
Age-Related Macular Degeneration and Mitochondria-Associated Autoantibodies: A Review of the Specific Pathogenesis and Therapeutic Strategies


Abstract
:1. Introduction
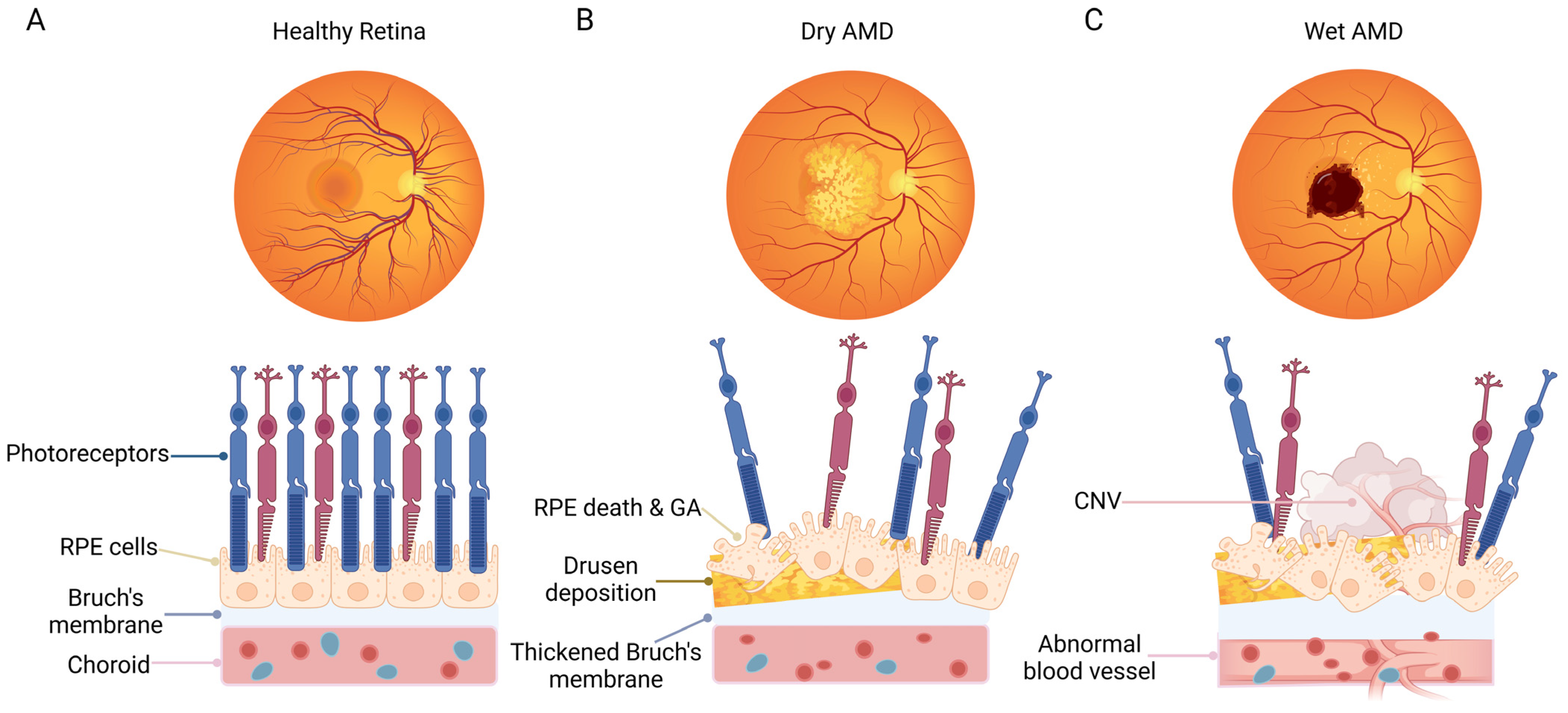
1.1. Overview of AMD
1.2. Functions of RPE Cells
2. Autoimmune Processes in AMD
3. Mitochondria-Related RPE Physiology and AMD Pathology
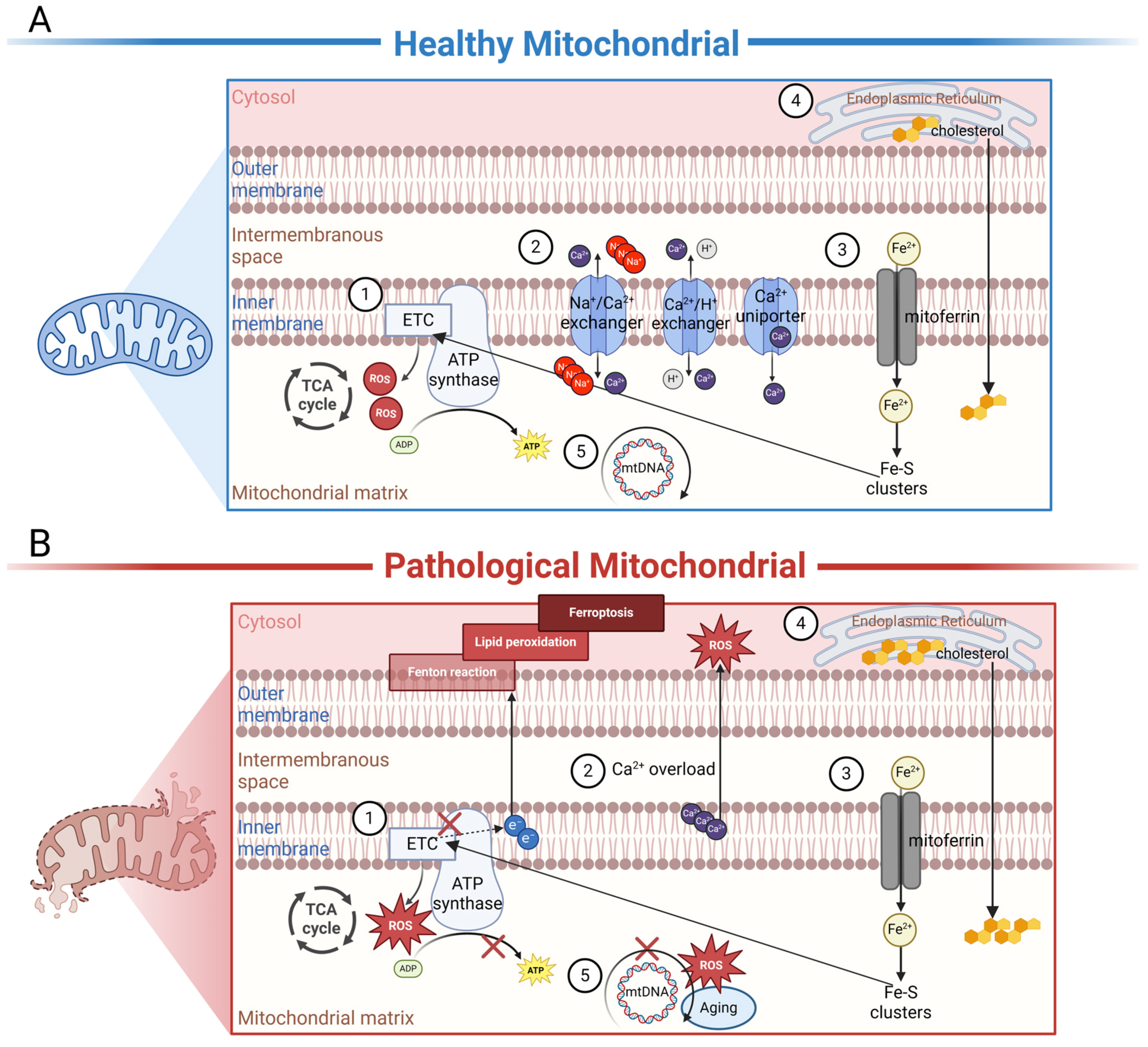
3.1. The Function of Mitochondria in the Normal RPE
3.2. The Mitochondria-Related Pathogenesis in AMD
4. The Crosstalk between Mitochondria and Other Organelles
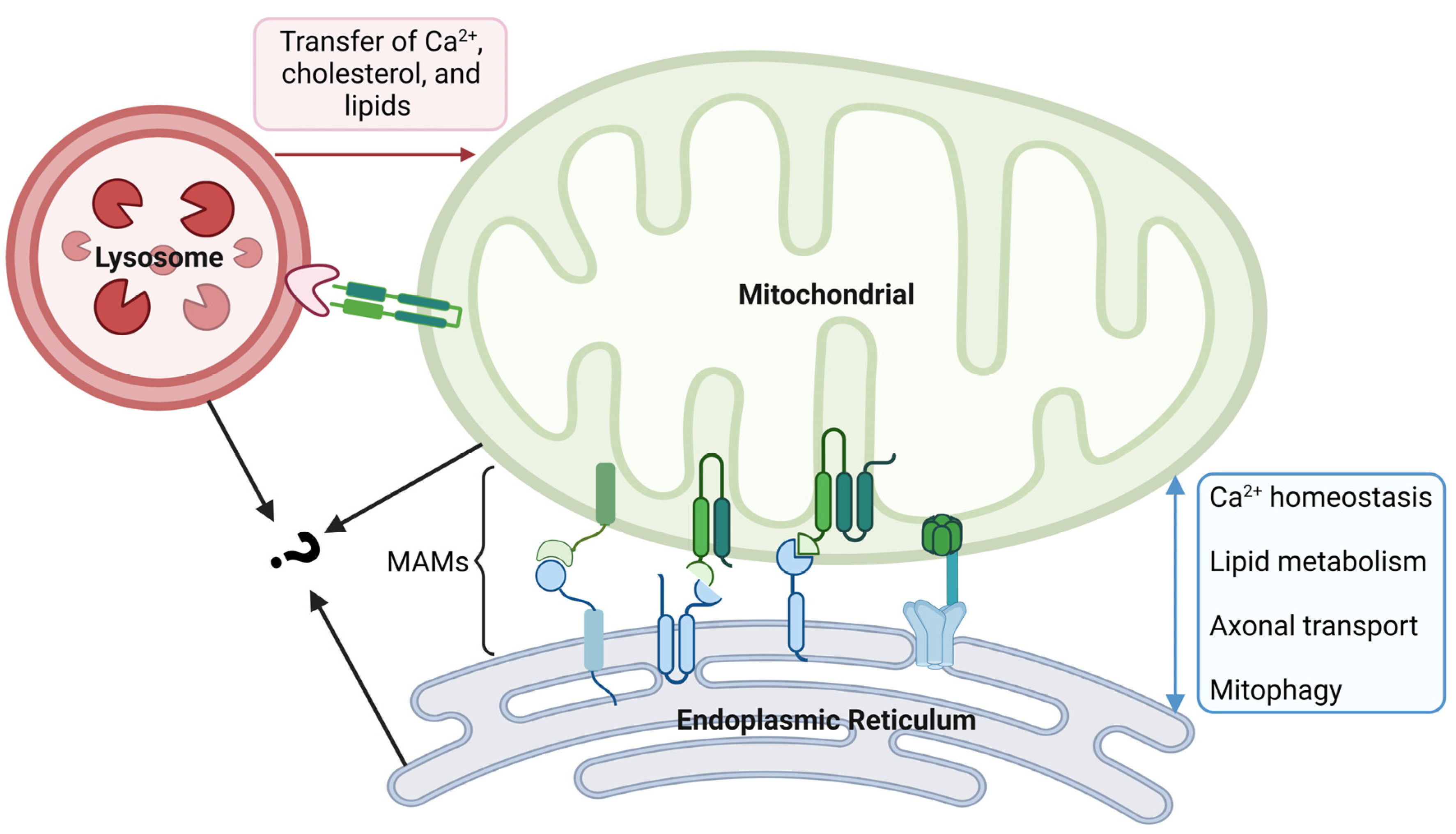
4.1. Mitochondria and the Endoplasmic Reticulum (ER)
4.2. Mitochondria and Lysosomes
5. Therapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid β |
| AD | Alzheimer’s disease |
| Alpha-enolase | α-enolase |
| Alpha-synuclein | α-syn |
| AMD | age-related macular degeneration |
| APO | apolipoprotein |
| ATP | adenosine triphosphate |
| CNV | choroidal neovascularization |
| DNA | deoxyribonucleic acid |
| ER | endoplasmic reticulum |
| ETC | electron transport chain |
| Fe-S | iron–sulfur |
| GA | geographic atrophy |
| GFAP | glial fibrillary acid protein |
| H2O2 | hydrogen peroxide |
| IMM | inner mitochondrial membrane |
| IMS | intermembrane space |
| MAMs | mitochondria-associated membranes |
| MDPs | mitochondria-derived peptides |
| mtDNA | mitochondrial DNA |
| mTOR | mammalian target of rapamycin |
| mPTP | mitochondrial permeability transition pore |
| NDI1 | NADH–ubiquinone oxidoreductase |
| NRFs | nuclear respiratory factors |
| O2− | superoxide anion radical |
| OH· | hydroxyl radicals |
| OxPhos | oxidative phosphorylation |
| PGC | peroxisome proliferator-activated receptor γ coactivator |
| PR | photoreceptors |
| RNA | ribonucleic acid |
| RO· | alkoxy radical |
| ROO· | peroxyl radical |
| ROS | reactive oxygen species |
| RPE | retinal pigment epithelium |
| SHLPs | small humanin-like peptides |
| TCA | tricarboxylic acid cycle |
| TF | transferrin |
| TFAM | mitochondrial transcription factor A |
| TFR | transferrin receptor |
| TSPO | transporters |
| VEGF | vascular endothelial growth factor |
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Jacob, L.; Spiess, A.; Kostev, K. Prevalence of depression, anxiety, adjustment disorders, and somatoform disorders in patients with age-related macular degeneration in Germany. Ger. Med. Sci. 2017, 15, Doc04. [Google Scholar] [CrossRef]
- Ferris, F.L., 3rd; Wilkinson, C.P.; Bird, A.; Chakravarthy, U.; Chew, E.; Csaky, K.; Sadda, S.R.; Beckman Initiative for Macular Research Classification Committee. Clinical classification of age-related macular degeneration. Ophthalmology 2013, 120, 844–851. [Google Scholar] [CrossRef]
- Mitchell, P.; Liew, G.; Gopinath, B.; Wong, T.Y. Age-related macular degeneration. Lancet 2018, 392, 1147–1159. [Google Scholar] [CrossRef]
- Blasiak, J.; Pawlowska, E.; Sobczuk, A.; Szczepanska, J.; Kaarniranta, K. The Aging Stress Response and Its Implication for AMD Pathogenesis. Int. J. Mol. Sci. 2020, 21, 8840. [Google Scholar] [CrossRef]
- Deangelis, M.M.; Silveira, A.C.; Carr, E.A.; Kim, I.K. Genetics of age-related macular degeneration: Current concepts, future directions. Semin. Ophthalmol. 2011, 26, 77–93. [Google Scholar] [CrossRef]
