
Conventional Cytogenetic Analysis and Array CGH + SNP Identify Essential Thrombocythemia and Prefibrotic Primary Myelofibrosis Patients Who Are at Risk for Disease Progression
 ,
,
Abstract
:1. Introduction
2. Results
2.1. Baseline Characteristics
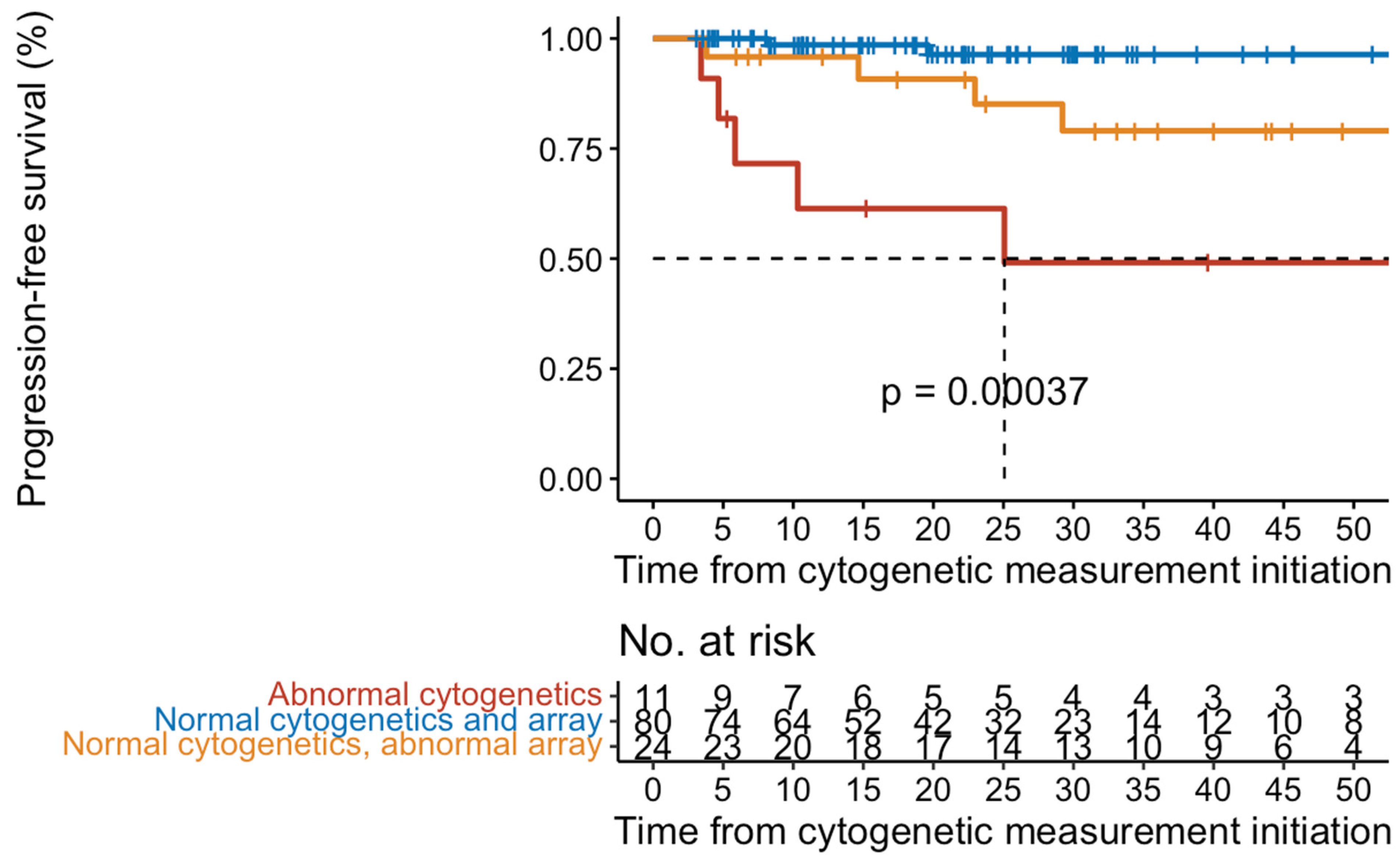
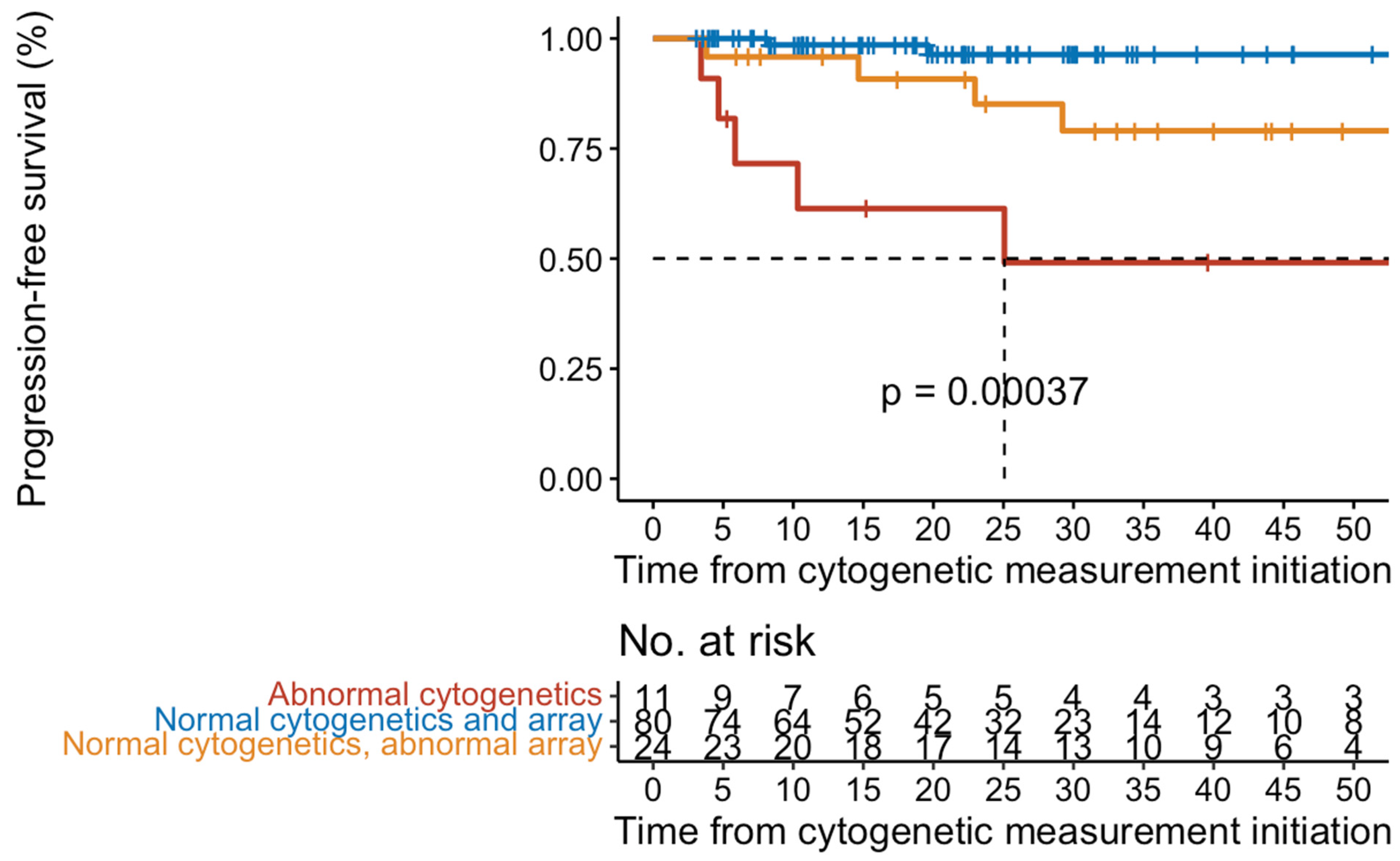
2.2. Disease Progression
2.3. Conventional Cytogenetic Results
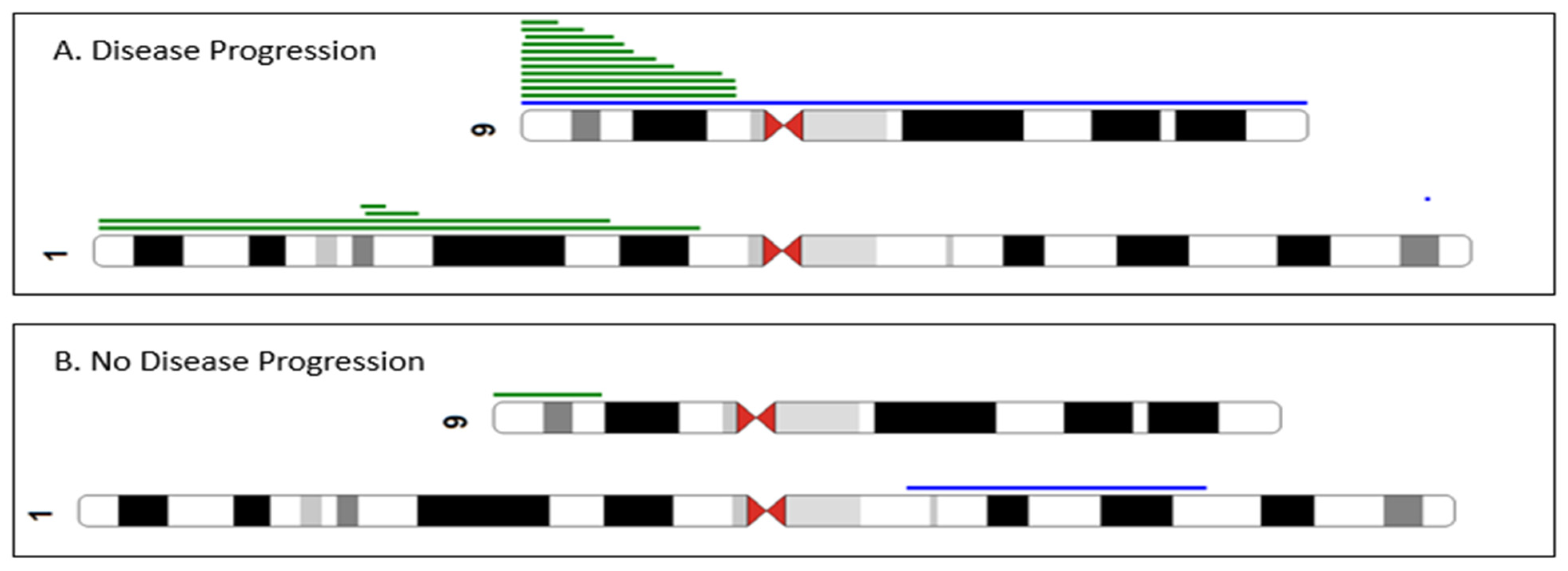
2.4. Array-CGH + SNP Results
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Conventional Cytogenetics/Chromosomal Microarray
4.3. Next Generation Sequencing
4.4. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Gangat, N.; Jadoon, Y.; Szuber, N.; Hanson, C.A.; Wolanskyj-Spinner, A.P.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. Cytogenetic Abnormalities in Essential Thrombocythemia: Clinical and Molecular Correlates and Prognostic Relevance in 809 Informative Cases. Blood Cancer J. 2022, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-Y.; Kwag, D.; Lee, J.-H.; Lee, J.; Min, G.-J.; Park, S.-S.; Park, S.; Jeon, Y.-W.; Yoon, J.-H.; Shin, S.-H.; et al. Clinical Features, Gene Alterations, and Outcomes in Prefibrotic and Overt Primary and Secondary Myelofibrotic Patients. Cancers 2022, 14, 4485. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-Term Survival and Blast Transformation in Molecularly Annotated Essential Thrombocythemia, Polycythemia Vera, and Myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef] [PubMed]
- Yogarajah, M.; Tefferi, A. Leukemic Transformation in Myeloproliferative Neoplasms A Literature Review on Risk, Characteristics, and Outcome. Mayo Clin. Proc. 2017, 92, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Gangat, N.; Wolanskyj, A.P.; McClure, R.F.; Li, C.