166. [Google Scholar] [CrossRef]
- Tremblay, D.; Winters, A.; Beckman, J.D.; Naymagon, L.; Patel, R.; Mascarenhas, J.; Schiano, T.D. Splanchnic vein thrombosis associated with myeloproliferative neoplasms. Thromb. Res. 2022, 218, 8–16. [Google Scholar] [CrossRef]
- Hoekstra, J.; Bresser, E.L.; Smalberg, J.H.; Spaander, M.C.W.; Leebeek, F.W.G.; Janssen, H.L.A. Long-term follow-up of patients with portal vein thrombosis and myeloproliferative neoplasms. J. Thromb. Haemost. 2011, 9, 2208–2214. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Larrán, A.; Pereira, A.; Magaz, M.; Hernández-Boluda, J.C.; Garrote, M.; Cuevas, B.; Ferrer-Marín, F.; Gómez-Casares, M.T.; García-Gutiérrez, V.; Mata-Vázquez, M.I.; et al. Natural history of polycythemia vera and essential thrombocythemia presenting with splanchnic vein thrombosis. Ann. Hematol. 2020, 99, 791–798. [Google Scholar] [CrossRef]
- Dentali, F.; Ageno, W.; Rumi, E.; Casetti, I.; Poli, D.; Scoditti, U.; Maffioli, M.; di Minno, M.N.D.; Caramazza, D.; Pietra, D.; et al. Cerebral venous thrombosis and myeloproliferative neoplasms: Results from two large databases. Thromb. Res. 2014, 134, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Rupoli, S.; Fiorentini, A.; Morsia, E.; Svegliati-Baroni, G.; Micucci, G.; Maroni, L.; Garvey, K.B.; Riva, A.; Da Lio, L.; Benedetti, A.; et al. Anticoagulation and Vessel Recanalization in Cirrhotic Patients with Splanchnic Vein Thrombosis: A Multidisciplinary “Real Life” Experience. Vasc. Health Risk Manag. 2021, 17, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Marneth, A.E.; Mullally, A. The Molecular Genetics of Myeloproliferative Neoplasms. Cold Spring Harb. Perspect. Med. 2020, 10, a034876. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.W.Y.; Hannah, R.; Green, A.R.; Göttgens, B. The JAK-STAT signaling pathway is differentially activated in CALR-positive compared with JAK2V617F-positive ET patients. Blood 2015, 125, 1679–1681. [Google Scholar] [CrossRef]
- Szybinski, J.; Meyer, S.C. Genetics of Myeloproliferative Neoplasms. Hematol. Oncol. Clin. N. Am. 2021, 35, 217–236. [Google Scholar] [CrossRef]
- Jensen, M.K.; de Nully Brown, P.; Lund, B.V.; Nielsen, O.J.; Hasselbalch, H.C. Increased platelet activation and abnormal membrane glycoprotein content and redistribution in myeloproliferative disorders. Br. J. Haematol. 2000, 110, 116–124. [Google Scholar] [CrossRef]
- Falanga, A.; Marchetti, M.; Barbui, T.; Smith, C.W. Pathogenesis of thrombosis in essential thrombocythemia and polycythemia vera: The role of neutrophils. Semin. Hematol. 2005, 42, 239–247. [Google Scholar] [CrossRef]
- Wautier, M.-P.; El Nemer, W.; Gane, P.; Rain, J.-D.; Cartron, J.-P.; Colin, Y.; Le Van Kim, C.; Wautier, J.-L. Increased adhesion to endothelial cells of erythrocytes from patients with polycythemia vera is mediated by laminin alpha5 chain and Lu/BCAM. Blood 2007, 110, 894–901. [Google Scholar] [CrossRef]
- Sozer, S.; Fiel, M.I.; Schiano, T.; Xu, M.; Mascarenhas, J.; Hoffman, R. The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood 2009, 113, 5246–5249. [Google Scholar] [CrossRef]
- Rosti, V.; Villani, L.; Riboni, R.; Poletto, V.; Bonetti, E.; Tozzi, L.; Bergamaschi, G.; Catarsi, P.; Dallera, E.; Novara, F.; et al. Spleen endothelial cells from patients with myelofibrosis harbor the JAK2V617F mutation. Blood 2013, 121, 360–368. [Google Scholar] [CrossRef]
