
The Lectin Pathway of the Complement System—Activation, Regulation, Disease Connections and Interplay with Other (Proteolytic) Systems
 , ,
, ,
Abstract
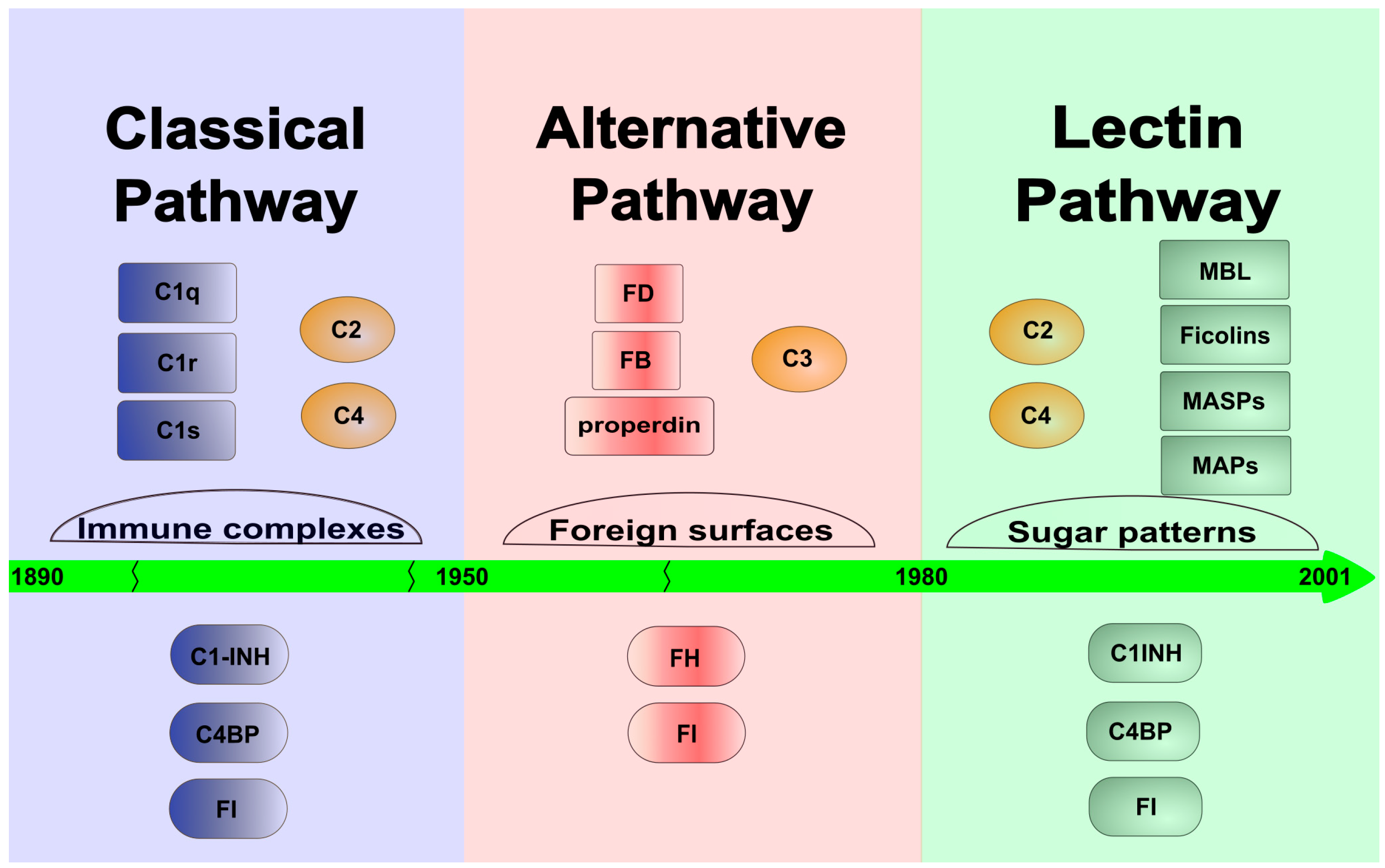
:1. Introduction and Brief History of the Lectin Pathway
2. Genes and Components
2.1. Pattern Recognition Molecules (PRMs)
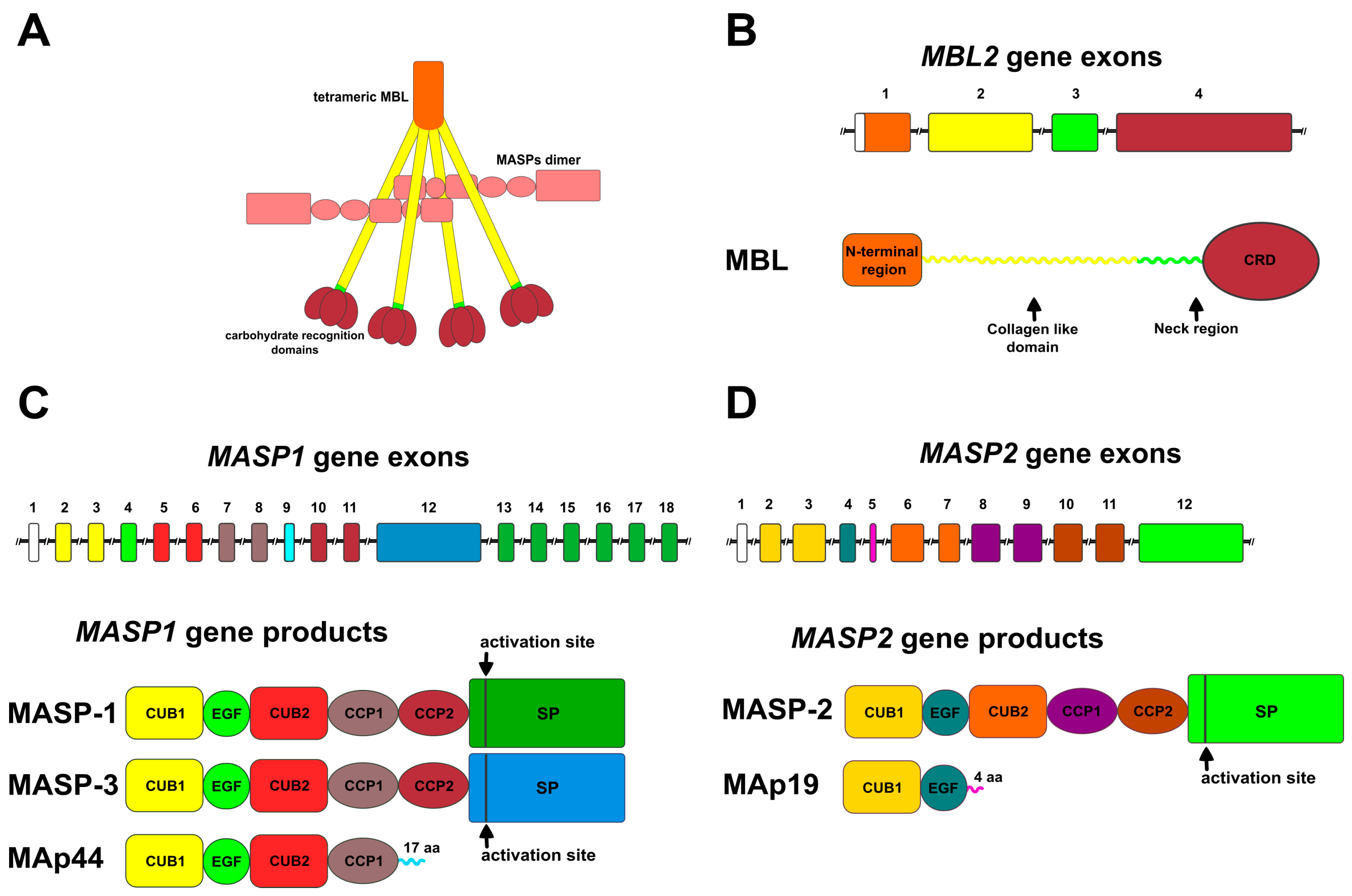
2.1.1. Mannose-Binding Lectin (MBL)
2.1.2. Other Collectins
2.1.3. Ficolins
2.2. MBL-Associated Serine Proteases (MASPs) and Proteins (MAps)
2.2.1. Products of the MASP1 Gene
2.2.2. Products of the MASP2 Gene
2.3. The Sites of Synthesis
3. Activation Mechanism of the Lectin Pathway
4. Connection to the Other Pathways of Complement
4.1. Connection to the Classical Pathway (CP)
4.2. Connection to the Alternative Pathway (AP)
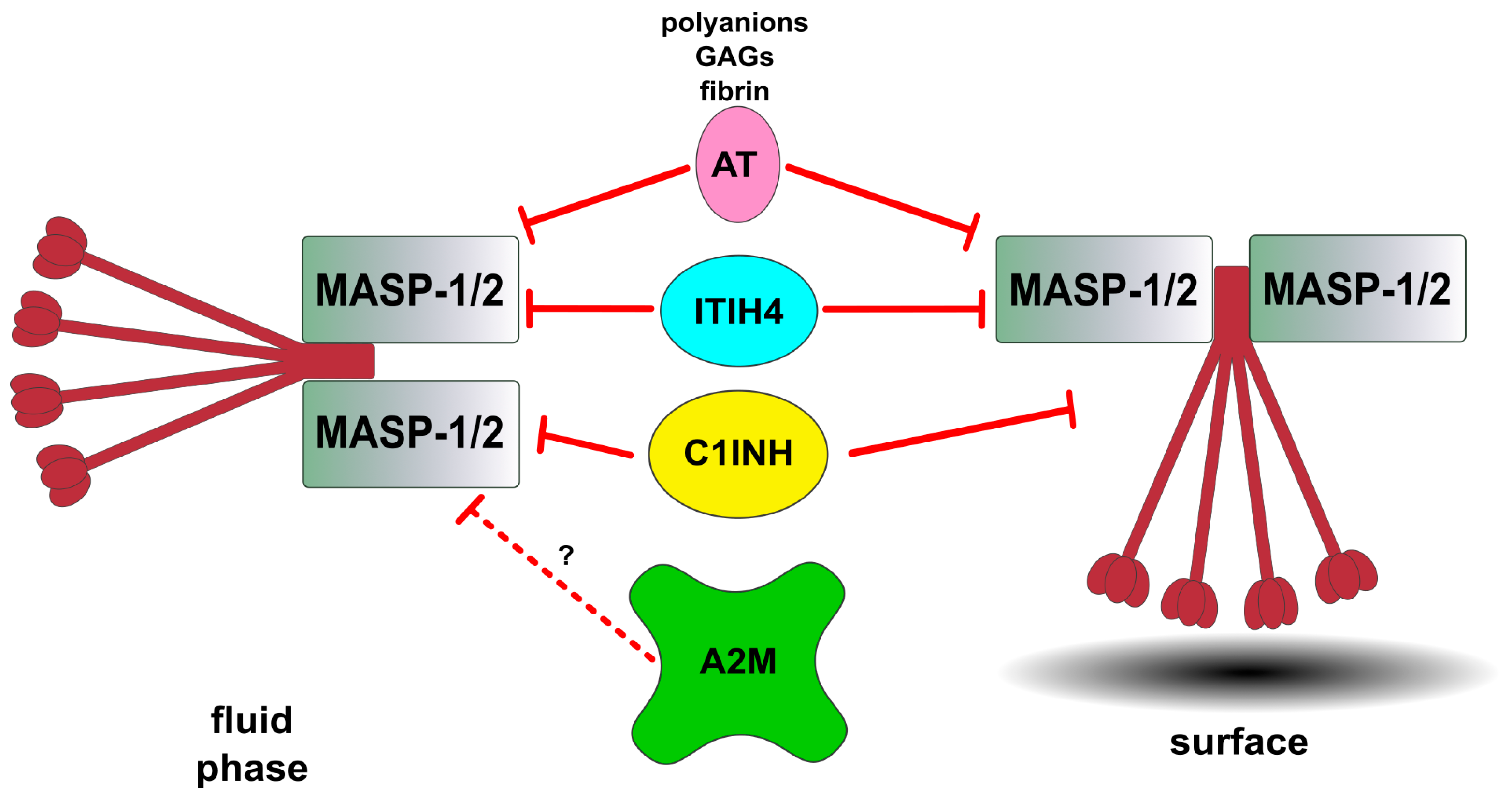
5. Regulation of the Lectin Pathway by Natural Inhibitors
5.1. Inhibitors of Lectin Pathway Proteases
5.2. Factor I, the Common Regulator of the Three Pathways
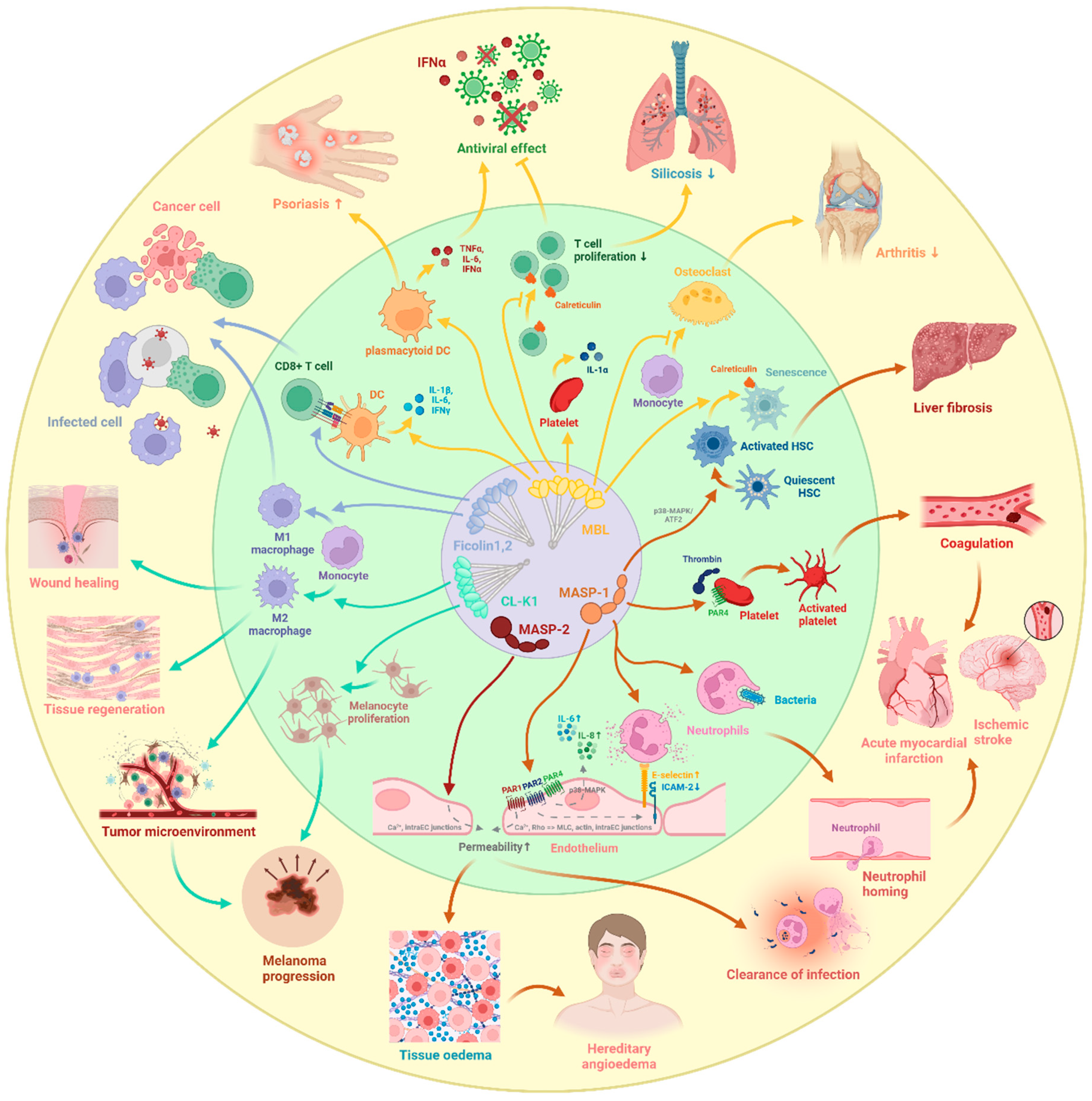
6. Direct Cellular Effects of Lectin Pathway Components
6.1. Cellular Effects of PRMs
6.2. Cellular Effects of MASPs
7. Influence of Lectin Pathway Components on Coagulation
8. Diseases with Potential Lectin Pathway Involvement
8.1. Renal Diseases
8.1.1. IgA Nephropathy
8.1.2. Membranous Nephropathy
8.1.3. Diabetic Kidney Disease
8.2. Ischemia/Reperfusion Injury
8.2.1. Renal Ischemia/Reperfusion Injury
8.2.2. Myocardial Ischemia/Reperfusion Injury
8.2.3. Ischemic Stroke
8.3. Atherosclerosis
8.4. COVID-19
8.5. Hereditary Angioedema (HAE)
8.6. Autoimmune Diseases: Systemic Lupus Erythematosus and Rheumatoid Arthritis
8.7. Schizophrenia
8.8. Influence of Aging
9. Therapeutic Inhibition and Testing of the Lectin Pathway
9.1. Therapeutic Inhibition of the Lectin Pathway
9.2. Measurement of the Activity of Lectin Pathway
10. Non-Canonical Functions of the Lectin Pathway
10.1. Role in Embryonal and Brain Development
10.2. Role in Cancer
11. Summary/Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Pryzdial, E.L.G.; Leatherdale, A.; Conway, E.M. Coagulation and Complement: Key Innate Defense Participants in a Seamless Web. Front. Immunol. 2022, 13, 918775. [Google Scholar] [CrossRef]
- Gavriilaki, E.; Brodsky, R.A. Complementopathies and Precision Medicine. J. Clin. Investig. 2020, 130, 2152–2163. [Google Scholar] [CrossRef]
- Kawakami, M.; Ihara, I.; Suzuki, A.; Harada, Y. Properties of a New Complement-Dependent Bactericidal Factor Specific for Ra Chemotype Salmonella in Sera of Conventional and Germ-Free Mice. J. Immunol. 1982, 129, 2198–2201. [Google Scholar] [CrossRef]
- Ikeda, K.; Sannoh, T.; Kawasaki, N.; Kawasaki, T.; Yamashina, I. Serum Lectin with Known Structure Activates Complement through the Classical Pathway. J. Biol. Chem. 1987, 262, 7451–7454. [Google Scholar] [CrossRef]
- Matsushita, M.; Fujita, T. Activation of the Classical Complement Pathway by Mannose-Binding Protein in Association with a Novel C1s-like Serine Protease. J. Exp. Med. 1992, 176, 1497–1502. [Google Scholar] [CrossRef]
- Thiel, S.; Vorup-Jensen, T.; Stover, C.M.; Schwaeble, W.; Laursen, S.B.; Poulsen, K.; Willis, A.C.; Eggleton, P.; Hansen, S.; Holmskov, U.; et al. A Second Serine Protease Associated with Mannan-Binding Lectin That Activates Complement. Nature 1997, 386, 506–510. [Google Scholar] [CrossRef]
- Vorup-Jensen, T.; Petersen, S.V.; Hansen, A.G.; Poulsen, K.; Schwaeble, W.; Sim, R.B.; Reid, K.B.; Davis, S.J.; Thiel, S.; Jensenius, J.C. Distinct Pathways of Mannan-Binding Lectin (MBL)- and C1-Complex Autoactivation Revealed by Reconstitution of MBL with Recombinant MBL-Associated Serine Protease-2. J. Immunol. 2000, 165, 2093–2100. [Google Scholar] [CrossRef]
- Dahl, M.R.; Thiel, S.; Matsushita, M.; Fujita, T.; Willis, A.C.; Christensen, T.; Vorup-Jensen, T.; Jensenius, J.C. MASP-3 and Its Association with Distinct Complexes of the Mannan-Binding Lectin Complement Activation Pathway. Immunity 2001, 15, 127–135. [Google Scholar] [CrossRef]
- Stover, C.M.; Thiel, S.; Thelen, M.; Lynch, N.J.; Vorup-Jensen, T.; Jensenius, J.C.; Schwaeble, W.J. Two Constituents of the Initiation Complex of the Mannan-Binding Lectin Activation Pathway of Complement Are Encoded by a Single Structural Gene. J. Immunol. 1999, 162, 3481–3490. [Google Scholar] [CrossRef]
- Degn, S.E.; Hansen, A.G.; Steffensen, R.; Jacobsen, C.; Jensenius, J.C.; Thiel, S. MAp44, a Human Protein Associated with Pattern Recognition Molecules of the Complement System and Regulating the Lectin Pathway of Complement Activation. J. Immunol. 2009, 183, 7371–7378. [Google Scholar] [CrossRef]
- Endo, Y.; Matsushita, M.; Fujita, T. New Insights into the Role of Ficolins in the Lectin Pathway of Innate Immunity. Int. Rev. Cell Mol. Biol. 2015, 316, 49–110. [Google Scholar] [CrossRef]
- Henriksen, M.L.; Brandt, J.; Andrieu, J.-P.; Nielsen, C.; Jensen, P.H.; Holmskov, U.; Jorgensen, T.J.D.; Palarasah, Y.; Thielens, N.M.; Hansen, S. Heteromeric Complexes of Native Collectin Kidney 1 and Collectin Liver 1 Are Found in the Circulation with MASPs and Activate the Complement System. J. Immunol. 2013, 191, 6117–6127. [Google Scholar] [CrossRef]
- Kjaer, T.R.; Jensen, L.; Hansen, A.; Dani, R.; Jensenius, J.C.; Dobó, J.; Gál, P.; Thiel, S. Oligomerization of Mannan-Binding Lectin Dictates Binding Properties and Complement Activation. Scand. J. Immunol. 2016, 84, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Paréj, K.; Hermann, A.; Donáth, N.; Závodszky, P.; Gál, P.; Dobó, J. Dissociation and Re-Association Studies on the Interaction Domains of Mannan-Binding Lectin (MBL)-Associated Serine Proteases, MASP-1 and MASP-2, Provide Evidence for Heterodimer Formation. Mol. Immunol. 2014, 59, 1–9. [Google Scholar] [CrossRef]
- Rosbjerg, A.; Munthe-Fog, L.; Garred, P.; Skjoedt, M.-O. Heterocomplex Formation between MBL/Ficolin/CL-11-Associated Serine Protease-1 and -3 and MBL/Ficolin/CL-11-Associated Protein-1. J. Immunol. 2014, 192, 4352–4360. [Google Scholar] [CrossRef]
- Super, M.; Thiel, S.; Lu, J.; Levinsky, R.J.; Turner, M.W. Association of Low Levels of Mannan-Binding Protein with a Common Defect of Opsonisation. Lancet 1989, 2, 1236–1239. [Google Scholar] [CrossRef]
- Turner, M.W. Mannose-Binding Lectin: The Pluripotent Molecule of the Innate Immune System. Immunol. Today 1996, 17, 532–540. [Google Scholar] [CrossRef]
- Garred, P.; Larsen, F.; Seyfarth, J.; Fujita, R.; Madsen, H.O. Mannose-Binding Lectin and Its Genetic Variants. Genes Immun. 2006, 7, 85–94. [Google Scholar] [CrossRef]
- Murugaiah, V.; Tsolaki, A.G.; Kishore, U. Collectins: Innate Immune Pattern Recognition Molecules. Adv. Exp. Med. Biol. 2020, 1204, 75–127. [Google Scholar] [CrossRef]
- Teillet, F.; Dublet, B.; Andrieu, J.-P.; Gaboriaud, C.; Arlaud, G.J.; Thielens, N.M. The Two Major Oligomeric Forms of Human Mannan-Binding Lectin: Chemical Characterization, Carbohydrate-Binding Properties, and Interaction with MBL-Associated Serine Proteases. J. Immunol. 2005, 174, 2870–2877. [Google Scholar] [CrossRef]
- Sheriff, S.; Chang, C.Y.; Ezekowitz, R.A. Human Mannose-Binding Protein Carbohydrate Recognition Domain Trimerizes through a Triple Alpha-Helical Coiled-Coil. Nat. Struct. Biol. 1994, 1, 789–794. [Google Scholar] [CrossRef]
- Ohtani, K.; Suzuki, Y.; Eda, S.; Kawai, T.; Kase, T.; Yamazaki, H.; Shimada, T.; Keshi, H.; Sakai, Y.; Fukuoh, A.; et al. Molecular Cloning of a Novel Human Collectin from Liver (CL-L1). J. Biol. Chem. 1999, 274, 13681–13689. [Google Scholar] [CrossRef]
- Hansen, S.; Selman, L.; Palaniyar, N.; Ziegler, K.; Brandt, J.; Kliem, A.; Jonasson, M.; Skjoedt, M.-O.; Nielsen, O.; Hartshorn, K.; et al. Collectin 11 (CL-11, CL-K1) Is a MASP-1/3-Associated Plasma Collectin with Microbial-Binding Activity. J. Immunol. 2010, 185, 6096–6104. [Google Scholar] [CrossRef]
- Hansen, S.W.K.; Ohtani, K.; Roy, N.; Wakamiya, N. The Collectins CL-L1, CL-K1 and CL-P1, and Their Roles in Complement and Innate Immunity. Immunobiology 2016, 221, 1058–1067. [Google Scholar] [CrossRef]
- Henriksen, M.L.; Madsen, K.L.; Skjoedt, K.; Hansen, S. Calcium-Sensitive Immunoaffinity Chromatography: Gentle and Highly Specific Retrieval of a Scarce Plasma Antigen, Collectin-LK (CL-LK). J. Immunol. Methods 2014, 413, 25–31. [Google Scholar] [CrossRef]
- Ma, Y.J.; Hein, E.; Munthe-Fog, L.; Skjoedt, M.-O.; Bayarri-Olmos, R.; Romani, L.; Garred, P. Soluble Collectin-12 (CL-12) Is a Pattern Recognition Molecule Initiating Complement Activation via the Alternative Pathway. J. Immunol. 2015, 195, 3365–3373. [Google Scholar] [CrossRef]