-Y.; Schwager, S.; Wu, W.; Tefferi, A. Risk Stratification for Survival and Leukemic Transformation in Essential Thrombocythemia: A Single Institutional Study of 605 Patients. Leukemia 2007, 21, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, G.; Caruso, V.; Marchioli, R.; Capnist, G.; Chisesi, T.; Finelli, C.; Gugliotta, L.; Landolfi, R.; Kutti, J.; Gisslinger, H.; et al. Acute Leukemia in Polycythemia Vera: An Analysis of 1638 Patients Enrolled in a Prospective Observational Study. Blood 2005, 105, 2664–2670. [Google Scholar] [CrossRef] [PubMed]
- Marcellino, B.K.; Verstovsek, S.; Mascarenhas, J. The Myelodepletive Phenotype in Myelofibrosis: Clinical Relevance and Therapeutic Implication. Clin. Lymphoma Myeloma Leuk. 2020, 20, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Grinfeld, J.; Nangalia, J.; Baxter, J.E.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Masarova, L.; Verstovsek, S. The Evolving Understanding of Prognosis in Post-Essential Thrombocythemia Myelofibrosis and Post-Polycythemia Vera Myelofibrosis vs Primary Myelofibrosis. Clin. Adv. Hematol. Oncol. 2019, 17, 299–307. [Google Scholar]
- López, J.E.H.; Carballo-Zarate, A.; Verstovsek, S.; Wang, S.A.; Hu, S.; Li, S.; Xu, J.; Zuo, W.; Tang, Z.; Yin, C.C.; et al. Bone Marrow Findings in Blast Phase of Polycythemia Vera. Ann. Hematol. 2018, 97, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A. Primary Myelofibrosis: 2023 Update on Diagnosis, Risk-stratification, and Management. Am. J. Hematol. 2023, 98, 801–821. [Google Scholar] [CrossRef] [PubMed]
- Gangat, N.; Tefferi, A.; Thanarajasingam, G.; Patnaik, M.; Schwager, S.; Ketterling, R.; Wolanskyj, A.P. Cytogenetic Abnormalities in Essential Thrombocythemia: Prevalence and Prognostic Significance. Eur. J. Haematol. 2009, 83, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wheeler, D.A.; Prchal, J.T. Acquired Uniparental Disomy of Chromosome 9p in Hematologic Malignancies. Exp. Hematol. 