
Immunopathology of Pulmonary Mycobacterium tuberculosis Infection in a Humanized Mouse Model




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Validation of the Humanized Mice
2.2. Body Weight Changes in Mtb-Infected Humanized Mice
2.3. Kinetics of Bacterial Growth in the Organs of Mtb-Infected Humanized Mice
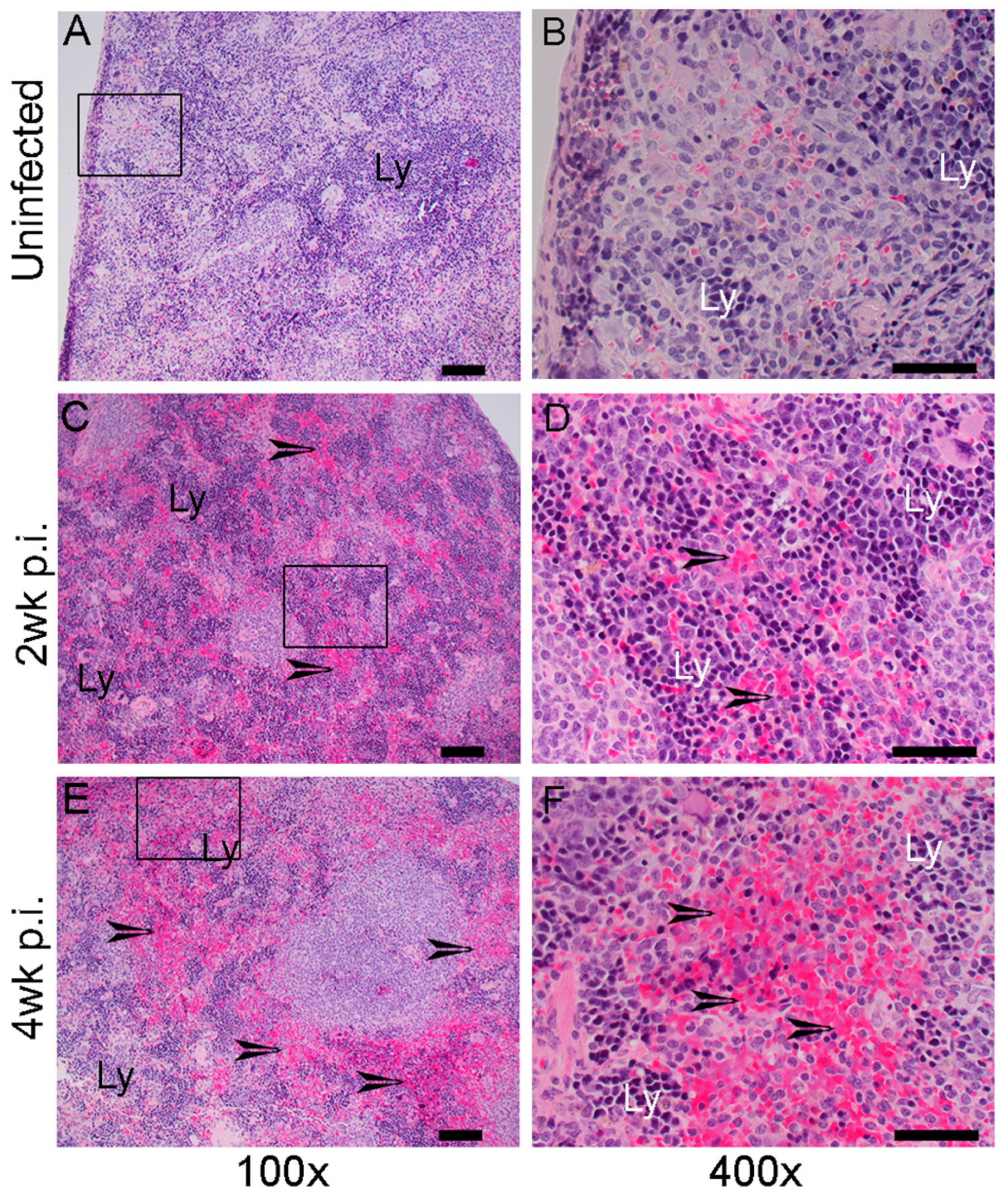
2.4. Histopathologic Changes in the Organs of Mtb-Infected Humanized Mice
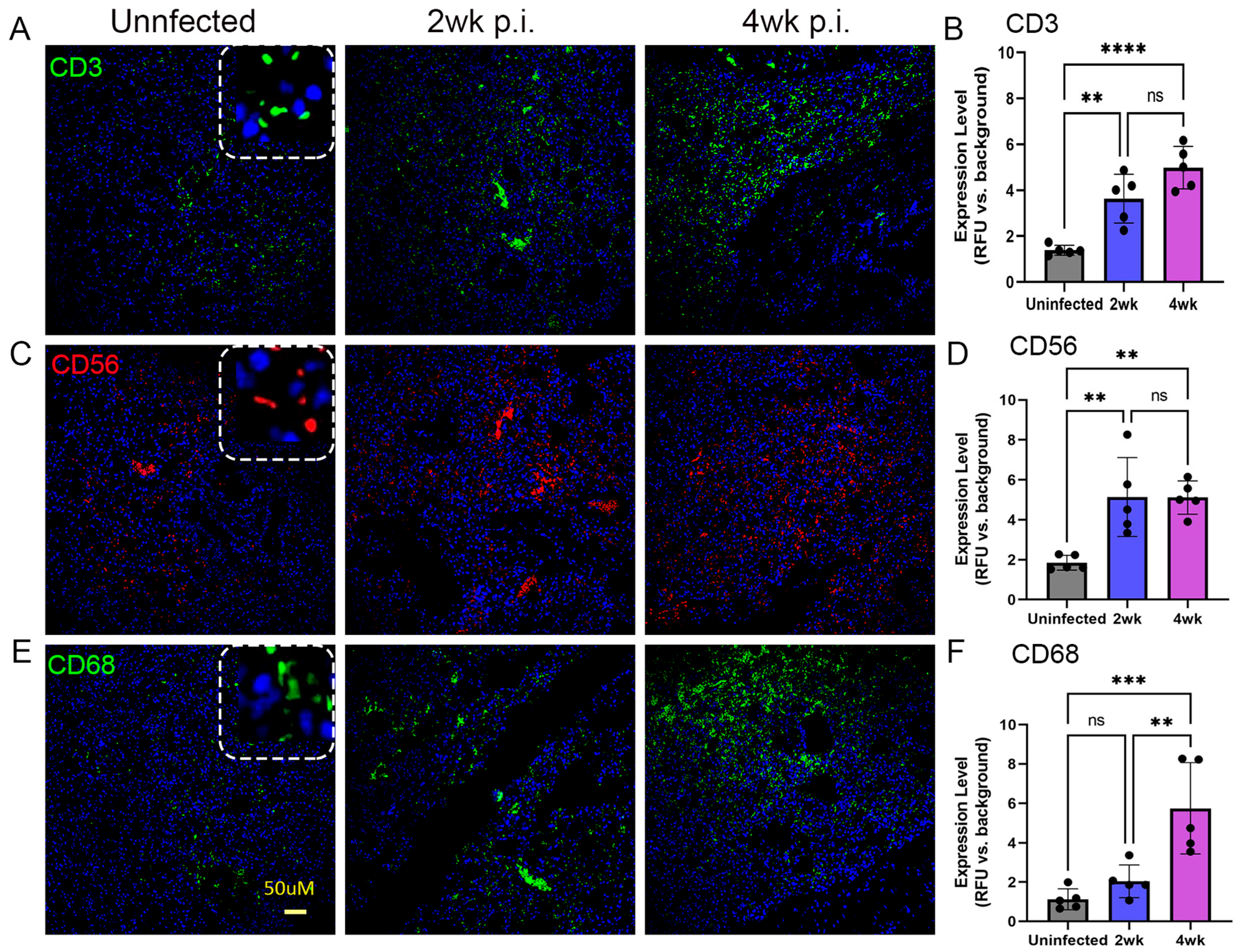