- Matsushita, M.; Endo, Y.; Fujita, T. Cutting Edge: Complement-Activating Complex of Ficolin and Mannose-Binding Lectin-Associated Serine Protease. J. Immunol. 2000, 164, 2281–2284. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Kuraya, M.; Hamasaki, N.; Tsujimura, M.; Shiraki, H.; Fujita, T. Activation of the Lectin Complement Pathway by H-Ficolin (Hakata Antigen). J. Immunol. 2002, 168, 3502–3506. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Endo, Y.; Iwaki, D.; Nakata, M.; Matsushita, M.; Wada, I.; Inoue, K.; Munakata, M.; Fujita, T. Human M-Ficolin Is a Secretory Protein That Activates the Lectin Complement Pathway. J. Immunol. 2005, 175, 3150–3156. [Google Scholar] [CrossRef]
- Krarup, A.; Thiel, S.; Hansen, A.; Fujita, T.; Jensenius, J.C. L-Ficolin Is a Pattern Recognition Molecule Specific for Acetyl Groups. J. Biol. Chem. 2004, 279, 47513–47519. [Google Scholar] [CrossRef]
- Frederiksen, P.D.; Thiel, S.; Larsen, C.B.; Jensenius, J.C. M-Ficolin, an Innate Immune Defence Molecule, Binds Patterns of Acetyl Groups and Activates Complement. Scand. J. Immunol. 2005, 62, 462–473. [Google Scholar] [CrossRef]
- Garlatti, V.; Belloy, N.; Martin, L.; Lacroix, M.; Matsushita, M.; Endo, Y.; Fujita, T.; Fontecilla-Camps, J.C.; Arlaud, G.J.; Thielens, N.M.; et al. Structural Insights into the Innate Immune Recognition Specificities of L- and H-Ficolins. EMBO J. 2007, 26, 623–633. [Google Scholar] [CrossRef]
- Le, Y.; Lee, S.H.; Kon, O.L.; Lu, J. Human L-Ficolin: Plasma Levels, Sugar Specificity, and Assignment of Its Lectin Activity to the Fibrinogen-like (FBG) Domain. FEBS Lett. 1998, 425, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Krarup, A.; Sørensen, U.B.S.; Matsushita, M.; Jensenius, J.C.; Thiel, S. Effect of Capsulation of Opportunistic Pathogenic Bacteria on Binding of the Pattern Recognition Molecules Mannan-Binding Lectin, L-Ficolin, and H-Ficolin. Infect. Immun. 2005, 73, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Yae, Y.; Inaba, S.; Sato, H.; Okochi, K.; Tokunaga, F.; Iwanaga, S. Isolation and Characterization of a Thermolabile Beta-2 Macroglycoprotein (“thermolabile Substance” or ‘Hakata Antigen’) Detected by Precipitating (Auto) Antibody in Sera of Patients with Systemic Lupus Erythematosus. Biochim. Biophys. Acta 1991, 1078, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Erickson, H.P. Two Oligomeric Forms of Plasma Ficolin Have Differential Lectin Activity. J. Biol. Chem. 1997, 272, 14220–14226. [Google Scholar] [CrossRef]
- Ohashi, T.; Erickson, H.P. Oligomeric Structure and Tissue Distribution of Ficolins from Mouse, Pig and Human. Arch. Biochem. Biophys. 1998, 360, 223–232. [Google Scholar] [CrossRef]
- Zacho, R.M.; Jensen, L.; Terp, R.; Jensenius, J.C.; Thiel, S. Studies of the Pattern Recognition Molecule H-Ficolin: Specificity and Purification. J. Biol. Chem. 2012, 287, 8071–8081. [Google Scholar] [CrossRef]
- Troldborg, A.; Hansen, A.; Hansen, S.W.K.; Jensenius, J.C.; Stengaard-Pedersen, K.; Thiel, S. Lectin Complement Pathway Proteins in Healthy Individuals. Clin. Exp. Immunol. 2017, 188, 138–147. [Google Scholar] [CrossRef]
- Takahashi, A.; Takayama, Y.; Hatsuse, H.; Kawakami, M. Presence of a Serine Protease in the Complement-Activating Component of the Complement-Dependent Bactericidal Factor, RaRF, in Mouse Serum. Biochem. Biophys. Res. Commun. 1993, 190, 681–687. [Google Scholar] [CrossRef]
- Skjoedt, M.-O.; Palarasah, Y.; Munthe-Fog, L.; Jie Ma, Y.; Weiss, G.; Skjodt, K.; Koch, C.; Garred, P. MBL-Associated Serine Protease-3 Circulates in High Serum Concentrations Predominantly in Complex with Ficolin-3 and Regulates Ficolin-3 Mediated Complement Activation. Immunobiology 2010, 215, 921–931. [Google Scholar] [CrossRef]
- Degn, S.E.; Jensenius, J.C.; Thiel, S. Disease-Causing Mutations in Genes of the Complement System. Am. J. Hum. Genet. 2011, 88, 689–705. [Google Scholar] [CrossRef]
- Takahashi, M.; Endo, Y.; Fujita, T.; Matsushita, M. A Truncated Form of Mannose-Binding Lectin-Associated Serine Protease (MASP)-2 Expressed by Alternative Polyadenylation Is a Component of the Lectin Complement Pathway. Int. Immunol. 1999, 11, 859–863. [Google Scholar] [CrossRef]
- Ambrus, G.; Gál, P.; Kojima, M.; Szilágyi, K.; Balczer, J.; Antal, J.; Gráf, L.; Laich, A.; Moffatt, B.E.; Schwaeble, W.; et al. Natural Substrates and Inhibitors of Mannan-Binding Lectin-Associated Serine Protease-1 and -2: A Study on Recombinant Catalytic Fragments. J. Immunol. 2003, 170, 1374–1382. [Google Scholar] [CrossRef]
- Howard, M.; Farrar, C.A.; Sacks, S.H. Structural and Functional Diversity of Collectins and Ficolins and Their Relationship to Disease. Semin. Immunopathol. 2018, 40, 75–85. [Google Scholar] [CrossRef]
- Uemura, K.; Saka, M.; Nakagawa, T.; Kawasaki, N.; Thiel, S.; Jensenius, J.C.; Kawasaki, T. L-MBP Is Expressed in Epithelial Cells of Mouse Small Intestine. J. Immunol. 2002, 169, 6945–6950. [Google Scholar] [CrossRef]
- Gajek, G.; Świerzko, A.S.; Cedzyński, M. Association of Polymorphisms of MASP1/3, COLEC10, and COLEC11 Genes with 3MC Syndrome. Int. J. Mol. Sci. 2020, 21, 5483. [Google Scholar] [CrossRef]
- Lynch, N.J.; Khan, S.-H.; Stover, C.M.; Sandrini, S.M.; Marston, D.; Presanis, J.S.; Schwaeble, W.J. Composition of the Lectin Pathway of Complement in Gallus Gallus: Absence of Mannan-Binding Lectin-Associated Serine Protease-1 in Birds. J. Immunol. 2005, 174, 4998–5006. [Google Scholar] [CrossRef]
- Rossi, V.; Cseh, S.; Bally, I.; Thielens, N.M.; Jensenius, J.C.; Arlaud, G.J. Substrate Specificities of Recombinant Mannan-Binding Lectin-Associated Serine Proteases-1 and -2. J. Biol. Chem. 2001, 276, 40880–40887. [Google Scholar] [CrossRef]
- Kidmose, R.T.; Laursen, N.S.; Dobó, J.; Kjaer, T.R.; Sirotkina, S.; Yatime, L.; Sottrup-Jensen, L.; Thiel, S.; Gál, P.; Andersen, G.R. Structural Basis for Activation of the Complement System by Component C4 Cleavage. Proc. Natl. Acad. Sci. USA 2012, 109, 15425–15430. [Google Scholar] [CrossRef]
- Megyeri, M.; Harmat, V.; Major, B.; Végh, Á.; Balczer, J.; Héja, D.; Szilágyi, K.; Datz, D.; Pál, G.; Závodszky, P.; et al. Quantitative Characterization of the Activation Steps of Mannan-Binding Lectin (MBL)-Associated Serine Proteases (MASPs) Points to the Central Role of MASP-1 in the Initiation of the Complement Lectin Pathway. J. Biol. Chem. 2013, 288, 8922–8934. [Google Scholar] [CrossRef]
- Møller-Kristensen, M.; Thiel, S.; Sjöholm, A.; Matsushita, M.; Jensenius, J.C. Cooperation between MASP-1 and MASP-2 in the Generation of C3 Convertase through the MBL Pathway. Int. Immunol. 2007, 19, 141–149. [Google Scholar] [CrossRef]
- Héja, D.; Kocsis, A.; Dobó, J.; Szilágyi, K.; Szász, R.; Závodszky, P.; Pál, G.; Gál, P. Revised Mechanism of Complement Lectin-Pathway Activation Revealing the Role of Serine Protease MASP-1 as the Exclusive Activator of MASP-2. Proc. Natl. Acad. Sci. USA 2012, 109, 10498–10503. [Google Scholar] [CrossRef] [PubMed]
- Degn, S.E.; Jensen, L.; Hansen, A.G.; Duman, D.; Tekin, M.; Jensenius, J.C.; Thiel, S. Mannan-Binding Lectin-Associated Serine Protease (MASP)-1 Is Crucial for Lectin Pathway Activation in Human Serum, Whereas Neither MASP-1 nor MASP-3 Is Required for Alternative Pathway Function. J. Immunol. 2012, 189, 3957–3969. [Google Scholar] [CrossRef] [PubMed]
- Gál, P.; Harmat, V.; Kocsis, A.; Bián, T.; Barna, L.; Ambrus, G.; Végh, B.; Balczer, J.; Sim, R.B.; Náray-Szabó, G.; et al. A True Autoactivating Enzyme. Structural Insight into Mannose-Binding Lectin-Associated Serine Protease-2 Activations. J. Biol. Chem. 2005, 280, 33435–33444. [Google Scholar] [CrossRef]
- Takahashi, M.; Iwaki, D.; Kanno, K.; Ishida, Y.; Xiong, J.; Matsushita, M.; Endo, Y.; Miura, S.; Ishii, N.; Sugamura, K.; et al. Mannose-Binding Lectin (MBL)-Associated Serine Protease (MASP)-1 Contributes to Activation of the Lectin Complement Pathway. J. Immunol. 2008, 180, 6132–6138. [Google Scholar] [CrossRef]
- Hayashi, M.; Machida, T.; Ishida, Y.; Ogata, Y.; Omori, T.; Takasumi, M.; Endo, Y.; Suzuki, T.; Sekimata, M.; Homma, Y.; et al. Cutting Edge: Role of MASP-3 in the Physiological Activation of Factor D of the Alternative Complement Pathway. J. Immunol. 2019, 203, 1411–1416. [Google Scholar] [CrossRef]
- Degn, S.E.; Jensen, L.; Olszowski, T.; Jensenius, J.C.; Thiel, S. Co-Complexes of MASP-1 and MASP-2 Associated with the Soluble Pattern-Recognition Molecules Drive Lectin Pathway Activation in a Manner Inhibitable by MAp44. J. Immunol. 2013, 191, 1334–1345. [Google Scholar] [CrossRef] [PubMed]
- Mayilyan, K.R.; Presanis, J.S.; Arnold, J.N.; Hajela, K.; Sim, R.B. Heterogeneity of MBL-MASP Complexes. Mol. Immunol. 2006, 43, 1286–1292. [Google Scholar] [CrossRef]