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.; Edwards, R.; Mitchell, P.; Harrison, R.A.; Buchan, I.; Kelly, S.P. Smoking and age-related macular degeneration: A review of association. Eye 2005, 19, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Armento, A.; Ueffing, M.; Clark, S.J. The complement system in age-related macular degeneration. Cell. Mol. Life Sci. 2021, 78, 4487–4505. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. Primers 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Butcher, E.; Saint-Geniez, M. EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2020, 21, 4271. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Bandello, F.; Navarra, P.; Staurenghi, G.; Stumpp, M.; Zarbin, M. Neovascular Age-Related Macular Degeneration: Therapeutic Management and New-Upcoming Approaches. Int. J. Mol. Sci. 2020, 21, 8242. [Google Scholar] [CrossRef]
- Ammar, M.J.; Hsu, J.; Chiang, A.; Ho, A.C.; Regillo, C.D. Age-related macular degeneration therapy: A review. Curr. Opin. Ophthalmol. 2020, 31, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, G.M.; Kijlstra, A.; Peek, R.; de Vos, A.F. Retinal pigment epithelium-immune system interactions: Cytokine production and cytokine-induced changes. Prog. Retin. Eye Res. 2001, 20, 29–48. [Google Scholar] [CrossRef] [PubMed]
- McBee, J.K.; Van Hooser, J.P.; Jang, G.F.; Palczewski, K. Isomerization of 11-cis-retinoids to all-trans-retinoids in vitro and in vivo. J. Biol. Chem. 2001, 276, 48483–48493. [Google Scholar] [CrossRef]
- Wimmers, S.; Karl, M.O.; Strauss, O. Ion channels in the RPE. Prog. Retin. Eye Res. 2007, 26, 263–301. [Google Scholar] [CrossRef]
- Blasiak, J.; Sobczuk, P.; Pawlowska, E.; Kaarniranta, K. Interplay between aging and other factors of the pathogenesis of age-related macular degeneration. Ageing Res. Rev. 2022, 81, 101735. [Google Scholar] [CrossRef]
- Fisher, C.R.; Ferrington, D.A. Perspective on AMD Pathobiology: A Bioenergetic Crisis in the RPE. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD41–AMD47. [Google Scholar] [CrossRef]
- Allingham, M.J.; Loksztejn, A.; Cousins, S.W.; Mettu, P.S. Immunological Aspects of Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2021, 1256, 143–189. [Google Scholar] [CrossRef] [PubMed]
- Penfold, P.L.; Provis, J.M.; Furby, J.H.; Gatenby, P.A.; Billson, F.A. Autoantibodies to retinal astrocytes associated with age-related macular degeneration. Graefe’s Arch. Clin. Exp. Ophthalmol. 1990, 228, 270–274. [Google Scholar] [CrossRef]
- Ezzat, M.K.; Hann, C.R.; Vuk-Pavlovic, S.; Pulido, J.S. Immune cells in the human choroid. Br. J. Ophthalmol. 2008, 92, 976–980. [Google Scholar] [CrossRef]
- Moir, J.; Hyman, M.J.; Wang, J.; Shah, A.; Maatouk, C.; Flores, A.; Skondra, D. Associations Between Autoimmune Disease and the Development of Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2023, 64, 45. [Google Scholar] [CrossRef]
- Morohoshi, K.; Goodwin, A.M.; Ohbayashi, M.; Ono, S.J. Autoimmunity in retinal degeneration: Autoimmune retinopathy and age-related macular degeneration. J. Autoimmun. 2009, 33, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Korb, C.A.; Beck, S.; Wolters, D.; Lorenz, K.; Pfeiffer, N.; Grus, F.H. Serum Autoantibodies in Patients with Dry and Wet Age-Related Macular Degeneration. J. Clin. Med. 2023, 12, 1590. [Google Scholar] [CrossRef]
- Joachim, S.C.; Bruns, K.; Lackner, K.J.; Pfeiffer, N.; Grus, F.H. Analysis of IgG antibody patterns against retinal antigens and antibodies to alpha-crystallin, GFAP, and alpha-enolase in sera of patients with "wet" age-related macular degeneration. Graefe’s Arch. Clin. Exp. Ophthalmol. 2007, 245, 619–626. [Google Scholar] [CrossRef]