2016, 44, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Harutyunyan, A.; Elena, C.; Pietra, D.; Klampfl, T.; Bagienski, K.; Berg, T.; Casetti, I.; Pascutto, C.; Passamonti, F.; et al. Identification of Genomic Aberrations Associated with Disease Transformation by Means of High-resolution SNP Array Analysis in Patients with Myeloproliferative Neoplasm. Am. J. Hematol. 2011, 86, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Szuber, N.; Gangat, N.; Capecchi, G.; Maccari, C.; Harnois, M.; Karrar, O.; Abdelmagid, M.; Balliu, M.; Nacca, E.; et al. CALR Mutation Burden in Essential Thrombocythemia and Disease Outcome. Blood 2024, 143, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Ayres-Silva, J.P.; Bonamino, M.H.; Gouveia, M.E.; Monte-Mor, B.C.; Coutinho, D.F.; Daumas, A.H.; Solza, C.; Braggio, E.; Zalcberg, I. Genetic Alterations in Essential Thrombocythemia Progression to Acute Myeloid Leukemia: A Case Series and Review of the Literature. Front. Oncol. 2018, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Hahm, C.; Huh, H.; Mun, Y.; Seong, C.; Chung, W.; Huh, J. Genomic Aberrations of Myeloproliferative and Myelodysplastic/Myeloproliferative Neoplasms in Chronic Phase and during Disease Progression. Int. J. Lab. Hematol. 2015, 37, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Zimran, E.; Tripodi, J.; Rampal, R.; Rapaport, F.; Zirkiev, S.; Hoffman, R.; Najfeld, V. Genomic Characterization of Spleens in Patients with Myelofibrosis. Haematologica 2018, 103, e446–e449. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Variables | All Patients (n = 169) | No Progression (n = 119) | Progression (n = 50) |
|---|---|---|---|
| Age in years, median (range) | 62 (13–92) | 56 (13–92) | 67 (31–89) |
| Gender (female), n (%) | 96 (57) | 70 (59) | 26 (52) |
| Cytogenomic Results | |||
| Normal, n (%) | 108 (64) | 92 (77) | 16 (9) |
| Abnormal, n (%) | 61 (36) | 27 (23) | 34 (68) |
| Driver Mutations | |||
| N, evaluable | n = 166 | n = 116 | n = 50 |
| JAK2, n (%) | 99 (60) | 68 (59) | 31 (62) |