- Degn, S.E.; Kjaer, T.R.; Kidmose, R.T.; Jensen, L.; Hansen, A.G.; Tekin, M.; Jensenius, J.C.; Andersen, G.R.; Thiel, S. Complement Activation by Ligand-Driven Juxtaposition of Discrete Pattern Recognition Complexes. Proc. Natl. Acad. Sci. USA 2014, 111, 13445–13450. [Google Scholar] [CrossRef]
- Ohta, M.; Okada, M.; Yamashina, I.; Kawasaki, T. The Mechanism of Carbohydrate-Mediated Complement Activation by the Serum Mannan-Binding Protein. J. Biol. Chem. 1990, 265, 1980–1984. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.H.; Thiel, S.; Wiedemann, H.; Timpl, R.; Reid, K.B. Binding of the Pentamer/Hexamer Forms of Mannan-Binding Protein to Zymosan Activates the Proenzyme C1r2C1s2 Complex, of the Classical Pathway of Complement, without Involvement of C1q. J. Immunol. 1990, 144, 2287–2294. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.E.; Toth, J.; Dodds, A.W.; Girija, U.V.; Furze, C.M.; Pala, E.; Sim, R.B.; Reid, K.B.M.; Schwaeble, W.J.; Schmid, R.; et al. Analogous Interactions in Initiating Complexes of the Classical and Lectin Pathways of Complement. J. Immunol. 2009, 182, 7708–7717. [Google Scholar] [CrossRef] [PubMed]
- Garred, P.; Genster, N.; Pilely, K.; Bayarri-Olmos, R.; Rosbjerg, A.; Ma, Y.J.; Skjoedt, M.-O. A Journey through the Lectin Pathway of Complement-MBL and Beyond. Immunol. Rev. 2016, 274, 74–97. [Google Scholar] [CrossRef] [PubMed]
- Oroszlán, G.; Kortvely, E.; Szakács, D.; Kocsis, A.; Dammeier, S.; Zeck, A.; Ueffing, M.; Závodszky, P.; Pál, G.; Gál, P.; et al. MASP-1 and MASP-2 Do Not Activate Pro-Factor D in Resting Human Blood, Whereas MASP-3 Is a Potential Activator: Kinetic Analysis Involving Specific MASP-1 and MASP-2 Inhibitors. J. Immunol. 2016, 196, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Dobó, J.; Szakács, D.; Oroszlán, G.; Kortvely, E.; Kiss, B.; Boros, E.; Szász, R.; Závodszky, P.; Gál, P.; Pál, G. MASP-3 Is the Exclusive pro-Factor D Activator in Resting Blood: The Lectin and the Alternative Complement Pathways Are Fundamentally Linked. Sci. Rep. 2016, 6, 31877. [Google Scholar] [CrossRef]
- Pihl, R.; Jensen, L.; Hansen, A.G.; Thøgersen, I.B.; Andres, S.; Dagnæs-Hansen, F.; Oexle, K.; Enghild, J.J.; Thiel, S. Analysis of Factor D Isoforms in Malpuech-Michels-Mingarelli-Carnevale Patients Highlights the Role of MASP-3 as a Maturase in the Alternative Pathway of Complement. J. Immunol. 2017, 199, 2158–2170. [Google Scholar] [CrossRef]
- Oroszlán, G.; Dani, R.; Szilágyi, A.; Závodszky, P.; Thiel, S.; Gál, P.; Dobó, J. Extensive Basal Level Activation of Complement Mannose-Binding Lectin-Associated Serine Protease-3: Kinetic Modeling of Lectin Pathway Activation Provides Possible Mechanism. Front. Immunol. 2017, 8, 1821. [Google Scholar] [CrossRef]
- Gullipalli, D.; Miwa, T.; Golla, M.; Sato, S.; Angampalli, S.; Song, W.-C. MASP3 Deficiency in Mice Reduces but Does Not Abrogate Alternative Pathway Complement Activity Due to Intrinsic Profactor D Activity. J. Immunol. 2023, 210, 1543–1551. [Google Scholar] [CrossRef]
- Dani, R.; Oroszlán, G.; Martinusz, R.; Farkas, B.; Dobos, B.; Vadas, E.; Závodszky, P.; Gál, P.; Dobó, J. Quantification of the Zymogenicity and the Substrate-Induced Activity Enhancement of Complement Factor D. Front. Immunol. 2023, 14, 1197023. [Google Scholar] [CrossRef]
- Oroszlán, G.; Dani, R.; Végh, B.M.; Varga, D.; Ács, A.V.; Pál, G.; Závodszky, P.; Farkas, H.; Gál, P.; Dobó, J. Proprotein Convertase Is the Highest-Level Activator of the Alternative Complement Pathway in the Blood. J. Immunol. 2021, 206, 2198–2205. [Google Scholar] [CrossRef] [PubMed]
- Paréj, K.; Kocsis, A.; Enyingi, C.; Dani, R.; Oroszlán, G.; Beinrohr, L.; Dobó, J.; Závodszky, P.; Pál, G.; Gál, P. Cutting Edge: A New Player in the Alternative Complement Pathway, MASP-1 Is Essential for LPS-Induced, but Not for Zymosan-Induced, Alternative Pathway Activation. J. Immunol. 2018, 200, 2247–2252. [Google Scholar] [CrossRef]
- Matsushita, M.; Fujita, T. Cleavage of the Third Component of Complement (C3) by Mannose-Binding Protein-Associated Serine Protease (MASP) with Subsequent Complement Activation. Immunobiology 1995, 194, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Schweinle, J.E.; Ezekowitz, R.A.; Tenner, A.J.; Kuhlman, M.; Joiner, K.A. Human Mannose-Binding Protein Activates the Alternative Complement Pathway and Enhances Serum Bactericidal Activity on a Mannose-Rich Isolate of Salmonella. J. Clin. Investig. 1989, 84, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Selander, B.; Mårtensson, U.; Weintraub, A.; Holmström, E.; Matsushita, M.; Thiel, S.; Jensenius, J.C.; Truedsson, L.; Sjöholm, A.G. Mannan-Binding Lectin Activates C3 and the Alternative Complement Pathway without Involvement of C2. J. Clin. Investig. 2006, 116, 1425–1434. [Google Scholar] [CrossRef]
- Schwaeble, W.J.; Lynch, N.J.; Clark, J.E.; Marber, M.; Samani, N.J.; Ali, Y.M.; Dudler, T.; Parent, B.; Lhotta, K.; Wallis, R.; et al. Targeting of Mannan-Binding Lectin-Associated Serine Protease-2 Confers Protection from Myocardial and Gastrointestinal Ischemia/Reperfusion Injury. Proc. Natl. Acad. Sci. USA 2011, 108, 7523–7528. [Google Scholar] [CrossRef]
- Asgari, E.; Farrar, C.A.; Lynch, N.; Ali, Y.M.; Roscher, S.; Stover, C.; Zhou, W.; Schwaeble, W.J.; Sacks, S.H. Mannan-Binding Lectin-Associated Serine Protease 2 Is Critical for the Development of Renal Ischemia Reperfusion Injury and Mediates Tissue Injury in the Absence of Complement C4. FASEB J. 2014, 28, 3996–4003. [Google Scholar] [CrossRef]
- Yaseen, S.; Demopulos, G.; Dudler, T.; Yabuki, M.; Wood, C.L.; Cummings, W.J.; Tjoelker, L.W.; Fujita, T.; Sacks, S.; Garred, P.; et al. Lectin Pathway Effector Enzyme Mannan-Binding Lectin-Associated Serine Protease-2 can Activate Native Complement C3 in Absence of C4 and/or C2. FASEB J. 2017, 31, 2210–2219. [Google Scholar] [CrossRef]
- Paréj, K.; Dobó, J.; Závodszky, P.; Gál, P. The Control of the Complement Lectin Pathway Activation Revisited: Both C1-Inhibitor and Antithrombin Are likely Physiological Inhibitors, While A2-Macroglobulin Is Not. Mol. Immunol. 2013, 54, 415–422. [Google Scholar] [CrossRef]
- Pihl, R.; Jensen, R.K.; Poulsen, E.C.; Jensen, L.; Hansen, A.G.; Thøgersen, I.B.; Dobó, J.; Gál, P.; Andersen, G.R.; Enghild, J.J.; et al. ITIH4 Acts as a Protease Inhibitor by a Novel Inhibitory Mechanism. Sci. Adv. 2021, 7, eaba7381. [Google Scholar] [CrossRef]
- Drouet, C.; López-Lera, A.; Ghannam, A.; López-Trascasa, M.; Cichon, S.; Ponard, D.; Parsopoulou, F.; Grombirikova, H.; Freiberger, T.; Rijavec, M.; et al. SERPING1 Variants and C1-INH Biological Function: A Close Relationship with C1-INH-HAE. Front. Allergy 2022, 3, 835503. [Google Scholar] [CrossRef] [PubMed]
- Hurler, L.; Toonen, E.J.M.; Kajdácsi, E.; van Bree, B.; Brandwijk, R.J.M.G.E.; de Bruin, W.; Lyons, P.A.; Bergamaschi, L.; Sinkovits, G.; Cervenak, L.; et al. Distinction of Early Complement Classical and Lectin Pathway Activation via Quantification of C1s/C1-INH and MASP-1/C1-INH Complexes Using Novel ELISAs. Front. Immunol. 2022, 13, 1039765. [Google Scholar] [CrossRef] [PubMed]
- Kajdácsi, E.; Jandrasics, Z.; Veszeli, N.; Makó, V.; Koncz, A.; Gulyás, D.; Köhalmi, K.V.; Temesszentandrási, G.; Cervenak, L.; Gál, P.; et al. Patterns of C1-Inhibitor/Plasma Serine Protease Complexes in Healthy Humans and in Hereditary Angioedema Patients. Front. Immunol. 2020, 11, 794. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.P.; Mackman, N. Anticoagulant SERPINs: Endogenous Regulators of Hemostasis and Thrombosis. Front. Cardiovasc. Med. 2022, 9, 878199. [Google Scholar] [CrossRef]
- Dobó, J.; Harmat, V.; Beinrohr, L.; Sebestyén, E.; Závodszky, P.; Gál, P. MASP-1, a Promiscuous Complement Protease: Structure of Its Catalytic Region Reveals the Basis of Its Broad Specificity. J. Immunol. 2009, 183, 1207–1214. [Google Scholar] [CrossRef]
- Jenny, L.; Dobó, J.; Gál, P.; Schroeder, V. MASP-1 Induced Clotting--The First Model of Prothrombin Activation by MASP-1. PLoS ONE 2015, 10, e0144633. [Google Scholar] [CrossRef]
- Krarup, A.; Wallis, R.; Presanis, J.S.; Gál, P.; Sim, R.B. Simultaneous Activation of Complement and Coagulation by MBL-Associated Serine Protease 2. PLoS ONE 2007, 2, e623. [Google Scholar] [CrossRef]
- Gál, P.; Dobó, J.; Beinrohr, L.; Pál, G.; Závodszky, P. Inhibition of the Serine Proteases of the Complement System. Adv. Exp. Med. Biol. 2013, 735, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Vandooren, J.; Itoh, Y. Alpha-2-Macroglobulin in Inflammation, Immunity and Infections. Front. Immunol. 2021, 12, 803244. [Google Scholar] [CrossRef]