- Gonzalez, A.; Pariente, J.A.; Salido, G.M. Ethanol stimulates ROS generation by mitochondria through Ca2+ mobilization and increases GFAP content in rat hippocampal astrocytes. Brain Res. 2007, 1178, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhang, Z.; Lu, J.; Pei, G. Mitochondria Are Dynamically Transferring Between Human Neural Cells and Alexander Disease-Associated GFAP Mutations Impair the Astrocytic Transfer. Front. Cell. Neurosci. 2019, 13, 316. [Google Scholar] [CrossRef]
- Iida, H.a.I.Y. Yeast heat-shock protein of Mr 48,000 is an isoprotein of enolase. Nature 1985, 315, 688–690. [Google Scholar] [CrossRef]
- Magrys, A.; Anekonda, T.; Ren, G.; Adamus, G. The role of anti-alpha-enolase autoantibodies in pathogenicity of autoimmune-mediated retinopathy. J. Clin. Immunol. 2007, 27, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Li, H.; Cai, Y.; Ye, J.T.; Liu, Z.P.; Lu, J.; Huang, X.Y.; Feng, X.J.; Gao, H.; Chen, S.R.; et al. Mitochondrial binding of alpha-enolase stabilizes mitochondrial membrane: Its role in doxorubicin-induced cardiomyocyte apoptosis. Arch. Biochem. Biophys. 2014, 542, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Baksi, S.; Tripathi, A.K.; Singh, N. Alpha-synuclein modulates retinal iron homeostasis by facilitating the uptake of transferrin-bound iron: Implications for visual manifestations of Parkinson’s disease. Free. Radic. Biol. Med. 2016, 97, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Munoz, L.E.; Mallavarapu, M.; Herrmann, M.; Finnemann, S.C. Annexin A5 regulates surface alphavbeta5 integrin for retinal clearance phagocytosis. J. Cell Sci. 2019, 132, jcs232439. [Google Scholar] [CrossRef] [PubMed]
- La Cunza, N.; Tan, L.X.; Thamban, T.; Germer, C.J.; Rathnasamy, G.; Toops, K.A.; Lakkaraju, A. Mitochondria-dependent phase separation of disease-relevant proteins drives pathological features of age-related macular degeneration. JCI Insight 2021, 6, e142254. [Google Scholar] [CrossRef]
- Brown, E.E.; Lewin, A.S.; Ash, J.D. Mitochondria: Potential Targets for Protection in Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2018, 1074, 11–17. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Diaz, F.; Kotarsky, H.; Fellman, V.; Moraes, C.T. Mitochondrial disorders caused by mutations in respiratory chain assembly factors. Semin. Fetal Neonatal Med. 2011, 16, 197–204. [Google Scholar] [CrossRef]
- Riazi-Esfahani, M.; Kuppermann, B.D.; Kenney, M.C. The Role of Mitochondria in AMD: Current Knowledge and Future Applications. J. Ophthalmic Vis. Res. 2017, 12, 424–428. [Google Scholar] [CrossRef]
- Burgoyne, T.; Toms, M.; Way, C.; Tracey-White, D.; Futter, C.E.; Moosajee, M. Changes in Mitochondrial Size and Morphology in the RPE and Photoreceptors of the Developing and Ageing Zebrafish. Cells 2022, 11, 3542. [Google Scholar] [CrossRef] [PubMed]
- Adijanto, J.; Du, J.; Moffat, C.; Seifert, E.L.; Hurle, J.B.; Philp, N.J. The retinal pigment epithelium utilizes fatty acids for ketogenesis. J. Biol. Chem. 2014, 289, 20570–20582. [Google Scholar] [CrossRef]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Fiskum, G.; Lehninger, A.L. Regulated release of Ca2+ from respiring mitochondria by Ca2+/2H+ antiport. J. Biol. Chem. 1979, 254, 6236–6239. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef]
- Olson, M.L.; Chalmers, S.; McCarron, J.G. Mitochondrial organization and Ca2+ uptake. Biochem. Soc. Trans. 2012, 40, 158–167. [Google Scholar] [CrossRef]