- Guy, A.; Danaee, A.; Paschalaki, K.; Boureau, L.; Rivière, E.; Etienne, G.; Mansier, O.; Laffan, M.; Sekhar, M.; James, C. Absence of JAK2V617F Mutated Endothelial Colony-Forming Cells in Patients With JAK2V617F Myeloproliferative Neoplasms and Splanchnic Vein Thrombosis. HemaSphere 2020, 4, e364. [Google Scholar] [CrossRef]
- Farina, M.; Bernardi, S.; Polverelli, N.; D’adda, M.; Malagola, M.; Bosio, K.; Re, F.; Almici, C.; Dunbar, A.; Levine, R.L.; et al. Comparative Mutational Profiling of Hematopoietic Progenitor Cells and Circulating Endothelial Cells (CECs) in Patients with Primary Myelofibrosis. Cells 2021, 10, 2764. [Google Scholar] [CrossRef] [PubMed]
- Morsia, E.; Torre, E.; Poloni, A.; Olivieri, A.; Rupoli, S. Molecular Pathogenesis of Myeloproliferative Neoplasms: From Molecular Landscape to Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 4573. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Screening for ASXL1 and SRSF2 mutations is imperative for treatment decision-making in otherwise low or intermediate-1 risk patients with myelofibrosis. Br. J. Haematol. 2018, 183, 678–681. [Google Scholar] [CrossRef] [PubMed]
- Morsia, E.; Gangat, N. Myelofibrosis: Challenges for preclinical models and emerging therapeutic targets. Expert Opin. Ther. Targets 2021, 25, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.-J.; Cervantes, F.; Leebeek, F.W.G.; Marzac, C.; Cassinat, B.; Chevret, S.; Cazals-Hatem, D.; Plessier, A.; Garcia-Pagan, J.-C.; Murad, S.D.; et al. The impact of JAK2 and MPL mutations on diagnosis and prognosis of splanchnic vein thrombosis: A report on 241 cases. Blood 2008, 111, 4922–4929. [Google Scholar] [CrossRef] [PubMed]
- Stein, B.L.; Saraf, S.; Sobol, U.; Halpern, A.; Shammo, J.; Rondelli, D.; Michaelis, L.; Odenike, O.; Rademaker, A.; Zakarija, A.; et al. Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk. Lymphoma 2013, 54, 1989–1995. [Google Scholar] [CrossRef]
- Stein, B.L.; Rademaker, A.; Spivak, J.L.; Moliterno, A.R. Gender and Vascular Complications in the JAK2 V617F-Positive Myeloproliferative Neoplasms. Thrombosis 2011, 2011, 874146. [Google Scholar] [CrossRef]
- Shukla, A.; Giri, S. Portal Vein Thrombosis in Cirrhosis. J. Clin. Exp. Hepatol. 2022, 12, 965–979. [Google Scholar] [CrossRef] [PubMed]
- Valla, D. Splanchnic Vein Thrombosis. Semin. Thromb. Hemost. 2015, 41, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Van Bijnen, S.T.A.; Van Heerde, W.L.; Muus, P. Mechanisms and clinical implications of thrombosis in paroxysmal nocturnal hemoglobinuria. J. Thromb. Haemost. 2012, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lamy, T.; Devillers, A.; Bernard, M.; Moisan, A.; Grulois, I.; Drenou, B.; Amiot, L.; Fauchet, R.; Le Prise, P. Inapparent polycythemia vera: An unrecognized diagnosis. Am. J. Med. 1997, 102, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Lavu, S.; Szuber, N.; Mudireddy, M.; Yogarajah, M.; Gangat, N.; Pardanani, A.; Hanson, C.A.; Ketterling, R.P.; Ashrani, A.A.; Kamath, P.S.; et al. Splanchnic vein thrombosis in patients with myeloproliferative neoplasms: The Mayo clinic experience with 84 consecutive cases. Am. J. Hematol. 2018, 93, E61–E64. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, V.; Vannucchi, A.M.; Ruggeri, M.; Cervantes, F.; Alvarez-Larrán, A.; Iurlo, A.; Randi, M.L.; Pieri, L.; Rossi, E.; Guglielmelli, P.; et al. Splanchnic vein thrombosis in myeloproliferative neoplasms: Risk factors for recurrences in a cohort of 181 patients. Blood Cancer J. 2016, 6, e493. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, V.; Qi, X.; Betti, S.; Rossi, E. Splanchnic vein thrombosis and myeloproliferative neoplasms: Molecular-driven diagnosis and long-term treatment. Thromb. Haemost. 2016, 115, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Green, T.R. The evolving genomic landscape of myeloproliferative neoplasms. Hematol. Am. Soc. Hematol. Educ. Progr. 2014, 2014, 287–296. [Google Scholar] [CrossRef]