| MPL, n (%) | 13 (8) | 9 (8) | 4 (8) |
| CALR, n (%) | 35 (21) | 23 (20) | 12 (24) |
| JAK2/CALR, n (%) | 2 (1) | 2 (2) | 0 (0) |
| Triple Negative, n (%) | 17 (10) | 14 (12) | 3 (6) |
| Next Generation Sequencing | |||
| Adverse Molecular Risk (MIPSS-ET) | |||
| N, evaluable | n = 165 | n = 115 | n = 50 |
| 1 gene, n (%) | 13 (8) | 3 (3) | 10 (20) |
| ≥2 gene, n (%) | 1 (<1) | 0 (0) | 1 (2) |
| ID# | Age | Diagnosis | Gender | Tissue | Cytogenetics Karyotype | Array | Mutations | Progression to: |
|---|---|---|---|---|---|---|---|---|
| 1 | 75 | ET | M | BM | 45,XY,add(4)(p14),der(5;17)(p10;q10),der(8)add(8)(p11.2)add(8)(q13),add(13)(p11.2),der(15)t(15;16)(p11.2;q11.2),-16,-18,add(19)(q13.3),del(21)(q21q22),+2mar[cp19]/ 46,XY[1].ish der(4)del(4)(p14p16)ins(4;8)(p14;q24.3q23)(MYC+),der(5;17)(p10;q10)(D17Z1+),der(8)(?::8p11.2->8q12::8q24.1->8q22::8q23->8q22::8q23->8q24.1::?)(D8Z2+,MYC++),der(13)(8qter::8q22->8q24.3::13p11.2->13qter)(MYC+) | ND | JAK2 | AP/BP |
| 2 | 55 | ET | M | BM | 47,XY,+Y,t(11;19)(q23;p13.1)[16]/ 46,XY[4] | ND | Triple Negative | AP/BP |
| 3 | 74 | ET | M | BM | 42~43,X,-Y,add(5)(q11.2),add(7)(p13),?t(9;17)(q21;p13),-15,-16,der(19)t(15;19)(q11.2;p13.3),-21[cp3]/46,XY[17] | ND | JAK2 | AP/BP |
| 4 | 73 | ET | F | PB | 46,XX,del(12)(p12p13)[14]/47,idem,+8[2]/46,XX,t(1;9)(p13;q32),del(12)(p12p13)[cp2]/46,XX[3] ADDENDUM on 8/21/2019: 46,XX,del(12)(p12p13).ish t(7;12)(q36;p13)(5′ETV6+;3′ETV6+) | 4q24(106036993_106456054)x1, 9p24.3p13.1(0_38694064)x2 hmz | JAK2 | AP/BP |
| 5 | 65 | ET | F | BM | 46,XX,der(5;19)(p10;q10),+19[5]/45,XX,der(5)t(5;9)(q13;p22),der(7)add(7)(p12)add(7)(q22),add(9)(p22),t(12;19)(p13;q13.1),-13,-15,add(17)(q21.3),+mar[4]/44,sl,-9,der(14)t(9;14)(q22;p24),der(16)t(9;16)(p22;p13.1)[2]/46,XX[18]/47,XX,i(9)(p10),+mar[1] | ND | JAK2 | AP/BP |
| 6 | 67 | ET | F | BM | 48,XX,+8,+8[20] | 8p23.3q24.3(0_146293435)x4, 9p24.3p21.3(0_24234310)x2 hmz | JAK2, IDH1, SRSF2 | AP/BP |