- Rosenthal, R.; Strauss, O. Ca2+-channels in the RPE. Adv. Exp. Med. Biol. 2002, 514, 225–235. [Google Scholar] [PubMed]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A human mitochondrial ferritin encoded by an intronless gene. J. Biol. Chem. 2001, 276, 24437–24440. [Google Scholar] [CrossRef] [PubMed]
- Yefimova, M.G.; Jeanny, J.C.; Guillonneau, X.; Keller, N.; Nguyen-Legros, J.; Sergeant, C.; Guillou, F.; Courtois, Y. Iron, ferritin, transferrin, and transferrin receptor in the adult rat retina. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2343–2351. [Google Scholar]
- Hunt, R.C.; Dewey, A.; Davis, A.A. Transferrin receptors on the surfaces of retinal pigment epithelial cells are associated with the cytoskeleton. J. Cell Sci. 1989, 92 Pt 4, 655–666. [Google Scholar] [CrossRef]
- Martin, L.A.; Kennedy, B.E.; Karten, B. Mitochondrial cholesterol: Mechanisms of import and effects on mitochondrial function. J. Bioenerg. Biomembr. 2016, 48, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, E. Mechanisms for cellular cholesterol transport: Defects and human disease. Physiol. Rev. 2006, 86, 1237–1261. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Rupprecht, R.; Papadopoulos, V.; Rammes, G.; Baghai, T.C.; Fan, J.; Akula, N.; Groyer, G.; Adams, D.; Schumacher, M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2010, 9, 971–988. [Google Scholar] [CrossRef]
- Storti, F.; Raphael, G.; Griesser, V.; Klee, K.; Drawnel, F.; Willburger, C.; Scholz, R.; Langmann, T.; von Eckardstein, A.; Fingerle, J.; et al. Regulated efflux of photoreceptor outer segment-derived cholesterol by human RPE cells. Exp. Eye Res. 2017, 165, 65–77. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Garcia, I.; Jones, E.; Ramos, M.; Innis-Whitehouse, W.; Gilkerson, R. The little big genome: The organization of mitochondrial DNA. Front. Biosci. 2017, 22, 710–721. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef]
- Yan, C.; Duanmu, X.; Zeng, L.; Liu, B.; Song, Z. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells 2019, 8, 379. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.T.; Viiri, J.; Kaarniranta, K.; Blasiak, J. Mitochondrial quality control in AMD: Does mitophagy play a pivotal role? Cell. Mol. Life Sci. 2018, 75, 2991–3008. [Google Scholar] [CrossRef]
- Lewis Lujan, L.M.; McCarty, M.F.; Di Nicolantonio, J.J.; Galvez Ruiz, J.C.; Rosas-Burgos, E.C.; Plascencia-Jatomea, M.; Iloki Assanga, S.B. Nutraceuticals/Drugs Promoting Mitophagy and Mitochondrial Biogenesis May Combat the Mitochondrial Dysfunction Driving Progression of Dry Age-Related Macular Degeneration. Nutrients 2022, 14, 1985. [Google Scholar] [CrossRef]
- Wei, Q.; Hu, W.; Lou, Q.; Yu, J. NAD+ inhibits the metabolic reprogramming of RPE cells in early AMD by upregulating mitophagy. Discov. Med. 2019, 27, 189–196. [Google Scholar]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Mehrzadi, S.; Hemati, K.; Reiter, R.J.; Hosseinzadeh, A. Mitochondrial dysfunction in age-related macular degeneration: Melatonin as a potential treatment. Expert Opin. Ther. Targets 2020, 24, 359–378. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1alpha Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Jeong, Y.; Son, S.; Kim, D.E. AMPK-induced mitochondrial biogenesis decelerates retinal pigment epithelial cell degeneration under nutrient starvation. BMB Rep. 2023, 56, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Ma, J.; Xu, G.; Sun, Z. SHP-1 knockdown suppresses mitochondrial biogenesis and aggravates mitochondria-dependent apoptosis induced by all trans retinal through the STING/AMPK pathways. Mol. Med. 2022, 28, 125. [Google Scholar] [CrossRef] [PubMed]