- Smalberg, J.H.; Arends, L.R.; Valla, D.C.; Kiladjian, J.-J.; Janssen, H.L.A.; Leebeek, F.W.G. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: A meta-analysis. Blood 2012, 120, 4921–4928. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, V.; Za, T.; Rossi, E.; Vannucchi, A.M.; Ruggeri, M.; Elli, E.; Micò, C.; Tieghi, A.; Cacciola, R.R.; Santoro, C.; et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: Incidence, risk factors, and effect of treatments. Haematologica 2008, 93, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Gangat, N.; Guglielmelli, P.; Betti, S.; Farrukh, F.; Carobbio, A.; Barbui, T.; Vannucchi, A.M.; De Stefano, V.; Tefferi, A. Cerebral venous thrombosis and myeloproliferative neoplasms: A three-center study of 74 consecutive cases. Am. J. Hematol. 2021, 96, 1580–1586. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, V.; Fiorini, A.; Rossi, E.; Za, T.; Farina, G.; Chiusolo, P.; Sica, S.; Leone, G. Incidence of the JAK2 V617F mutation among patients with splanchnic or cerebral venous thrombosis and without overt chronic myeloproliferative disorders. J. Thromb. Haemost. 2007, 5, 708–714. [Google Scholar] [CrossRef]
- Finazzi, G.; De Stefano, V.; Barbui, T. Splanchnic vein thrombosis in myeloproliferative neoplasms: Treatment algorithm 2018. Blood Cancer J. 2018, 8, 64. [Google Scholar] [CrossRef]
- Dentali, F.; Squizzato, A.; Brivio, L.; Appio, L.; Campiotti, L.; Crowther, M.; Grandi, A.M.; Ageno, W. JAK2V617F mutation for the early diagnosis of Ph- myeloproliferative neoplasms in patients with venous thromboembolism: A meta-analysis. Blood 2009, 113, 5617–5623. [Google Scholar] [CrossRef]
- How, J.; Zhou, A.; Oh, S.T. Splanchnic vein thrombosis in myeloproliferative neoplasms: Pathophysiology and molecular mechanisms of disease. Ther. Adv. Hematol. 2017, 8, 107–118. [Google Scholar] [CrossRef]
- Passamonti, F.; Rumi, E.; Arcaini, L.; Boveri, E.; Elena, C.; Pietra, D.; Boggi, S.; Astori, C.; Bernasconi, P.; Varettoni, M.; et al. Prognostic factors for thrombosis, myelofibrosis, and leukemia in essential thrombocythemia: A study of 605 patients. Haematologica 2008, 93, 1645–1651. [Google Scholar] [CrossRef]
- Benlabiod, C.; Dagher, T.; Marty, C.; Villeval, J.-L. Lessons from mouse models of MPN. Int. Rev. Cell Mol. Biol. 2022, 366, 125–185. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Loscocco, G.G.; Mannarelli, C.; Rossi, E.; Mannelli, F.; Ramundo, F.; Coltro, G.; Betti, S.; Maccari, C.; Ceglie, S.; et al. JAK2V617F variant allele frequency >50% identifies patients with polycythemia vera at high risk for venous thrombosis. Blood Cancer J. 2021, 11, 199. [Google Scholar] [CrossRef] [PubMed]
- Soudet, S.; Le Roy, G.; Cadet, E.; Michaud, A.; Morel, P.; Marolleau, J.P.; Sevestre, M.A. JAK2 allele burden is correlated with a risk of venous but not arterial thrombosis. Thromb. Res. 2022, 211, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, Y.; Wang, Y.; Teng, G.; Li, D.; Wang, Y.; Du, C.; Chen, Y.; Zhang, H.; Li, Y.; et al. Thrombosis among 1537 patients with JAK2(V617F) -mutated myeloproliferative neoplasms: Risk factors and development of a predictive model. Cancer Med. 2020, 9, 2096–2105. [Google Scholar] [CrossRef] [PubMed]