| 7 | 51 | ET | M | BM | 46,XY,del(15)(q15q23),t(17;18)(p12;q11.2) [7]/46,XY[13] | ND | CALR, TET2 | MF |
| 8 | 45 | ET | F | PB | 46,XX,del(13)(q12q14)[4]/ 46,XX,del(13)(q12q32)[4]/ 46,XX,dup(1)(q21q42)[3]/ 48,XX,+der(1)t(1;19)(p12;p12),+8[2]/ 46,XX[7] | ND | Triple Negative, SH2B3 | MF |
| 9 | 39 | ET | F | PB | 46,XX,del(13)(q12q14)[8]/ 46,XX[7] | ND | Triple Negative | MF |
| 10 | 80 | ET | M | BM | 45,X,-Y[16]/46,idem,+8[3]/ 45,XY,-3[1] | ND | JAK2, SRSF2 | No Progression |
| 11 | 74 | ET | F | BM | 46,XX,del(20)(q11.2q13.3)[11]/ 46,XX[9] | ND | JAK2, TET2 | MF |
| 12 | 54 | ET | M | PB | 47,XY,+Y[20] | Yp11.32q12(0_59373566)x3 | JAK2 | No Progression |
| 13 | 81 | ET | M | #N/A | 45,X,-Y[18]/ 46,XY[2] | Yp11.32q12(0_59373566)x0 | JAK2 | No Progression |
| 14 | 73 | ET | M | BM | 45,X,-Y[7]/ 46,XY[22]/ 46,XY,?del(8)(q22)[1] | ND | Not Performed | No Progression |
| 15 | 38 | ET | M | PB | 46,XY,+9,-14[1]/ 46,XY[6] | 9p24.3q34.3(0_141213431)x3 | JAK2 | MF |
| 16 | 70 | ET | M | BM | 46,XY,der(6)t(3;6)(q25;p22)[20] | Xq26.2q26.3(133506194_134184567)x0, 1p36.13p22.1(1089699_93720070)x2 hmz, 3q25.32q29(157780129_197861598)x3, 6p25.3p22.1(204009_27534514)x1 | MPL, TET2 | MF |
| 17 | 83 | ET | F | BM | 46,XX,del(13)(q12q14)[3]/ 46,XX[17] | ND | JAK2 | No Progression |
| 18 | 72 | ET | F | BM | 46,XX,del(20)(q11.2q13.3)[1]/ 46,XX[19] | 9p24.3p13.1(0_38694064)x2 hmz | JAK2, DNMT3A | MF |
| 19 | 89 | ET | M | BM | 46,XY,del(13)(q12q14)[5]/ 46,XY,del(13)(q12q22)[3]/ 46,XY[12] | ND | MPL | MF |
| 20 | 67 | ET | F | PB | 46,XX,der(6)t(1;6)(q21;p25),t(9;11)(p24;q21),del(13)(q12q22)[1]/ 46,idem,del(11)(q14q23)[8]/ 46,idem,del(12)(p13p11.2)[7]/ 46,XX,t(11;17)(p15;q11.2)[4] | ND | JAK2, DNMT3A, EZH2 | AP/BP |
| 21 | 81 | ET | M | BM | 46,XY,t(9;22)(q34;q11.2)[20] | ND | JAK2 | CML |
| 22 | 52 | ET | M | BM | 46,XY,del(13)(q12q22)[9]/ 46,XY[11] | ND | JAK2, GATA1 | MF |