- Rozing, M.P.; Durhuus, J.A.; Krogh Nielsen, M.; Subhi, Y.; Kirkwood, T.B.; Westendorp, R.G.; Sorensen, T.L. Age-related macular degeneration: A two-level model hypothesis. Prog. Retin. Eye Res. 2020, 76, 100825. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.; van Driel, D.; Valter, K.; Rees, S.; Provis, J. The locations of mitochondria in mammalian photoreceptors: Relation to retinal vasculature. Brain Res. 2008, 1189, 58–69. [Google Scholar] [CrossRef]
- Hiona, A.; Leeuwenburgh, C. The role of mitochondrial DNA mutations in aging and sarcopenia: Implications for the mitochondrial vicious cycle theory of aging. Exp. Gerontol. 2008, 43, 24–33. [Google Scholar] [CrossRef]
- Wong, R.; Steenbergen, C.; Murphy, E. Mitochondrial permeability transition pore and calcium handling. Methods Mol. Biol. 2012, 810, 235–242. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef]
- Smaili, S.S.; Hsu, Y.T.; Youle, R.J.; Russell, J.T. Mitochondria in Ca2+ signaling and apoptosis. J. Bioenerg. Biomembr. 2000, 32, 35–46. [Google Scholar] [CrossRef]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. The plasma membrane calcium pump in health and disease. FEBS J. 2013, 280, 5385–5397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hui, Y.N.; Wang, Y.S.; Ma, J.X.; Wang, J.B.; Ma, L.N. Calcium overload is associated with lipofuscin formation in human retinal pigment epithelial cells fed with photoreceptor outer segments. Eye 2011, 25, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Biesemeier, A.; Yoeruek, E.; Eibl, O.; Schraermeyer, U. Iron accumulation in Bruch’s membrane and melanosomes of donor eyes with age-related macular degeneration. Exp. Eye Res. 2015, 137, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Junemann, A.G.; Stopa, P.; Michalke, B.; Chaudhri, A.; Reulbach, U.; Huchzermeyer, C.; Schlotzer-Schrehardt, U.; Kruse, F.E.; Zrenner, E.; Rejdak, R. Levels of aqueous humor trace elements in patients with non-exsudative age-related macular degeneration: A case-control study. PLoS ONE 2013, 8, e56734. [Google Scholar] [CrossRef] [PubMed]
- Chowers, I.; Wong, R.; Dentchev, T.; Farkas, R.H.; Iacovelli, J.; Gunatilaka, T.L.; Medeiros, N.E.; Presley, J.B.; Campochiaro, P.A.; Curcio, C.A.; et al. The iron carrier transferrin is upregulated in retinas from patients with age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2135–2140. [Google Scholar] [CrossRef] [PubMed]
- Colak, E.; Zoric, L.; Radosavljevic, A.; Ignjatovic, S. The Association of Serum Iron-Binding Proteins and the Antioxidant Parameter Levels in Age-Related Macular Degeneration. Curr. Eye Res. 2018, 43, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Dentchev, T.; Hahn, P.; Dunaief, J.L. Strong labeling for iron and the iron-handling proteins ferritin and ferroportin in the photoreceptor layer in age-related macular degeneration. Arch. Ophthalmol. 2005, 123, 1745–1746. [Google Scholar] [CrossRef] [PubMed]
- Wysokinski, D.; Danisz, K.; Pawlowska, E.; Dorecka, M.; Romaniuk, D.; Robaszkiewicz, J.; Szaflik, M.; Szaflik, J.; Blasiak, J.; Szaflik, J.P. Transferrin receptor levels and polymorphism of its gene in age-related macular degeneration. Acta Biochim. Pol. 2015, 62, 177–184. [Google Scholar] [CrossRef]