- Song, J.; Patel, M.; Rosenzweig, C.N.; Chan-Li, Y.; Sokoll, L.J.; Fung, E.T.; Choi-Miura, N.-H.; Goggins, M.; Chan, D.W.; Zhang, Z. Quantification of Fragments of Human Serum Inter-Alpha-Trypsin Inhibitor Heavy Chain 4 by a Surface-Enhanced Laser Desorption/Ionization-Based Immunoassay. Clin. Chem. 2006, 52, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.C.; Sim, R.B.; Lea, S.M.; Fremeaux-Bacchi, V.; Blom, A.M. Complement Factor I in Health and Disease. Mol. Immunol. 2011, 48, 1611–1620. [Google Scholar] [CrossRef]
- Hallam, T.M.; Sharp, S.J.; Andreadi, A.; Kavanagh, D. Complement Factor I: Regulatory Nexus, Driver of Immunopathology, and Therapeutic. Immunobiology 2023, 228, 152410. [Google Scholar] [CrossRef]
- Dobó, J.; Kocsis, A.; Dani, R.; Gál, P. Proprotein Convertases and the Complement System. Front. Immunol. 2022, 13, 958121. [Google Scholar] [CrossRef] [PubMed]
- Roversi, P.; Johnson, S.; Caesar, J.J.E.; McLean, F.; Leath, K.J.; Tsiftsoglou, S.A.; Morgan, B.P.; Harris, C.L.; Sim, R.B.; Lea, S.M. Structural Basis for Complement Factor I Control and Its Disease-Associated Sequence Polymorphisms. Proc. Natl. Acad. Sci. USA 2011, 108, 12839–12844. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Wu, J.; Ricklin, D.; Forneris, F.; Di Crescenzio, P.; Schmidt, C.Q.; Granneman, J.; Sharp, T.H.; Lambris, J.D.; Gros, P. Regulator-Dependent Mechanisms of C3b Processing by Factor I Allow Differentiation of Immune Responses. Nat. Struct. Mol. Biol. 2017, 24, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Werner, L.M.; Criss, A.K. Diverse Functions of C4b-Binding Protein in Health and Disease. J. Immunol. 2023, 211, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Alba-Domínguez, M.; López-Lera, A.; Garrido, S.; Nozal, P.; González-Granado, I.; Melero, J.; Soler-Palacín, P.; Cámara, C.; López-Trascasa, M. Complement Factor I Deficiency: A Not so Rare Immune Defect: Characterization of New Mutations and the First Large Gene Deletion. Orphanet J. Rare Dis. 2012, 7, 42. [Google Scholar] [CrossRef]
- Kavanagh, D.; Yu, Y.; Schramm, E.C.; Triebwasser, M.; Wagner, E.K.; Raychaudhuri, S.; Daly, M.J.; Atkinson, J.P.; Seddon, J.M. Rare Genetic Variants in the CFI Gene Are Associated with Advanced Age-Related Macular Degeneration and Commonly Result in Reduced Serum Factor I Levels. Hum. Mol. Genet. 2015, 24, 3861–3870. [Google Scholar] [CrossRef]
- de Jong, S.; de Breuk, A.; Bakker, B.; Katti, S.; Hoyng, C.B.; Nilsson, S.C.; Blom, A.M.; van den Heuvel, L.P.; den Hollander, A.I.; Volokhina, E.B. Functional Analysis of Variants in Complement Factor I Identified in Age-Related Macular Degeneration and Atypical Hemolytic Uremic Syndrome. Front. Immunol. 2021, 12, 789897. [Google Scholar] [CrossRef]
- Nepomuceno, R.R.; Henschen-Edman, A.H.; Burgess, W.H.; Tenner, A.J. cDNA Cloning and Primary Structure Analysis of C1qR(P), the Human C1q/MBL/SPA Receptor That Mediates Enhanced Phagocytosis in Vitro. Immunity 1997, 6, 119–129. [Google Scholar] [CrossRef]
- Zhao, N.; Wu, J.; Xiong, S.; Zhang, L.; Lu, X.; Chen, S.; Wu, Q.; Wang, H.; Liu, Y.; Chen, Z.; et al. Mannan-Binding Lectin, a Serum Collectin, Suppresses T-Cell Proliferation via Direct Interaction with Cell Surface Calreticulin and Inhibition of Proximal T-Cell Receptor Signaling. FASEB J. 2017, 31, 2405–2417. [Google Scholar] [CrossRef]
- Luo, J.; Li, L.; Chang, B.; Zhu, Z.; Deng, F.; Hu, M.; Yu, Y.; Lu, X.; Chen, Z.; Zuo, D.; et al. Mannan-Binding Lectin via Interaction with Cell Surface Calreticulin Promotes Senescence of Activated Hepatic Stellate Cells to Limit Liver Fibrosis Progression. Cell Mol. Gastroenterol. Hepatol. 2022, 14, 75–99. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, N.; Xu, Q.; Deng, F.; Wang, P.; Dong, L.; Lu, X.; Xia, L.; Wang, M.; Chen, Z.; et al. MBL Binding with AhR Controls Th17 Immunity in Silicosis-Associated Lung Inflammation and Fibrosis. J. Inflamm. Res. 2022, 15, 4315–4329. [Google Scholar] [CrossRef]
- Fraser, D.A.; Bohlson, S.S.; Jasinskiene, N.; Rawal, N.; Palmarini, G.; Ruiz, S.; Rochford, R.; Tenner, A.J. C1q and MBL, Components of the Innate Immune System, Influence Monocyte Cytokine Expression. J. Leukoc. Biol. 2006, 80, 107–116. [Google Scholar] [CrossRef]
- Liu, L.; Dang, Y. Antimicrobial Activity of Mannose Binding Lectin in Grass Carp (Ctenopharyngodon Idella) In Vivo and In Vitro. Fish. Shellfish. Immunol. 2020, 98, 25–33. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, S.L.; Downing, I.; Atkinson, A.P.M.; Gallagher, R.C.J.; Turner, M.L.; Kilpatrick, D.C. Dendritic Cells Previously Exposed to Mannan-Binding Lectin Enhance Cytokine Production in Allogeneic Mononuclear Cell Cultures. Hum. Immunol. 2010, 71, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Wang, D.; Luo, J.; Li, L.; Lin, L.; Li, J.; Zheng, W.; Zuo, D.; Yang, B. Mannan-Binding Lectin Exacerbates the Severity of Psoriasis by Promoting Plasmacytoid Dendritic Cell Differentiation via the Signal Transducer and Activator of Transcription 3-Interferon Regulatory Factor 8 Axis. J. Dermatol. 2022, 49, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Fumagalli, S.; Császár, E.; Tóth, K.; De Blasio, D.; Zangari, R.; Lénárt, N.; Dénes, Á.; De Simoni, M.-G. Mannose-Binding Lectin Drives Platelet Inflammatory Phenotype and Vascular Damage After Cerebral Ischemia in Mice via IL (Interleukin)-1α. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2678–2690. [Google Scholar] [CrossRef]
- Peng, X.; Zhao, G.; Lin, J.; Li, C. Interaction of Mannose Binding Lectin and Other Pattern Recognition Receptors in Human Corneal Epithelial Cells during Aspergillus Fumigatus Infection. Int. Immunopharmacol. 2018, 63, 161–169. [Google Scholar] [CrossRef] [PubMed]
- van der Pol, P.; Schlagwein, N.; van Gijlswijk, D.J.; Berger, S.P.; Roos, A.; Bajema, I.M.; de Boer, H.C.; de Fijter, J.W.; Stahl, G.L.; Daha, M.R.; et al. Mannan-Binding Lectin Mediates Renal Ischemia/Reperfusion Injury Independent of Complement Activation. Am. J. Transplant. 2012, 12, 877–887. [Google Scholar] [CrossRef]
- Dong, L.; Wu, J.; Chen, K.; Xie, J.; Wang, Y.; Li, D.; Liu, Y.; Yin, A.; Zhao, Y.; Han, Y.; et al. Mannan-Binding Lectin Attenuates Inflammatory Arthritis through the Suppression of Osteoclastogenesis. Front. Immunol. 2019, 10, 1239. [Google Scholar] [CrossRef]
- Oroszlán, M.; Daha, M.R.; Cervenak, L.; Prohászka, Z.; Füst, G.; Roos, A. MBL and C1q Compete for Interaction with Human Endothelial Cells. Mol. Immunol. 2007, 44, 1150–1158. [Google Scholar] [CrossRef]
- Yang, Y.-F.; Zhou, Y.-D.; Hu, J.-C.; Luo, F.-L.; Xie, Y.; Shen, Y.-Y.; Bian, W.-X.; Yin, Z.-N.; Li, H.-L.; Zhang, X.-L. Ficolin-A/2, Acting as a New Regulator of Macrophage Polarization, Mediates the Inflammatory Response in Experimental Mouse Colitis. Immunology 2017, 151, 433–450. [Google Scholar] [CrossRef]
- Macarie, R.D.; Tucureanu, M.M.; Ciortan, L.; Gan, A.-M.; Butoi, E.; Mânduțeanu, I. Ficolin-2 Amplifies Inflammation in Macrophage-Smooth Muscle Cell Cross-Talk and Increases Monocyte Transmigration by Mechanisms Involving IL-1β and IL-6. Sci. Rep. 2023, 13, 19431. [Google Scholar] [CrossRef]
- Ding, Q.; Shen, Y.; Li, D.; Yang, J.; Yu, J.; Yin, Z.; Zhang, X.-L. Ficolin-2 Triggers Antitumor Effect by Activating Macrophages and CD8+ T Cells. Clin. Immunol. 2017, 183, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-X.; Cao, B.; Ma, N.; Wu, K.-Y.; Chen, W.-B.; Wu, W.; Dong, X.; Liu, C.-F.; Gao, Y.-F.; Diao, T.-Y.; et al. Collectin-11 Promotes Cancer Cell Proliferation and Tumor Growth. JCI Insight 2023, 8, e159452. [Google Scholar] [CrossRef] [PubMed]
- Megyeri, M.; Makó, V.; Beinrohr, L.; Doleschall, Z.; Prohászka, Z.; Cervenak, L.; Závodszky, P.; Gál, P. Complement Protease MASP-1 Activates Human Endothelial Cells: PAR4 Activation Is a Link between Complement and Endothelial Function. J. Immunol. 2009, 183, 3409–3416. [Google Scholar] [CrossRef] [PubMed]
- Jani, P.K.; Kajdácsi, E.; Megyeri, M.; Dobó, J.; Doleschall, Z.; Futosi, K.; Tímár, C.I.; Mócsai, A.; Makó, V.; Gál, P.; et al. MASP-1 Induces a Unique Cytokine Pattern in Endothelial Cells: A Novel Link between Complement System and Neutrophil Granulocytes. PLoS ONE 2014, 9, e87104. [Google Scholar] [CrossRef] [PubMed]
- Jani, P.K.; Schwaner, E.; Kajdácsi, E.; Debreczeni, M.L.; Ungai-Salánki, R.; Dobó, J.; Doleschall, Z.; Rigó, J.; Geiszt, M.; Szabó, B.; et al. Complement MASP-1 Enhances Adhesion between Endothelial Cells and Neutrophils by up-Regulating E-Selectin Expression. Mol. Immunol. 2016, 75, 38–47. [Google Scholar] [CrossRef]
- Patel, P.K.; Choudhary, K.; Patidar, P.; Sharma, S.; Hajela, K. Mannose-Binding Lectin and Associate Serine Protease Complex Modulates Neutrophil Respiratory Burst and Gene Expression in Capra Hircus. Immunobiology 2020, 225, 151972. [Google Scholar] [CrossRef]