- Debureaux, P.-E.; Cassinat, B.; Soret-Dulphy, J.; Mora, B.; Verger, E.; Maslah, N.; Plessier, A.; Rautou, P.-E.; Ollivier-Hourman, I.; De Ledinghen, V.; et al. Molecular profiling and risk classification of patients with myeloproliferative neoplasms and splanchnic vein thromboses. Blood Adv. 2020, 4, 3708–3715. [Google Scholar] [CrossRef]
- Galimberti, S.; Balducci, S.; Guerrini, F.; Del Re, M.; Cacciola, R. Digital Droplet PCR in Hematologic Malignancies: A New Useful Molecular Tool. Diagnostics 2022, 12, 1305. [Google Scholar] [CrossRef]
- Zheng, C.-F.; Zhao, X.-X.; Chen, X.-H.; Liu, Z.; Wang, W.; Luo, M.; Ren, Y.; Wang, H. Quantification of JAK2V617F mutation load by droplet digital PCR can aid in diagnosis of myeloproliferative neoplasms. Int. J. Lab. Hematol. 2021, 43, 645–650. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, Q.; Luo, J.; Xing, S.; Li, Q.; Krantz, S.B.; Fu, X.; Zhao, Z.J. JAK2(V617F): Prevalence in a large Chinese hospital population. Blood 2007, 109, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.; Birgens, H.S.; Nordestgaard, B.G.; Bojesen, S.E. Diagnostic value of JAK2 V617F somatic mutation for myeloproliferative cancer in 49 488 individuals from the general population. Br. J. Haematol. 2013, 160, 70–79. [Google Scholar] [CrossRef]
- Nielsen, C.; Birgens, H.S.; Nordestgaard, B.G.; Kjaer, L.; Bojesen, S.E. The JAK2 V617F somatic mutation, mortality and cancer risk in the general population. Haematologica 2011, 96, 450–453. [Google Scholar] [CrossRef]
- Cordua, S.; Kjaer, L.; Skov, V.; Pallisgaard, N.; Hasselbalch, H.C.; Ellervik, C. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood 2019, 134, 469–479. [Google Scholar] [CrossRef]
- Stein, B.L.; Moliterno, A.R. Primary myelofibrosis and the myeloproliferative neoplasms: The role of individual variation. JAMA 2010, 303, 2513–2518. [Google Scholar] [CrossRef] [PubMed]
- De Grandis, M.; Cambot, M.; Wautier, M.-P.; Cassinat, B.; Chomienne, C.; Colin, Y.; Wautier, J.-L.; Le Van Kim, C.; El Nemer, W. JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independent Rap1/Akt pathway. Blood 2013, 121, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; De Stefano, V.; Song, T.; Zhou, X.; Guo, Z.; Zhu, J.; Qi, X. Prevalence of CALR mutations in splanchnic vein thrombosis: A systematic review and meta-analysis. Thromb. Res. 2018, 167, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Segura-Díaz, A.; Stuckey, R.; Florido, Y.; González-Martín, J.M.; López-Rodríguez, J.F.; Sánchez-Sosa, S.; González-Pérez, E.; Perdomo, M.N.S.; Perera, M.d.M.; de la Iglesia, S.; et al. Thrombotic Risk Detection in Patients with Polycythemia Vera: The Predictive Role of DNMT3A/TET2/ASXL1 Mutations. Cancers 2020, 12, 934. [Google Scholar] [CrossRef]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, aan8292. [Google Scholar] [CrossRef]
- Dunbar, A.; Bolton, K.L.; Devlin, S.M.; Sanchez-Vega, F.; Gao, J.; Mones, J.V.; Wills, J.; Kelly, D.; Farina, M.; Cordner, K.B.; et al. Genomic profiling identifies somatic mutations predicting thromboembolic risk in patients with solid tumors. Blood 2021, 137, 2103–2113. [Google Scholar] [CrossRef]
- Soudet, S.; Jedraszak, G.; Evrard, O.; Marolleau, J.P.; Garcon, L.; Pietri, M.A.S. Is Hematopoietic Clonality of Indetermined Potential a Risk Factor for Pulmonary Embolism? TH Open Companion J. Thromb. Haemost. 2021, 5, e338–e342. [Google Scholar] [CrossRef]