- Liu, Y.; Bell, B.A.; Song, Y.; Kim, H.J.; Sterling, J.K.; Kim, B.J.; Poli, M.; Guo, M.; Zhang, K.; Rao, A.; et al. Intraocular iron injection induces oxidative stress followed by elements of geographic atrophy and sympathetic ophthalmia. Aging Cell 2021, 20, e13490. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Gan, B. Mitochondrial regulation of ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef] [PubMed]
- Krainz, T.; Gaschler, M.M.; Lim, C.; Sacher, J.R.; Stockwell, B.R.; Wipf, P. A Mitochondrial-Targeted Nitroxide Is a Potent Inhibitor of Ferroptosis. ACS Cent. Sci. 2016, 2, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef] [PubMed]
- Curcio, C.A.; Millican, C.L.; Bailey, T.; Kruth, H.S. Accumulation of cholesterol with age in human Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 2001, 42, 265–274. [Google Scholar]
- Curcio, C.A.; Johnson, M.; Rudolf, M.; Huang, J.D. The oil spill in ageing Bruch membrane. Br. J. Ophthalmol. 2011, 95, 1638–1645. [Google Scholar] [CrossRef]
- Chen, W.; Stambolian, D.; Edwards, A.O.; Branham, K.E.; Othman, M.; Jakobsdottir, J.; Tosakulwong, N.; Pericak-Vance, M.A.; Campochiaro, P.A.; Klein, M.L.; et al. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 7401–7406. [Google Scholar] [CrossRef]
- Fernandez-Checa, J.C.; Kaplowitz, N. Hepatic mitochondrial glutathione: Transport and role in disease and toxicity. Toxicol. Appl. Pharmacol. 2005, 204, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Biswas, L.; Zhou, X.; Dhillon, B.; Graham, A.; Shu, X. Retinal pigment epithelium cholesterol efflux mediated by the 18 kDa translocator protein, TSPO, a potential target for treating age-related macular degeneration. Hum. Mol. Genet. 2017, 26, 4327–4339. [Google Scholar] [CrossRef]
- Ananth, S.; Gnana-Prakasam, J.P.; Bhutia, Y.D.; Veeranan-Karmegam, R.; Martin, P.M.; Smith, S.B.; Ganapathy, V. Regulation of the cholesterol efflux transporters ABCA1 and ABCG1 in retina in hemochromatosis and by the endogenous siderophore 2,5-dihydroxybenzoic acid. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 603–612. [Google Scholar] [CrossRef]
- Lin, H.; Xu, H.; Liang, F.Q.; Liang, H.; Gupta, P.; Havey, A.N.; Boulton, M.E.; Godley, B.F. Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3521–3529. [Google Scholar] [CrossRef]
- Udar, N.; Atilano, S.R.; Memarzadeh, M.; Boyer, D.S.; Chwa, M.; Lu, S.; Maguen, B.; Langberg, J.; Coskun, P.; Wallace, D.C.; et al. Mitochondrial DNA haplogroups associated with age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2966–2974. [Google Scholar] [CrossRef] [PubMed]
- Leuthner, T.C.; Meyer, J.N. Mitochondrial DNA Mutagenesis: Feature of and Biomarker for Environmental Exposures and Aging. Curr. Environ. Health Rep. 2021, 8, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Chen, W.; Chen, L.; Li, L. Potential role of mitochondria-associated endoplasmic reticulum membrane proteins in diseases. Biochem. Pharmacol. 2022, 199, 115011. [Google Scholar] [CrossRef] [PubMed]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.W.; Mueller, S.; Miller, T.; Miller, C.C.J. There’s Something Wrong with my MAM; the ER-Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Csordas, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnoczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Ronnback, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef]
- Anderson, D.H.; Talaga, K.C.; Rivest, A.J.; Barron, E.; Hageman, G.S.; Johnson, L.V. Characterization of beta amyloid assemblies in drusen: The deposits associated with aging and age-related macular degeneration. Exp. Eye Res. 2004, 78, 243–256. [Google Scholar] [CrossRef]