- Megyeri, M.; Jani, P.K.; Kajdácsi, E.; Dobó, J.; Schwaner, E.; Major, B.; Rigó, J.; Závodszky, P.; Thiel, S.; Cervenak, L.; et al. Serum MASP-1 in Complex with MBL Activates Endothelial Cells. Mol. Immunol. 2014, 59, 39–45. [Google Scholar] [CrossRef]
- Debreczeni, M.L.; Németh, Z.; Kajdácsi, E.; Schwaner, E.; Makó, V.; Masszi, A.; Doleschall, Z.; Rigó, J.; Walter, F.R.; Deli, M.A.; et al. MASP-1 Increases Endothelial Permeability. Front. Immunol. 2019, 10, 991. [Google Scholar] [CrossRef]
- Debreczeni, M.L.; Németh, Z.; Kajdácsi, E.; Farkas, H.; Cervenak, L. Molecular Dambusters: What Is Behind Hyperpermeability in Bradykinin-Mediated Angioedema? Clin. Rev. Allergy Immunol. 2021, 60, 318–347. [Google Scholar] [CrossRef] [PubMed]
- Németh, Z.; Debreczeni, M.L.; Kajdácsi, E.; Dobó, J.; Gál, P.; Cervenak, L. Cooperation of Complement MASP-1 with Other Proinflammatory Factors to Enhance the Activation of Endothelial Cells. Int. J. Mol. Sci. 2023, 24, 9181. [Google Scholar] [CrossRef]
- Saeed, A.; Baloch, K.; Brown, R.J.P.; Wallis, R.; Chen, L.; Dexter, L.; McClure, C.P.; Shakesheff, K.; Thomson, B.J. Mannan Binding Lectin-Associated Serine Protease 1 Is Induced by Hepatitis C Virus Infection and Activates Human Hepatic Stellate Cells. Clin. Exp. Immunol. 2013, 174, 265–273. [Google Scholar] [CrossRef]
- Liu, X.; Tan, S.; Liu, H.; Jiang, J.; Wang, X.; Li, L.; Wu, B. Hepatocyte-Derived MASP1-Enriched Small Extracellular Vesicles Activate HSCs to Promote Liver Fibrosis. Hepatology 2023, 77, 1181–1197. [Google Scholar] [CrossRef]
- Mu, L.; Yin, X.; Wu, H.; Han, K.; Wu, L.; Ding, M.; Bian, X.; Li, B.; Fu, S.; Liang, F.; et al. Expression and Functional Characterization of a Mannose-Binding Lectin-Associated Serine Protease-1 (MASP-1) from Nile Tilapia (Oreochromis Niloticus) in Host Defense against Bacterial Infection. Fish. Shellfish. Immunol. 2019, 91, 68–77. [Google Scholar] [CrossRef]
- Golomingi, M.; Kohler, J.; Jenny, L.; Hardy, E.T.; Dobó, J.; Gál, P.; Pál, G.; Kiss, B.; Lam, W.A.; Schroeder, V. Complement Lectin Pathway Components MBL and MASP-1 Promote Haemostasis upon Vessel Injury in a Microvascular Bleeding Model. Front. Immunol. 2022, 13, 948190. [Google Scholar] [CrossRef]
- Debreczeni, M.L.; Szekacs, I.; Kovacs, B.; Saftics, A.; Kurunczi, S.; Gál, P.; Dobó, J.; Cervenak, L.; Horvath, R. Human Primary Endothelial Label-Free Biochip Assay Reveals Unpredicted Functions of Plasma Serine Proteases. Sci. Rep. 2020, 10, 3303. [Google Scholar] [CrossRef]
- Elhadad, S.; Chapin, J.; Copertino, D.; Van Besien, K.; Ahamed, J.; Laurence, J. MASP2 Levels Are Elevated in Thrombotic Microangiopathies: Association with Microvascular Endothelial Cell Injury and Suppression by Anti-MASP2 Antibody Narsoplimab. Clin. Exp. Immunol. 2021, 203, 96–104. [Google Scholar] [CrossRef]
- Elhadad, S.; Redmond, D.; Huang, J.; Tan, A.; Laurence, J. MASP2 Inhibition by Narsoplimab Suppresses Endotheliopathies Characteristic of Transplant-Associated Thrombotic Microangiopathy: In Vitro and Ex Vivo Evidence. Clin. Exp. Immunol. 2023, 213, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Howes, J.-M.; Richardson, V.R.; Smith, K.A.; Schroeder, V.; Somani, R.; Shore, A.; Hess, K.; Ajjan, R.; Pease, R.J.; Keen, J.N.; et al. Complement C3 Is a Novel Plasma Clot Component with Anti-Fibrinolytic Properties. Diabetes Vasc. Dis. Res. 2012, 9, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Golomingi, M.; Kohler, J.; Lamers, C.; Pouw, R.B.; Ricklin, D.; Dobó, J.; Gál, P.; Pál, G.; Kiss, B.; Dopler, A.; et al. Complement Inhibition Can Decrease the Haemostatic Response in a Microvascular Bleeding Model at Multiple Levels. Front. Immunol. 2023, 14, 1226832. [Google Scholar] [CrossRef] [PubMed]
- Krarup, A.; Gulla, K.C.; Gál, P.; Hajela, K.; Sim, R.B. The Action of MBL-Associated Serine Protease 1 (MASP1) on Factor XIII and Fibrinogen. Biochim. Biophys. Acta 2008, 1784, 1294–1300. [Google Scholar] [CrossRef]
- Hess, K.; Ajjan, R.; Phoenix, F.; Dobó, J.; Gál, P.; Schroeder, V. Effects of MASP-1 of the Complement System on Activation of Coagulation Factors and Plasma Clot Formation. PLoS ONE 2012, 7, e35690. [Google Scholar] [CrossRef] [PubMed]
- Jenny, L.; Dobó, J.; Gál, P.; Schroeder, V. MASP-1 of the Complement System Promotes Clotting via Prothrombin Activation. Mol. Immunol. 2015, 65, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.; Kita, A.; Leach, J.; Rounsevell, R.; Huang, J.N.; Moake, J.; Ware, R.E.; Fletcher, D.A.; Lam, W.A. In Vitro Modeling of the Microvascular Occlusion and Thrombosis That Occur in Hematologic Diseases Using Microfluidic Technology. J. Clin. Investig. 2012, 122, 408–418. [Google Scholar] [CrossRef]
- Jenny, L.; Dobó, J.; Gál, P.; Pál, G.; Lam, W.A.; Schroeder, V. MASP-1 of the Complement System Enhances Clot Formation in a Microvascular Whole Blood Flow Model. PLoS ONE 2018, 13, e0191292. [Google Scholar] [CrossRef]
- Jenny, L.; Noser, D.; Larsen, J.B.; Dobó, J.; Gál, P.; Pál, G.; Schroeder, V. MASP-1 of the Complement System Alters Fibrinolytic Behaviour of Blood Clots. Mol. Immunol. 2019, 114, 1–9. [Google Scholar] [CrossRef]
- Kozarcanin, H.; Lood, C.; Munthe-Fog, L.; Sandholm, K.; Hamad, O.A.; Bengtsson, A.A.; Skjoedt, M.-O.; Huber-Lang, M.; Garred, P.; Ekdahl, K.N.; et al. The Lectin Complement Pathway Serine Proteases (MASPs) Represent a Possible Crossroad between the Coagulation and Complement Systems in Thromboinflammation. J. Thromb. Haemost. 2016, 14, 531–545. [Google Scholar] [CrossRef]
- Takahashi, K.; Chang, W.-C.; Takahashi, M.; Pavlov, V.; Ishida, Y.; La Bonte, L.; Shi, L.; Fujita, T.; Stahl, G.L.; Van Cott, E.M. Mannose-Binding Lectin and Its Associated Proteases (MASPs) Mediate Coagulation and Its Deficiency Is a Risk Factor in Developing Complications from Infection, Including Disseminated Intravascular Coagulation. Immunobiology 2011, 216, 96–102. [Google Scholar] [CrossRef]
- La Bonte, L.R.; Pavlov, V.I.; Tan, Y.S.; Takahashi, K.; Takahashi, M.; Banda, N.K.; Zou, C.; Fujita, T.; Stahl, G.L. Mannose-Binding Lectin-Associated Serine Protease-1 Is a Significant Contributor to Coagulation in a Murine Model of Occlusive Thrombosis. J. Immunol. 2012, 188, 885–891. [Google Scholar] [CrossRef]
- Pavlov, V.I.; Skjoedt, M.-O.; Siow Tan, Y.; Rosbjerg, A.; Garred, P.; Stahl, G.L. Endogenous and Natural Complement Inhibitor Attenuates Myocardial Injury and Arterial Thrombogenesis. Circulation 2012, 126, 2227–2235. [Google Scholar] [CrossRef]
- Liang, R.A.; Høiland, I.I.; Ueland, T.; Aukrust, P.; Snir, O.; Hindberg, K.; Braekkan, S.K.; Garred, P.; Mollnes, T.E.; Hansen, J.-B. Plasma Levels of Mannose-Binding Lectin and Future Risk of Venous Thromboembolism. J. Thromb. Haemost. 2019, 17, 1661–1669. [Google Scholar] [CrossRef]
- Daha, M.R.; van Kooten, C. Role of Complement in IgA Nephropathy. J. Nephrol. 2016, 29, 1–4. [Google Scholar] [CrossRef]
- Endo, M.; Ohi, H.; Ohsawa, I.; Fujita, T.; Matsushita, M.; Fujita, T. Glomerular Deposition of Mannose-Binding Lectin (MBL) Indicates a Novel Mechanism of Complement Activation in IgA Nephropathy. Nephrol. Dial. Transplant. 1998, 13, 1984–1990. [Google Scholar] [CrossRef]
- Roos, A.; Rastaldi, M.P.; Calvaresi, N.; Oortwijn, B.D.; Schlagwein, N.; van Gijlswijk-Janssen, D.J.; Stahl, G.L.; Matsushita, M.; Fujita, T.; van Kooten, C.; et al. Glomerular Activation of the Lectin Pathway of Complement in IgA Nephropathy Is Associated with More Severe Renal Disease. J. Am. Soc. Nephrol. 2006, 17, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.-Y.; Zhu, L.; Meng, S.-J.; Shi, S.-F.; Liu, L.-J.; Lv, J.-C.; Zhang, H. Mannose-Binding Lectin Levels Could Predict Prognosis in IgA Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 3175–3181. [Google Scholar] [CrossRef] [PubMed]
- Medjeral-Thomas, N.R.; Troldborg, A.; Constantinou, N.; Lomax-Browne, H.J.; Hansen, A.G.; Willicombe, M.; Pusey, C.D.; Cook, H.T.; Thiel, S.; Pickering, M.C. Progressive IgA Nephropathy Is Associated With Low Circulating Mannan-Binding Lectin-Associated Serine Protease-3 (MASP-3) and Increased Glomerular Factor H-Related Protein-5 (FHR5) Deposition. Kidney Int. Rep. 2018, 3, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.E.; Dudler, T.; Marber, M.S.; Schwaeble, W. Cardioprotection by an Anti-MASP-2 Antibody in a Murine Model of Myocardial Infarction. Open Heart 2018, 5, e000652. [Google Scholar] [CrossRef] [PubMed]
- Omeros Corporation—Omeros Corporation Provides Update on Interim Analysis of ARTEMIS-IGAN Phase 3 Trial of Narsoplimab in IgA Nephropathy. Available online: https://investor.omeros.com/news-releases/news-release-details/omeros-corporation-provides-update-interim-analysis-artemis-igan (accessed on 19 December 2023).