- Kimishima, Y.; Misaka, T.; Yokokawa, T.; Wada, K.; Ueda, K.; Sugimoto, K.; Minakawa, K.; Nakazato, K.; Ishida, T.; Oshima, M.; et al. Clonal hematopoiesis with JAK2V617F promotes pulmonary hypertension with ALK1 upregulation in lung neutrophils. Nat. Commun. 2021, 12, 6177. [Google Scholar] [CrossRef]
- Montani, D.; Thoré, P.; Mignard, X.; Jaïs, X.; Boucly, A.; Jevnikar, M.; Seferian, A.; Jutant, E.-M.; Cottin, V.; Fadel, E.; et al. Clinical Phenotype and Outcomes of Pulmonary Hypertension Associated with Myeloproliferative Neoplasms: A Population-based Study. Am. J. Respir. Crit. Care Med. 2023, 208, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Gangat, N.; Coltro, G.; Lasho, T.L.; Loscocco, G.G.; Finke, C.M.; Morsia, E.; Sordi, B.; Szuber, N.; Hanson, C.A.; et al. Mutations and thrombosis in essential thrombocythemia. Blood Cancer J. 2021, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Thiele, J.; Passamonti, F.; Rumi, E.; Boveri, E.; Ruggeri, M.; Rodeghiero, F.; D’Amore, E.S.; Randi, M.L.; Bertozzi, I.; et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: An international study. J. Clin. Oncol. 2011, 29, 3179–3184. [Google Scholar] [CrossRef]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef]
- Cattaneo, D.; Bucelli, C.; Marchetti, A.; Lionetti, M.; Fermo, E.; Bellani, V.; De Magistris, C.; Maeda, A.; Marella, A.; Primignani, M.; et al. Pathological and genomic features of myeloproliferative neoplasms associated with splanchnic vein thrombosis in a single-center cohort. Ann. Hematol. 2023, 102, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Colaizzo, D.; Tiscia, G.L.; Pisanelli, D.; Bafunno, V.; Amitrano, L.; Grandone, E.; Guardascione, M.A.; Margaglione, M. New TET2 gene mutations in patients with myeloproliferative neoplasms and splanchnic vein thrombosis. J. Thromb. Haemost. 2010, 8, 1142–1144. [Google Scholar] [CrossRef]
- Solary, E.; Bernard, O.A.; Tefferi, A.; Fuks, F.; Vainchenker, W. The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia 2014, 28, 485–496. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.-L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef]
- Sousos, N.; Ní Leathlobhair, M.; Simoglou Karali, C.; Louka, E.; Bienz, N.; Royston, D.; Clark, S.-A.; Hamblin, A.; Howard, K.; Mathews, V.; et al. In utero origin of myelofibrosis presenting in adult monozygotic twins. Nat. Med. 2022, 28, 1207–1211. [Google Scholar] [CrossRef] [PubMed]
- Magaz, M.; Alvarez-Larrán, A.; Colomer, D.; López-Guerra, M.; García-Criado, M.; Mezzano, G.; Belmonte, E.; Olivas, P.; Soy, G.; Cervantes, F.; et al. Next-generation sequencing in the diagnosis of non-cirrhotic splanchnic vein thrombosis. J. Hepatol. 2021, 74, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Carrà, G.; Giugliano, E.; Camerlo, S.; Rosati, G.; Branca, E.; Maffeo, B.; Russo, I.; Piazza, R.; Cilloni, D.; Morotti, A. Clonal hematopoiesis by DNMT3A mutations as a common finding in idiopathic splanchnic vein thrombosis. Haematologica 2023, 108, 1447–1449. [Google Scholar] [CrossRef] [PubMed]
- de Franchis, R.; Bosch, J.; Garcia-Tsao, G.; Reiberger, T.; Ripoll, C. Baveno VII—Renewing consensus in portal hypertension. J. Hepatol. 2022, 76, 959–974. [Google Scholar] [CrossRef] [PubMed]