- Csordas, G.; Weaver, D.; Hajnoczky, G. Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Janikiewicz, J.; Szymanski, J.; Malinska, D.; Patalas-Krawczyk, P.; Michalska, B.; Duszynski, J.; Giorgi, C.; Bonora, M.; Dobrzyn, A.; Wieckowski, M.R. Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell Death Dis. 2018, 9, 332. [Google Scholar] [CrossRef]
- Cisneros, J.; Belton, T.B.; Shum, G.C.; Molakal, C.G.; Wong, Y.C. Mitochondria-lysosome contact site dynamics and misregulation in neurodegenerative diseases. Trends Neurosci. 2022, 45, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef]
- Wong, Y.C.; Kim, S.; Peng, W.; Krainc, D. Regulation and Function of Mitochondria-Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol. 2019, 29, 500–513. [Google Scholar] [CrossRef]
- Martins, W.K.; Santos, N.F.; Rocha, C.S.; Bacellar, I.O.L.; Tsubone, T.M.; Viotto, A.C.; Matsukuma, A.Y.; Abrantes, A.B.P.; Siani, P.; Dias, L.G.; et al. Parallel damage in mitochondria and lysosomes is an efficient way to photoinduce cell death. Autophagy 2019, 15, 259–279. [Google Scholar] [CrossRef]
- Deane, K.D. Preclinical rheumatoid arthritis (autoantibodies): An updated review. Curr. Rheumatol. Rep. 2014, 16, 419. [Google Scholar] [CrossRef]
- Kubicka-Trzaska, A.; Wilanska, J.; Romanowska-Dixon, B.; Sanak, M. Circulating anti-retinal antibodies in response to anti-angiogenic therapy in exudative age-related macular degeneration. Acta Ophthalmol. 2014, 92, e610–e614. [Google Scholar] [CrossRef]
- Kubicka-Trzaska, A.; Wilanska, J.; Romanowska-Dixon, B.; Sanak, M. Circulating antiretinal antibodies predict the outcome of anti-VEGF therapy in patients with exudative age-related macular degeneration. Acta Ophthalmol. 2012, 90, e21–e24. [Google Scholar] [CrossRef]
- Evans, J.R.; Lawrenson, J.G. Antioxidant vitamin and mineral supplements for preventing age-related macular degeneration. Cochrane Database Syst. Rev. 2017, 7, CD000253. [Google Scholar] [CrossRef]
- Tokarz, P.; Kaarniranta, K.; Blasiak, J. Role of antioxidant enzymes and small molecular weight antioxidants in the pathogenesis of age-related macular degeneration (AMD). Biogerontology 2013, 14, 461–482. [Google Scholar] [CrossRef]
- Lu, L.; Oveson, B.C.; Jo, Y.J.; Lauer, T.W.; Usui, S.; Komeima, K.; Xie, B.; Campochiaro, P.A. Increased expression of glutathione peroxidase 4 strongly protects retina from oxidative damage. Antioxid. Redox Signal. 2009, 11, 715–724. [Google Scholar] [CrossRef]
- Rex, T.S.; Tsui, I.; Hahn, P.; Maguire, A.M.; Duan, D.; Bennett, J.; Dunaief, J.L. Adenovirus-mediated delivery of catalase to retinal pigment epithelial cells protects neighboring photoreceptors from photo-oxidative stress. Hum. Gene Ther. 2004, 15, 960–967. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, L.; Jiang, Y.; Silva, M.; Zhen, X.; Zheng, W. Protective Effect of Metformin against Hydrogen Peroxide-Induced Oxidative Damage in Human Retinal Pigment Epithelial (RPE) Cells by Enhancing Autophagy through Activation of AMPK Pathway. Oxidative Med. Cell. Longev. 2020, 2020, 2524174. [Google Scholar] [CrossRef] [PubMed]
- Landowski, M.; Bowes Rickman, C. Targeting Lipid Metabolism for the Treatment of Age-Related Macular Degeneration: Insights from Preclinical Mouse Models. J. Ocul. Pharmacol. Ther. 2022, 38, 3–32. [Google Scholar] [CrossRef]
- Ana, R.D.; Gliszczynska, A.; Sanchez-Lopez, E.; Garcia, M.L.; Krambeck, K.; Kovacevic, A.; Souto, E.B. Precision Medicines for Retinal Lipid Metabolism-Related Pathologies. J. Pers. Med. 2023, 13, 635. [Google Scholar] [CrossRef] [PubMed]