- FDA Grants Orphan Drug Designation to Omeros’ MASP-3 Inhibitor OMS906 for Treatment of Paroxysmal Nocturnal Hemoglobinuria. Available online: https://www.businesswire.com/news/home/20220729005194/en/FDA-Grants-Orphan-Drug-Designation-to-Omeros%E2%80%99-MASP-3-Inhibitor-OMS906-for-Treatment-of-Paroxysmal-Nocturnal-Hemoglobinuria (accessed on 19 December 2023).
- Haddad, G.; Lorenzen, J.M.; Ma, H.; de Haan, N.; Seeger, H.; Zaghrini, C.; Brandt, S.; Kölling, M.; Wegmann, U.; Kiss, B.; et al. Altered Glycosylation of IgG4 Promotes Lectin Complement Pathway Activation in Anti-PLA2R1-Associated Membranous Nephropathy. J. Clin. Investig. 2021, 131, e140453. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, J.; Wang, X.; Zheng, X.; Gao, H.; Jiang, S.; Li, W. Lectin Complement Pathway Activation Is Associated with Massive Proteinuria in PLA2R-Positive Membranous Nephropathy: A Retrospective Study. Int. J. Gen. Med. 2023, 16, 1879–1889. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, C.; Jin, L.; He, F.; Li, C.; Gao, Q.; Chen, G.; He, Z.; Song, M.; Zhou, Z.; et al. IgG4 Anti-Phospholipase A2 Receptor Might Activate Lectin and Alternative Complement Pathway Meanwhile in Idiopathic Membranous Nephropathy: An Inspiration from a Cross-Sectional Study. Immunol. Res. 2016, 64, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wen, L.; Dou, Y.; Zhao, Z. Human Anti-Thrombospondin Type 1 Domain-Containing 7A Antibodies Induce Membranous Nephropathy through Activation of Lectin Complement Pathway. Biosci. Rep. 2018, 38, BSR20180131. [Google Scholar] [CrossRef] [PubMed]
- So, B.Y.F.; Chan, G.C.W.; Yap, D.Y.H.; Chan, T.M. The Role of the Complement System in Primary Membranous Nephropathy: A Narrative Review in the Era of New Therapeutic Targets. Front. Immunol. 2022, 13, 1009864. [Google Scholar] [CrossRef] [PubMed]
- Bus, P.; Chua, J.S.; Klessens, C.Q.F.; Zandbergen, M.; Wolterbeek, R.; van Kooten, C.; Trouw, L.A.; Bruijn, J.A.; Baelde, H.J. Complement Activation in Patients With Diabetic Nephropathy. Kidney Int. Rep. 2018, 3, 302–313. [Google Scholar] [CrossRef]
- Li, X.-Q.; Chang, D.-Y.; Chen, M.; Zhao, M.-H. Complement Activation in Patients with Diabetic Nephropathy. Diabetes Metab. 2019, 45, 248–253. [Google Scholar] [CrossRef]
- La Bonte, L.R.; Dokken, B.; Davis-Gorman, G.; Stahl, G.L.; McDonagh, P.F. The Mannose-Binding Lectin Pathway Is a Significant Contributor to Reperfusion Injury in the Type 2 Diabetic Heart. Diabetes Vasc. Dis. Res. 2009, 6, 172–180. [Google Scholar] [CrossRef]
- Farrar, C.A.; Zhou, W.; Sacks, S.H. Role of the Lectin Complement Pathway in Kidney Transplantation. Immunobiology 2016, 221, 1068–1072. [Google Scholar] [CrossRef]
- Farrar, C.A.; Tran, D.; Li, K.; Wu, W.; Peng, Q.; Schwaeble, W.; Zhou, W.; Sacks, S.H. Collectin-11 Detects Stress-Induced L-Fucose Pattern to Trigger Renal Epithelial Injury. J. Clin. Investig. 2016, 126, 1911–1925. [Google Scholar] [CrossRef] [PubMed]
- Møller-Kristensen, M.; Wang, W.; Ruseva, M.; Thiel, S.; Nielsen, S.; Takahashi, K.; Shi, L.; Ezekowitz, A.; Jensenius, J.C.; Gadjeva, M. Mannan-Binding Lectin Recognizes Structures on Ischaemic Reperfused Mouse Kidneys and Is Implicated in Tissue Injury. Scand. J. Immunol. 2005, 61, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Farrar, C.A.; Abe, K.; Pratt, J.R.; Marsh, J.E.; Wang, Y.; Stahl, G.L.; Sacks, S.H. Predominant Role for C5b-9 in Renal Ischemia/Reperfusion Injury. J. Clin. Investig. 2000, 105, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.P.; Roos, A.; Mallat, M.J.K.; Fujita, T.; de Fijter, J.W.; Daha, M.R. Association between Mannose-Binding Lectin Levels and Graft Survival in Kidney Transplantation. Am. J. Transplant. 2005, 5, 1361–1366. [Google Scholar] [CrossRef]
- Danobeitia, J.S.; Ziemelis, M.; Ma, X.; Zitur, L.J.; Zens, T.; Chlebeck, P.J.; Van Amersfoort, E.S.; Fernandez, L.A. Complement Inhibition Attenuates Acute Kidney Injury after Ischemia-Reperfusion and Limits Progression to Renal Fibrosis in Mice. PLoS ONE 2017, 12, e0183701. [Google Scholar] [CrossRef] [PubMed]
- Eerhart, M.J.; Reyes, J.A.; Blanton, C.L.; Danobeitia, J.S.; Chlebeck, P.J.; Zitur, L.J.; Springer, M.; Polyak, E.; Coonen, J.; Capuano, S.; et al. Complement Blockade in Recipients Prevents Delayed Graft Function and Delays Antibody-Mediated Rejection in a Nonhuman Primate Model of Kidney Transplantation. Transplantation 2022, 106, 60–71. [Google Scholar] [CrossRef]
- Szakács, D.; Kocsis, A.; Szász, R.; Gál, P.; Pál, G. Novel MASP-2 Inhibitors Developed via Directed Evolution of Human TFPI1 Are Potent Lectin Pathway Inhibitors. J. Biol. Chem. 2019, 294, 8227–8237. [Google Scholar] [CrossRef]
- Howard, M.C.; Nauser, C.L.; Farrar, C.A.; Wallis, R.; Sacks, S.H. L-Fucose Prevention of Renal Ischaemia/Reperfusion Injury in Mice. FASEB J. 2020, 34, 822–834. [Google Scholar] [CrossRef]
- Collard, C.D.; Väkevä, A.; Morrissey, M.A.; Agah, A.; Rollins, S.A.; Reenstra, W.R.; Buras, J.A.; Meri, S.; Stahl, G.L. Complement Activation after Oxidative Stress: Role of the Lectin Complement Pathway. Am. J. Pathol. 2000, 156, 1549–1556. [Google Scholar] [CrossRef]
- Zhang, M.; Hou, Y.J.; Cavusoglu, E.; Lee, D.C.; Steffensen, R.; Yang, L.; Bashari, D.; Villamil, J.; Moussa, M.; Fernaine, G.; et al. MASP-2 Activation Is Involved in Ischemia-Related Necrotic Myocardial Injury in Humans. Int. J. Cardiol. 2013, 166, 499–504. [Google Scholar] [CrossRef]
- Frauenknecht, V.; Thiel, S.; Storm, L.; Meier, N.; Arnold, M.; Schmid, J.-P.; Saner, H.; Schroeder, V. Plasma Levels of Mannan-Binding Lectin (MBL)-Associated Serine Proteases (MASPs) and MBL-Associated Protein in Cardio- and Cerebrovascular Diseases. Clin. Exp. Immunol. 2013, 173, 112–120. [Google Scholar] [CrossRef]
- Schoos, M.M.; Munthe-Fog, L.; Skjoedt, M.-O.; Ripa, R.S.; Lønborg, J.; Kastrup, J.; Kelbæk, H.; Clemmensen, P.; Garred, P. Association between Lectin Complement Pathway Initiators, C-Reactive Protein and Left Ventricular Remodeling in Myocardial Infarction-a Magnetic Resonance Study. Mol. Immunol. 2013, 54, 408–414. [Google Scholar] [CrossRef]
- Trendelenburg, M.; Theroux, P.; Stebbins, A.; Granger, C.; Armstrong, P.; Pfisterer, M. Influence of Functional Deficiency of Complement Mannose-Binding Lectin on Outcome of Patients with Acute ST-Elevation Myocardial Infarction Undergoing Primary Percutaneous Coronary Intervention. Eur. Heart J. 2010, 31, 1181–1187. [Google Scholar] [CrossRef]
- Bonnemeier, H.; Wiegand, U.K.H.; Giannitsis, E.; Schulenburg, S.; Hartmann, F.; Kurowski, V.; Bode, F.; Tölg, R.; Katus, H.A.; Richardt, G. Temporal Repolarization Inhomogeneity and Reperfusion Arrhythmias in Patients Undergoing Successful Primary Percutaneous Coronary Intervention for Acute ST-Segment Elevation Myocardial Infarction: Impact of Admission Troponin T. Am. Heart J. 2003, 145, 484–492. [Google Scholar] [CrossRef]
- Holt, C.B.; Østergaard, J.A.; Thiel, S.; Hansen, T.K.; Mellbin, L.; Sörensson, P.; Bjerre, M. Circulating Lectin Pathway Proteins Do Not Predict Short-Term Cardiac Outcomes after Myocardial Infarction. Clin. Exp. Immunol. 2019, 198, 94–100. [Google Scholar] [CrossRef]
- Jordan, J.E.; Montalto, M.C.; Stahl, G.L. Inhibition of Mannose-Binding Lectin Reduces Postischemic Myocardial Reperfusion Injury. Circulation 2001, 104, 1413–1418. [Google Scholar] [CrossRef]
- Pavlov, V.I.; Tan, Y.S.; McClure, E.E.; La Bonte, L.R.; Zou, C.; Gorsuch, W.B.; Stahl, G.L. Human Mannose-Binding Lectin Inhibitor Prevents Myocardial Injury and Arterial Thrombogenesis in a Novel Animal Model. Am. J. Pathol. 2015, 185, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Cervera, A.; Planas, A.M.; Justicia, C.; Urra, X.; Jensenius, J.C.; Torres, F.; Lozano, F.; Chamorro, A. Genetically-Defined Deficiency of Mannose-Binding Lectin Is Associated with Protection after Experimental Stroke in Mice and Outcome in Human Stroke. PLoS ONE 2010, 5, e8433. [Google Scholar] [CrossRef] [PubMed]
- Osthoff, M.; Katan, M.; Fluri, F.; Schuetz, P.; Bingisser, R.; Kappos, L.; Steck, A.J.; Engelter, S.T.; Mueller, B.; Christ-Crain, M.; et al. Mannose-Binding Lectin Deficiency Is Associated with Smaller Infarction Size and Favorable Outcome in Ischemic Stroke Patients. PLoS ONE 2011, 6, e21338. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Chrysanthou, E.; Dudler, T.; Cummings, W.J.; Takahashi, M.; Fujita, T.; Demopulos, G.; De Simoni, M.-G.; Schwaeble, W. Mannan Binding Lectin-Associated Serine Protease-2 (MASP-2) Critically Contributes to Post-Ischemic Brain Injury Independent of MASP-1. J. Neuroinflamm. 2016, 13, 213. [Google Scholar] [CrossRef] [PubMed]
- Tsakanova, G.; Stepanyan, A.; Nahapetyan, K.; Sim, R.B.; Arakelyan, A.; Boyajyan, A. Serine Proteases of the Complement Lectin Pathway and Their Genetic Variations in Ischaemic Stroke. J. Clin. Pathol. 2018, 71, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Vengen, I.T.; Madsen, H.O.; Garred, P.; Platou, C.; Vatten, L.; Videm, V. Mannose-Binding Lectin Deficiency Is Associated with Myocardial Infarction: The HUNT2 Study in Norway. PLoS ONE 2012, 7, e42113. [Google Scholar] [CrossRef]
- Fumagalli, S.; Perego, C.; Zangari, R.; De Blasio, D.; Oggioni, M.; De Nigris, F.; Snider, F.; Garred, P.; Ferrante, A.M.R.; De Simoni, M.-G. Lectin Pathway of Complement Activation Is Associated with Vulnerability of Atherosclerotic Plaques. Front. Immunol. 2017, 8, 288. [Google Scholar] [CrossRef]
- Vengen, I.T.; Enger, T.B.; Videm, V.; Garred, P. Pentraxin 3, Ficolin-2 and Lectin Pathway Associated Serine Protease MASP-3 as Early Predictors of Myocardial Infarction—The HUNT2 Study. Sci. Rep. 2017, 7, 43045. [Google Scholar] [CrossRef]
- Hurler, L.; Szilágyi, Á.; Mescia, F.; Bergamaschi, L.; Mező, B.; Sinkovits, G.; Réti, M.; Müller, V.; Iványi, Z.; Gál, J.; et al. Complement Lectin Pathway Activation Is Associated with COVID-19 Disease Severity, Independent of MBL2 Genotype Subgroups. Front. Immunol. 2023, 14, 1162171. [Google Scholar] [CrossRef]