- Poisson, J.; Plessier, A.; Kiladjian, J.-J.; Turon, F.; Cassinat, B.; Andreoli, A.; De Raucourt, E.; Goria, O.; Zekrini, K.; Bureau, C.; et al. Selective testing for calreticulin gene mutations in patients with splanchnic vein thrombosis: A prospective cohort study. J. Hepatol. 2017, 67, 501–507. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Bagg, A.; Hasserjian, R.P.; DiNardo, C.D.; Godley, L.A.; Iacobucci, I.; Jaiswal, S.; Malcovati, L.; Vannucchi, A.M.; Patel, K.P.; et al. Genomic profiling for clinical decision making in myeloid neoplasms and acute leukemia. Blood 2022, 140, 2228–2247. [Google Scholar] [CrossRef]
- De Stefano, V.; Rossi, E.; Carobbio, A.; Ghirardi, A.; Betti, S.; Finazzi, G.; Vannucchi, A.M.; Barbui, T. Hydroxyurea prevents arterial and late venous thrombotic recurrences in patients with myeloproliferative neoplasms but fails in the splanchnic venous district. Pooled analysis of 1500 cases. Blood Cancer J. 2018, 8, 112. [Google Scholar] [CrossRef]
- Barbui, T.; Carobbio, A.; Vannucchi, A.M.; De Stefano, V. Are the available data sufficient to suggest cytoreductive agents for patients with CHIP and stroke? Blood Adv. 2023, 7, 7551–7553. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Corresponding Effect | Gene Mutated | Rate (%) | References |
|---|---|---|---|
| Driver mutation | JAK2 V617F VAF > 50% | 21–22 | [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76] |
| JAK2 V617F VAF < 50% | 74–62 | [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76] | |
| CALR | 5 | [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76] | |
| MPL | 7 | [76] | |
| DNA methylation | TET2 | 21–28 | [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76] |
| DNMT3A | 11–17 | [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76] | |
| IDH1-IDH2 | 6 | [53] | |
| Chromatin Spliceosome | ASXL1 | 8–11 | [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76] |
| EZH2 | 2–3 | [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76] | |
| SF3B1 | 3 | [53] | |
| SRSF2 | 1 | [53] | |
| U2AF1 | 1–4 | [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76] | |
| ZRSF2 | 1 | [53] | |
| Other | TP53 | 4 | [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morsia, E.; Torre, E.; Martini, F.; Morè, S.; Poloni, A.; Olivieri, A.; Rupoli, S. Exploring the Molecular Aspects of Myeloproliferative Neoplasms Associated with Unusual Site Vein Thrombosis: Review of the Literature and Latest Insights. Int. J. Mol. Sci. 2024, 25, 1524. https://doi.org/10.3390/ijms25031524
Morsia E, Torre E, Martini F, Morè S, Poloni A, Olivieri A, Rupoli S. Exploring the Molecular Aspects of Myeloproliferative Neoplasms Associated with Unusual Site Vein Thrombosis: Review of the Literature and Latest Insights. International Journal of Molecular Sciences. 2024; 25(3):1524. https://doi.org/10.3390/ijms25031524
Chicago/Turabian StyleMorsia, Erika, Elena Torre, Francesco Martini, Sonia Morè, Antonella Poloni, Attilio Olivieri, and Serena Rupoli. 2024. "Exploring the Molecular Aspects of Myeloproliferative Neoplasms Associated with Unusual Site Vein Thrombosis: Review of the Literature and Latest Insights" International Journal of Molecular Sciences 25, no. 3: 1524. https://doi.org/10.3390/ijms25031524
APA StyleMorsia, E., Torre, E., Martini, F., Morè, S., Poloni, A., Olivieri, A., & Rupoli, S. (2024). Exploring the Molecular Aspects of Myeloproliferative Neoplasms Associated with Unusual Site Vein Thrombosis: Review of the Literature and Latest Insights. International Journal of Molecular Sciences, 25(3), 1524. https://doi.org/10.3390/ijms25031524