- Caron, A.; Richard, D.; Laplante, M. The Roles of mTOR Complexes in Lipid Metabolism. Annu. Rev. Nutr. 2015, 35, 321–348. [Google Scholar] [CrossRef] [PubMed]
- Nashine, S.; Kenney, M.C. Effects of Mitochondrial-Derived Peptides (MDPs) on Mitochondrial and Cellular Health in AMD. Cells 2020, 9, 1102. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, W.; Yang, H.; Zhang, J.; Ma, J. S14G-humanin restored cellular homeostasis disturbed by amyloid-beta protein. Neural Regen. Res. 2013, 8, 2573–2580. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Ishikawa, K.; Spee, C.; Mehta, H.H.; Wan, J.; Yen, K.; Cohen, P.; Kannan, R.; Hinton, D.R. The Mitochondrial-Derived Peptide Humanin Protects RPE Cells From Oxidative Stress, Senescence, and Mitochondrial Dysfunction. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1238–1253. [Google Scholar] [CrossRef]
- Okada, A.K.; Teranishi, K.; Lobo, F.; Isas, J.M.; Xiao, J.; Yen, K.; Cohen, P.; Langen, R. The Mitochondrial-Derived Peptides, HumaninS14G and Small Humanin-like Peptide 2, Exhibit Chaperone-like Activity. Sci. Rep. 2017, 7, 7802. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, T.A.C.; Georgiou, M.; Bainbridge, J.W.B.; Michaelides, M. Gene therapy for neovascular age-related macular degeneration: Rationale, clinical trials and future directions. Br. J. Ophthalmol. 2021, 105, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Millington-Ward, S.; Chadderton, N.; Finnegan, L.K.; Post, I.J.M.; Carrigan, M.; Nixon, R.; Humphries, M.M.; Humphries, P.; Kenna, P.F.; Palfi, A.; et al. RPE-Directed Gene Therapy Improves Mitochondrial Function in Murine Dry AMD Models. Int. J. Mol. Sci. 2023, 24, 3847. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Protein Name | MW (kDa) | Autoantibody-Related Mitochondrial Function | References |
|---|---|---|---|
| Alpha-enolase | 46 | ATP depletion and increase in intracellular Ca2+ | [25] |
| Alpha-synuclein | 14.5 | Regulating iron homeostasis | [25,26] |
| Annexin V | 35.9 | Ca2+ binding | [26] |
| ATP synthase | 56.6 | ATP synthesis | [25] |
| Glial fibrillary acidic protein | 52 | ROS generation and Ca2+ release | [21,26] |
| Malate dehydrogenase | 35.5 | Marker of the mitochondrial matrix | [25] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, S.; Lin, H.; Pfeiffer, N.; Grus, F.H. Age-Related Macular Degeneration and Mitochondria-Associated Autoantibodies: A Review of the Specific Pathogenesis and Therapeutic Strategies. Int. J. Mol. Sci. 2024, 25, 1624. https://doi.org/10.3390/ijms25031624
Qu S, Lin H, Pfeiffer N, Grus FH. Age-Related Macular Degeneration and Mitochondria-Associated Autoantibodies: A Review of the Specific Pathogenesis and Therapeutic Strategies. International Journal of Molecular Sciences. 2024; 25(3):1624. https://doi.org/10.3390/ijms25031624
Chicago/Turabian StyleQu, Sichang, Hao Lin, Norbert Pfeiffer, and Franz H. Grus. 2024. "Age-Related Macular Degeneration and Mitochondria-Associated Autoantibodies: A Review of the Specific Pathogenesis and Therapeutic Strategies" International Journal of Molecular Sciences 25, no. 3: 1624. https://doi.org/10.3390/ijms25031624
APA StyleQu, S., Lin, H., Pfeiffer, N., & Grus, F. H. (2024). Age-Related Macular Degeneration and Mitochondria-Associated Autoantibodies: A Review of the Specific Pathogenesis and Therapeutic Strategies. International Journal of Molecular Sciences, 25(3), 1624. https://doi.org/10.3390/ijms25031624