| 90 | 65 | prePMF | F | BM | 45,XX,del(12)(p11.2p13),-18[1]/ 46,XX[17] | ND | JAK2, RUNX1, SF3B1*, STAG2 | MDS |
| 156 | 64 | ET | M | BM | 47,XY,+9[4]/ 46,XY[15]/ 47,XY,+18[1] | ND | JAK2 | PV |
| 163 | 77 | ET | F | BM | 46,XX,dup(1)(q21q32)[8]/ 46,XX[12] | 1q21.2q32.1(150189472_204513328)x3, 5q21.3q23.1(107501113_115990907)x1 | CALR | No Progression |
| 167 | 80 | ET | M | BM | 46,XX,del(20)(q11.2q13.1)[4]/ 46,XX[16] | 9p24.3p22.1(0_19585456)x2 hmz, 17p11.2(17803134_19293234)x1~2, 20q11.23q13.2(34975018_50468054)x1 | JAK2 | No Progression |
| 169 | 77 | ET | M | BM | 46,XY,del(20)(q11.2q13.3)[14]/ 46,XY[6] | 9p24.3p24.1(0_6651634)x2 hmz, 20q11.21q13.32(32060886_58171738)x1, Yp11.32q12(0_59373566)x0 | JAK2 | MF |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tripodi, J.; Hoffman, R.; Tremblay, D.; Ahire, D.; Mascarenhas, J.; Kremyanskaya, M.; Najfeld, V. Conventional Cytogenetic Analysis and Array CGH + SNP Identify Essential Thrombocythemia and Prefibrotic Primary Myelofibrosis Patients Who Are at Risk for Disease Progression. Int. J. Mol. Sci. 2024, 25, 4061. https://doi.org/10.3390/ijms25074061
Tripodi J, Hoffman R, Tremblay D, Ahire D, Mascarenhas J, Kremyanskaya M, Najfeld V. Conventional Cytogenetic Analysis and Array CGH + SNP Identify Essential Thrombocythemia and Prefibrotic Primary Myelofibrosis Patients Who Are at Risk for Disease Progression. International Journal of Molecular Sciences. 2024; 25(7):4061. https://doi.org/10.3390/ijms25074061
Chicago/Turabian StyleTripodi, Joseph, Ronald Hoffman, Douglas Tremblay, Daiva Ahire, John Mascarenhas, Marina Kremyanskaya, and Vesna Najfeld. 2024. "Conventional Cytogenetic Analysis and Array CGH + SNP Identify Essential Thrombocythemia and Prefibrotic Primary Myelofibrosis Patients Who Are at Risk for Disease Progression" International Journal of Molecular Sciences 25, no. 7: 4061. https://doi.org/10.3390/ijms25074061