- Götz, M.P.; Skjoedt, M.-O.; Bayarri-Olmos, R.; Hansen, C.B.; Pérez-Alós, L.; Jarlhelt, I.; Benfield, T.; Rosbjerg, A.; Garred, P. Lectin Pathway Enzyme MASP-2 and Downstream Complement Activation in COVID-19. J. Innate Immun. 2023, 15, 122–135. [Google Scholar] [CrossRef]
- Niederreiter, J.; Eck, C.; Ries, T.; Hartmann, A.; Märkl, B.; Büttner-Herold, M.; Amann, K.; Daniel, C. Complement Activation via the Lectin and Alternative Pathway in Patients with Severe COVID-19. Front. Immunol. 2022, 13, 835156. [Google Scholar] [CrossRef] [PubMed]
- Stravalaci, M.; Pagani, I.; Paraboschi, E.M.; Pedotti, M.; Doni, A.; Scavello, F.; Mapelli, S.N.; Sironi, M.; Perucchini, C.; Varani, L.; et al. Recognition and Inhibition of SARS-CoV-2 by Humoral Innate Immunity Pattern Recognition Molecules. Nat. Immunol. 2022, 23, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Zhu, L.; Liu, H.; Zhang, X.; Wang, T.; Fu, Y.; Li, H.; Dong, Q.; Hu, Y.; Zhang, Z.; et al. Highly Pathogenic Coronavirus N Protein Aggravates Inflammation by MASP-2-Mediated Lectin Complement Pathway Overactivation. Signal Transduct. Target. Ther. 2022, 7, 318. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.M.; Ferrari, M.; Lynch, N.J.; Yaseen, S.; Dudler, T.; Gragerov, S.; Demopulos, G.; Heeney, J.L.; Schwaeble, W.J. Lectin Pathway Mediates Complement Activation by SARS-CoV-2 Proteins. Front. Immunol. 2021, 12, 714511. [Google Scholar] [CrossRef]
- Dudler, T.; Yaseen, S.; Cummings, W.J. Development and Characterization of Narsoplimab, a Selective MASP-2 Inhibitor, for the Treatment of Lectin-Pathway-Mediated Disorders. Front. Immunol. 2023, 14, 1297352. [Google Scholar] [CrossRef] [PubMed]
- Dobó, J.; Major, B.; Kékesi, K.A.; Szabó, I.; Megyeri, M.; Hajela, K.; Juhász, G.; Závodszky, P.; Gál, P. Cleavage of Kininogen and Subsequent Bradykinin Release by the Complement Component: Mannose-Binding Lectin-Associated Serine Protease (MASP)-1. PLoS ONE 2011, 6, e20036. [Google Scholar] [CrossRef]
- Hansen, C.B.; Csuka, D.; Munthe-Fog, L.; Varga, L.; Farkas, H.; Hansen, K.M.; Koch, C.; Skjødt, K.; Garred, P.; Skjoedt, M.-O. The Levels of the Lectin Pathway Serine Protease MASP-1 and Its Complex Formation with C1 Inhibitor Are Linked to the Severity of Hereditary Angioedema. J. Immunol. 2015, 195, 3596–3604. [Google Scholar] [CrossRef] [PubMed]
- Farkas, H.; Jakab, L.; Temesszentandrási, G.; Visy, B.; Harmat, G.; Füst, G.; Széplaki, G.; Fekete, B.; Karádi, I.; Varga, L. Hereditary Angioedema: A Decade of Human C1-Inhibitor Concentrate Therapy. J. Allergy Clin. Immunol. 2007, 120, 941–947. [Google Scholar] [CrossRef]
- Mayilyan, K.R.; Krarup, A.; Soghoyan, A.F.; Jensenius, J.C.; Sim, R.B. L-Ficolin-MASP Arm of the Complement System in Schizophrenia. Immunobiology 2023, 228, 152349. [Google Scholar] [CrossRef] [PubMed]
- Petr, V.; Thurman, J.M. The Role of Complement in Kidney Disease. Nat. Rev. Nephrol. 2023, 19, 771–787. [Google Scholar] [CrossRef] [PubMed]
- Tortajada, A.; Gutierrez, E.; Pickering, M.C.; Praga Terente, M.; Medjeral-Thomas, N. The Role of Complement in IgA Nephropathy. Mol. Immunol. 2019, 114, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, M.; Ortega, R.; Sánchez, M.; Segarra, A.; Salcedo, M.T.; González, F.; Camacho, R.; Valdivia, M.A.; Cabrera, R.; López, K.; et al. Association of C4d Deposition with Clinical Outcomes in IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2014, 9, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Flyvbjerg, A. The Role of the Complement System in Diabetic Nephropathy. Nat. Rev. Nephrol. 2017, 13, 311–318. [Google Scholar] [CrossRef]
- Riedemann, N.C.; Ward, P.A. Complement in Ischemia Reperfusion Injury. Am. J. Pathol. 2003, 162, 363–367. [Google Scholar] [CrossRef]
- Panagiotou, A.; Trendelenburg, M.; Osthoff, M. The Lectin Pathway of Complement in Myocardial Ischemia/Reperfusion Injury-Review of Its Significance and the Potential Impact of Therapeutic Interference by C1 Esterase Inhibitor. Front. Immunol. 2018, 9, 1151. [Google Scholar] [CrossRef]
- Mocco, J.; Wilson, D.A.; Komotar, R.J.; Sughrue, M.E.; Coates, K.; Sacco, R.L.; Elkind, M.S.V.; Connolly, E.S. Alterations in Plasma Complement Levels after Human Ischemic Stroke. Neurosurgery 2006, 59, 28–33, discussion 28–33. [Google Scholar] [CrossRef]
- Bumiller-Bini, V.; de Freitas Oliveira-Toré, C.; Carvalho, T.M.; Kretzschmar, G.C.; Gonçalves, L.B.; Alencar, N.d.M.; Gasparetto Filho, M.A.; Beltrame, M.H.; Winter Boldt, A.B. MASPs at the Crossroad between the Complement and the Coagulation Cascades—The Case for COVID-19. Genet. Mol. Biol. 2021, 44, e20200199. [Google Scholar] [CrossRef]
- Ballanti, E.; Perricone, C.; Greco, E.; Ballanti, M.; Di Muzio, G.; Chimenti, M.S.; Perricone, R. Complement and Autoimmunity. Immunol. Res. 2013, 56, 477–491. [Google Scholar] [CrossRef]
- Troldborg, A.; Thiel, S.; Trendelenburg, M.; Friebus-Kardash, J.; Nehring, J.; Steffensen, R.; Hansen, S.W.K.; Laska, M.J.; Deleuran, B.; Jensenius, J.C.; et al. The Lectin Pathway of Complement Activation in Patients with Systemic Lupus Erythematosus. J. Rheumatol. 2018, 45, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.K.; Parida, J.R.; Tripathy, R.; Pattanaik, S.S.; Ravindran, B.; Das, B.K. Mannose Binding Lectin: A Biomarker of Systemic Lupus Erythematosus Disease Activity. Arthritis Res. Ther. 2012, 14, R218. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, Y.; Nozawa, K.; Matsushita, M.; Kusaoi, M.; Abe, Y.; Yamaji, K.; Tamura, N. Critical Role of Lectin Pathway Mediated by MBL-Associated Serine Proteases in Complement Activation for the Pathogenesis in Systemic Lupus Erythematosus. Heliyon 2023, 9, e19072. [Google Scholar] [CrossRef]
- García-González, M.; Gómez-Bernal, F.; Quevedo-Abeledo, J.C.; Fernández-Cladera, Y.; González-Rivero, A.F.; de Vera-González, A.; de la Rua-Figueroa, I.; López-Mejias, R.; Díaz-González, F.; González-Gay, M.Á.; et al. Full Characterization of the Three Pathways of the Complement System in Patients with Systemic Lupus Erythematosus. Front. Immunol. 2023, 14, 1167055. [Google Scholar] [CrossRef]
- Larsen, M.L.; Troldborg, A.; Toonen, E.J.M.; Hurler, L.; Prohaszka, Z.; Cervenak, L.; Gudmann Hansen, A.; Thiel, S. Differentiating between Activation via the Lectin or the Classical Complement Pathway in Patients with Systemic Lupus Erythematosus. Clin. Exp. Immunol. 2023, 214, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Holers, V.M.; Banda, N.K. Complement in the Initiation and Evolution of Rheumatoid Arthritis. Front. Immunol. 2018, 9, 1057. [Google Scholar] [CrossRef]
- Makinde, V.A.; Senaldi, G.; Jawad, A.S.; Berry, H.; Vergani, D. Reflection of Disease Activity in Rheumatoid Arthritis by Indices of Activation of the Classical Complement Pathway. Ann. Rheum. Dis. 1989, 48, 302–306. [Google Scholar] [CrossRef]
- Malhotra, R.; Wormald, M.R.; Rudd, P.M.; Fischer, P.B.; Dwek, R.A.; Sim, R.B. Glycosylation Changes of IgG Associated with Rheumatoid Arthritis Can Activate Complement via the Mannose-Binding Protein. Nat. Med. 1995, 1, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Ammitzboll, C.G.; Thiel, S.; Ellingsen, T.; Deleuran, B.; Jorgensen, A.; Jensenius, J.C.; Stengaard-Pedersen, K. Levels of Lectin Pathway Proteins in Plasma and Synovial Fluid of Rheumatoid Arthritis and Osteoarthritis. Rheumatol. Int. 2012, 32, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Pieczarka, C.; Andrade, F.A.; Catarino, S.J.; Lidani, K.C.F.; Bavia, L.; Tizzot, R.; Skare, T.; de Messias-Reason, I.J. Ficolin-1 and Ficolin-3 Polymorphisms and Susceptibility to Rheumatoid Arthritis. Autoimmunity 2020, 53, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Banda, N.K.; Desai, D.; Scheinman, R.I.; Pihl, R.; Sekine, H.; Fujita, T.; Sharma, V.; Hansen, A.G.; Garred, P.; Thiel, S.; et al. Targeting of Liver Mannan-Binding Lectin-Associated Serine Protease-3 with RNA Interference Ameliorates Disease in a Mouse Model of Rheumatoid Arthritis. Immunohorizons 2018, 2, 274–295. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Radewicz, K.; Garey, L.J.; Gentleman, S.M.; Reynolds, R. Increase in HLA-DR Immunoreactive Microglia in Frontal and Temporal Cortex of Chronic Schizophrenics. J. Neuropathol. Exp. Neurol. 2000, 59, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia Risk from Complex Variation of Complement Component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef]
- Bohlson, S.S.; Tenner, A.J. Complement in the Brain: Contributions to Neuroprotection, Neuronal Plasticity, and Neuroinflammation. Annu. Rev. Immunol. 2023, 41, 431–452. [Google Scholar] [CrossRef]
- Gorelik, A.; Sapir, T.; Haffner-Krausz, R.; Olender, T.; Woodruff, T.M.; Reiner, O. Developmental Activities of the Complement Pathway in Migrating Neurons. Nat. Commun. 2017, 8, 15096. [Google Scholar] [CrossRef]
- Cedzynski, M.; Szemraj, J.; Swierzko, A.S.; Bak-Romaniszyn, L.; Banasik, M.; Zeman, K.; Kilpatrick, D.C. Mannan-Binding Lectin Insufficiency in Children with Recurrent Infections of the Respiratory System. Clin. Exp. Immunol. 2004, 136, 304–311. [Google Scholar] [CrossRef]
- Schlapbach, L.J.; Aebi, C.; Otth, M.; Leibundgut, K.; Hirt, A.; Ammann, R.A. Deficiency of Mannose-Binding Lectin-Associated Serine Protease-2 Associated with Increased Risk of Fever and Neutropenia in Pediatric Cancer Patients. Pediatr. Infect. Dis. J. 2007, 26, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Schlapbach, L.J.; Aebi, C.; Hansen, A.G.; Hirt, A.; Jensenius, J.C.; Ammann, R.A. H-Ficolin Serum Concentration and Susceptibility to Fever and Neutropenia in Paediatric Cancer Patients. Clin. Exp. Immunol. 2009, 157, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Sallenbach, S.; Thiel, S.; Aebi, C.; Otth, M.; Bigler, S.; Jensenius, J.C.; Schlapbach, L.J.; Ammann, R.A. Serum Concentrations of Lectin-Pathway Components in Healthy Neonates, Children and Adults: Mannan-Binding Lectin (MBL), M-, L-, and H-Ficolin, and MBL-Associated Serine Protease-2 (MASP-2). Pediatr. Allergy Immunol. 2011, 22, 424–430. [Google Scholar] [CrossRef] [PubMed]
- West, E.E.; Woodruff, T.; Fremeaux-Bacchi, V.; Kemper, C. Complement in Human Disease: Approved and up-and-Coming Therapeutics. Lancet 2023. [Google Scholar] [CrossRef]
- Gadek, J.E.; Hosea, S.W.; Gelfand, J.A.; Santaella, M.; Wickerhauser, M.; Triantaphyllopoulos, D.C.; Frank, M.M. Replacement Therapy in Hereditary Angioedema: Successful Treatment of Acute Episodes of Angioedema with Partly Purified C1 Inhibitor. N. Engl. J. Med. 1980, 302, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Zuraw, B.L.; Busse, P.J.; White, M.; Jacobs, J.; Lumry, W.; Baker, J.; Craig, T.; Grant, J.A.; Hurewitz, D.; Bielory, L.; et al. Nanofiltered C1 Inhibitor Concentrate for Treatment of Hereditary Angioedema. N. Engl. J. Med. 2010, 363, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Delaura, I.F.; Gao, Q.; Anwar, I.J.; Abraham, N.; Kahan, R.; Hartwig, M.G.; Barbas, A.S. Complement-Targeting Therapeutics for Ischemia-Reperfusion Injury in Transplantation and the Potential for Ex Vivo Delivery. Front. Immunol. 2022, 13, 1000172. [Google Scholar] [CrossRef]
- Jordan, S.C.; Choi, J.; Aubert, O.; Haas, M.; Loupy, A.; Huang, E.; Peng, A.; Kim, I.; Louie, S.; Ammerman, N.; et al. A Phase I/II, Double-Blind, Placebo-Controlled Study Assessing Safety and Efficacy of C1 Esterase Inhibitor for Prevention of Delayed Graft Function in Deceased Donor Kidney Transplant Recipients. Am. J. Transplant. 2018, 18, 2955–2964. [Google Scholar] [CrossRef]
- Huang, E.; Vo, A.; Choi, J.; Ammerman, N.; Lim, K.; Sethi, S.; Kim, I.; Kumar, S.; Najjar, R.; Peng, A.; et al. Three-Year Outcomes of a Randomized, Double-Blind, Placebo-Controlled Study Assessing Safety and Efficacy of C1 Esterase Inhibitor for Prevention of Delayed Graft Function in Deceased Donor Kidney Transplant Recipients. Clin. J. Am. Soc. Nephrol. 2020, 15, 109–116. [Google Scholar] [CrossRef]
- Keizer, M.P.; Kamp, A.; van Mierlo, G.; Kuijpers, T.W.; Wouters, D. Substitution of Mannan-Binding Lectin (MBL)-Deficient Serum With Recombinant MBL Results in the Formation of New MBL/MBL-Associated Serine Protease Complexes. Front. Immunol. 2018, 9, 1406. [Google Scholar] [CrossRef]
- Willrich, M.A.V.; Braun, K.M.P.; Moyer, A.M.; Jeffrey, D.H.; Frazer-Abel, A. Complement Testing in the Clinical Laboratory. Crit. Rev. Clin. Lab. Sci. 2021, 58, 447–478. [Google Scholar] [CrossRef] [PubMed]
- Seelen, M.A.; Roos, A.; Wieslander, J.; Mollnes, T.E.; Sjöholm, A.G.; Wurzner, R.; Loos, M.; Tedesco, F.; Sim, R.B.; Garred, P.; et al. Functional Analysis of the Classical, Alternative, and MBL Pathways of the Complement System: Standardization and Validation of a Simple ELISA. J. Immunol. Methods 2005, 296, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Kilpatrick, D.; Shiraki, H.; Liu, Y.; Tateishi, K.; Tsujimura, M.; Endo, Y.; Fujita, T. Purification, Measurement of Concentration, and Functional Complement Assay of Human Ficolins. Methods Mol. Biol. 2014, 1100, 141–159. [Google Scholar] [CrossRef] [PubMed]
- Hein, E.; Bay, J.T.; Munthe-Fog, L.; Garred, P. Ficolin-2 Reveals Different Analytical and Biological Properties Dependent on Different Sample Handling Procedures. Mol. Immunol. 2013, 56, 406–412. [Google Scholar] [CrossRef]
- Inoshita, H.; Matsushita, M.; Koide, S.; Kusaba, G.; Ishii, M.; Onda, K.; Gi, M.J.; Nakata, M.; Ohsawa, I.; Horikoshi, S.; et al. A Novel Measurement Method for Activation of the Lectin Complement Pathway via Both Mannose-Binding Lectin (MBL) and L-Ficolin. J. Immunol. Methods 2009, 349, 9–17. [Google Scholar] [CrossRef]
- Sirmaci, A.; Walsh, T.; Akay, H.; Spiliopoulos, M.; Sakalar, Y.B.; Hasanefendioğlu-Bayrak, A.; Duman, D.; Farooq, A.; King, M.-C.; Tekin, M. MASP1 Mutations in Patients with Facial, Umbilical, Coccygeal, and Auditory Findings of Carnevale, Malpuech, OSA, and Michels Syndromes. Am. J. Hum. Genet. 2010, 87, 679–686. [Google Scholar] [CrossRef]
- Rooryck, C.; Diaz-Font, A.; Osborn, D.P.S.; Chabchoub, E.; Hernandez-Hernandez, V.; Shamseldin, H.; Kenny, J.; Waters, A.; Jenkins, D.; Kaissi, A.A.; et al. Mutations in Lectin Complement Pathway Genes COLEC11 and MASP1 Cause 3MC Syndrome. Nat. Genet. 2011, 43, 197–203. [Google Scholar] [CrossRef]
- Atik, T.; Koparir, A.; Bademci, G.; Foster, J.; Altunoglu, U.; Mutlu, G.Y.; Bowdin, S.; Elcioglu, N.; Tayfun, G.A.; Atik, S.S.; et al. Novel MASP1 Mutations Are Associated with an Expanded Phenotype in 3MC1 Syndrome. Orphanet J. Rare Dis. 2015, 10, 128. [Google Scholar] [CrossRef]
- Cortesio, C.L.; Jiang, W. Mannan-Binding Lectin-Associated Serine Protease 3 Cleaves Synthetic Peptides and Insulin-like Growth Factor-Binding Protein 5. Arch. Biochem. Biophys. 2006, 449, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Busby, W.H.; Nam, T.J.; Moralez, A.; Smith, C.; Jennings, M.; Clemmons, D.R. The Complement Component C1s Is the Protease That Accounts for Cleavage of Insulin-like Growth Factor-Binding Protein-5 in Fibroblast Medium. J. Biol. Chem. 2000, 275, 37638–37644. [Google Scholar] [CrossRef] [PubMed]
- Kusakari, K.; Machida, T.; Ishida, Y.; Omori, T.; Suzuki, T.; Sekimata, M.; Wada, I.; Fujita, T.; Sekine, H. The Complex Formation of MASP-3 with Pattern Recognition Molecules of the Lectin Complement Pathway Retains MASP-3 in the Circulation. Front. Immunol. 2022, 13, 907023. [Google Scholar] [CrossRef]
- Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Mantovani, A.; Lambris, J.D. Complement in Cancer: Untangling an Intricate Relationship. Nat. Rev. Immunol. 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautès-Fridman, C.; Fridman, W.H. Context-Dependent Roles of Complement in Cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef]
- DeRycke, M.S.; Gunawardena, S.R.; Middha, S.; Asmann, Y.W.; Schaid, D.J.; McDonnell, S.K.; Riska, S.M.; Eckloff, B.W.; Cunningham, J.M.; Fridley, B.L.; et al. Identification of Novel Variants in Colorectal Cancer Families by High-Throughput Exome Sequencing. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Fraile, J.M.; Ordóñez, G.R.; Quirós, P.M.; Astudillo, A.; Galván, J.A.; Colomer, D.; López-Otín, C.; Freije, J.M.P.; Puente, X.S. Identification of Novel Tumor Suppressor Proteases by Degradome Profiling of Colorectal Carcinomas. Oncotarget 2013, 4, 1919–1932. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Guo, H.; Gong, H.; Xue, T.; Wang, X.; Tang, C.; Xu, Y.; Dai, C.; Bao, Y.; Zhang, T.; et al. Comprehensive Transcriptome Analysis of Colorectal Cancer Risk of Hyperglycemia in Humans. J. Gastrointest. Oncol. 2021, 12, 602–619. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Matrisian, L.M. Emerging Roles of Proteases in Tumour Suppression. Nat. Rev. Cancer 2007, 7, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Cedzyński, M.; Świerzko, A.S. Components of the Lectin Pathway of Complement in Solid Tumour Cancers. Cancers 2022, 14, 1543. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Yu, T.-H.; Wu, C.-C.; Hung, W.-C.; Lee, T.-L.; Tang, W.-H.; Tsai, I.-T.; Chung, F.-M.; Lee, Y.-J.; Hsu, C.-C. Loss of Ficolin-3 Expression Is Associated with Poor Prognosis in Patients with Hepatocellular Carcinoma. Int. J. Med. Sci. 2023, 20, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Liu, P.; Wen, J.; Gu, Y.; Yang, Z.; Lan, J.; Fan, H.; Liu, Z.; Guo, D. FCN3 Inhibits the Progression of Hepatocellular Carcinoma by Suppressing SBDS-Mediated Blockade of the P53 Pathway. Int. J. Biol. Sci. 2023, 19, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The Fungal Mycobiome Promotes Pancreatic Oncogenesis via Activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorders | LP Components Involved | Drugs under Clinical Trial | Drug Candidates under Preclinical Trial |
|---|---|---|---|
| Renal disorders | |||
| IgA nephropathy | MBL, ficolin-2, MASP-1, MASP-2, MASP-3, MAP-19 [145,146,147,148,149] | Narsoplimab (recently discontinued) [150,151] | Anti-MASP-3 (OMS906) [152] |
| Membranous nephropathy | MBL, MASP-1, MASP-2 [153,154,155,156] | Narsoplimab [157] | |
| Diabetic kidney disease | MBL, ficolin-3 [158,159] | Anti-MBL MAb [160] | |
| Ischemia Reperfusion Injury | |||
| Renal IRI | MBL, CL-K1, MASP-2 [76,77,78,161,162,163,164,165] | CL-K1 inhibition by L-fucose; C1INH; TFMI-2 [166,167,168,169] | |
| Myocardial IRI | MBL, ficolin-2, MASP-1, MASP-2 [138,170,171,172,173,174,175,176] | C1INH; anti-MBL MAb [177,178] | |
| Ischemic stroke | MBL, MASP-1, MASP-2 [179,180,181,182] | ||
| Atherosclerosis | MBL, ficolin-1, ficolin-2, ficolin-3, MASP-3 [183,184,185] | ||
| COVID-19 | MBL, ficolin-2, ficolin-3, MASP-2 [186,187,188,189,190,191] | Narsoplimab * [192] | |
| Hereditary angioedema | MASP-1 [193,194] | C1INH (approved) ** [195] | |
| Schizophrenia | MBL, ficolin-2, MASP-2 [59,196] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobó, J.; Kocsis, A.; Farkas, B.; Demeter, F.; Cervenak, L.; Gál, P. The Lectin Pathway of the Complement System—Activation, Regulation, Disease Connections and Interplay with Other (Proteolytic) Systems. Int. J. Mol. Sci. 2024, 25, 1566. https://doi.org/10.3390/ijms25031566
Dobó J, Kocsis A, Farkas B, Demeter F, Cervenak L, Gál P. The Lectin Pathway of the Complement System—Activation, Regulation, Disease Connections and Interplay with Other (Proteolytic) Systems. International Journal of Molecular Sciences. 2024; 25(3):1566. https://doi.org/10.3390/ijms25031566
Chicago/Turabian StyleDobó, József, Andrea Kocsis, Bence Farkas, Flóra Demeter, László Cervenak, and Péter Gál. 2024. "The Lectin Pathway of the Complement System—Activation, Regulation, Disease Connections and Interplay with Other (Proteolytic) Systems" International Journal of Molecular Sciences 25, no. 3: 1566. https://doi.org/10.3390/ijms25031566
APA StyleDobó, J., Kocsis, A., Farkas, B., Demeter, F., Cervenak, L., & Gál, P. (2024). The Lectin Pathway of the Complement System—Activation, Regulation, Disease Connections and Interplay with Other (Proteolytic) Systems. International Journal of Molecular Sciences, 25(3), 1566. https://doi.org/10.3390/ijms25031566