2.5. Spatial Distribution of Immune Cells in the Lungs of Mtb-Infected Humanized Mice
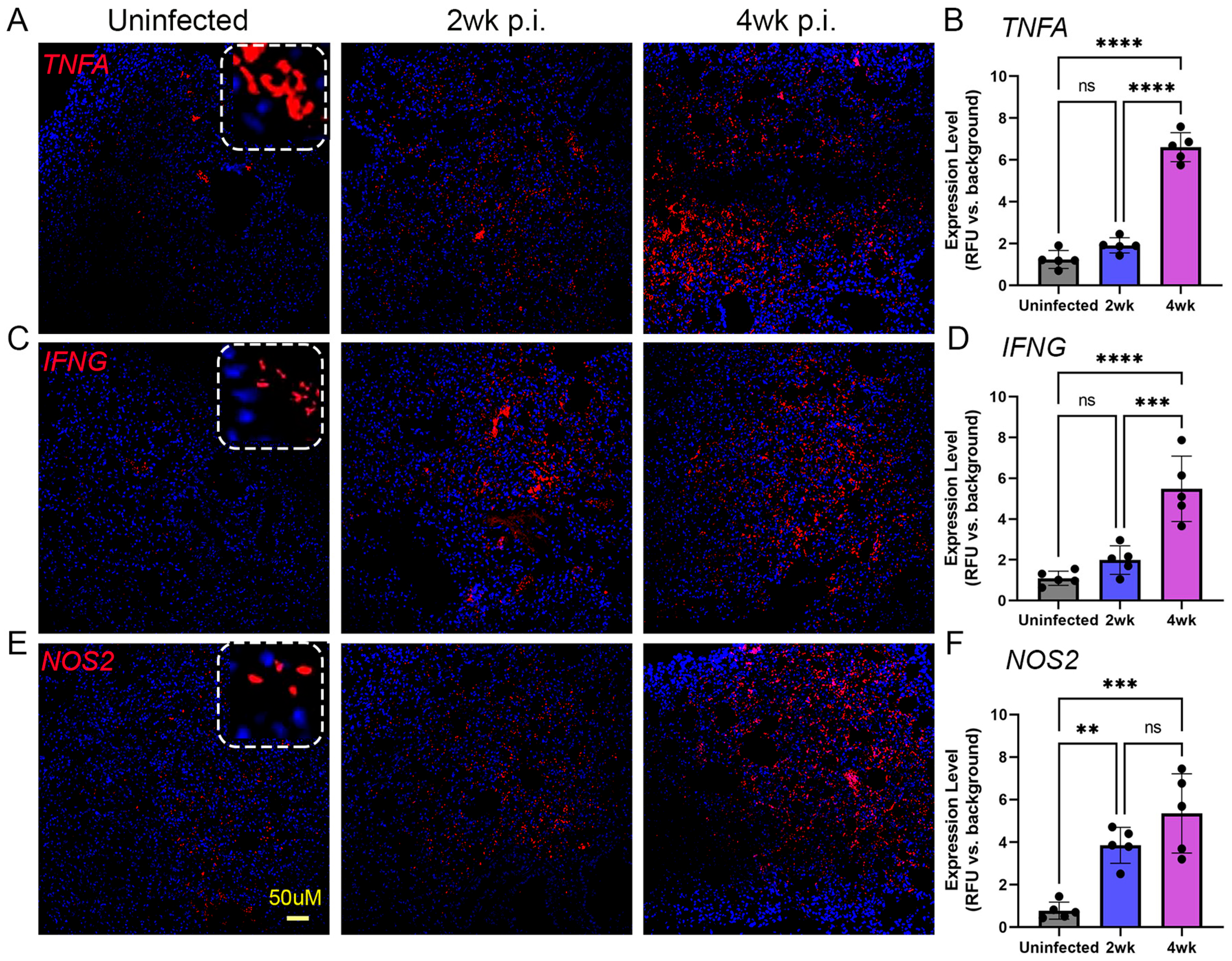
2.6. Spatial Expression of Immune Activation Markers in the Lungs of Mtb-Infected Humanized Mice
2.7. Spatial Expression of Antioxidant Markers in the Lungs of Mtb-Infected Humanized Mice
3. Discussion
4. Materials and Methods
4.1. Mycobacterium Tuberculosis Growth Conditions
4.2. Humanization of Mice
4.3. Humanized Mice Infection and Bacterial Load Determination
4.4. Histopathological Analysis
4.5. Spatial Immunohistochemistry Analysis
4.6. Spatial Gene Expression Analysis by Single-Molecule Fluorescent In Situ Hybridization (smFISH)
4.7. Spatial Imaging Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Global Tuberculosis (TB) Report; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Houben, R.M.; Dodd, P.J. The Global Burden of Latent Tuberculosis Infection: A Re-estimation Using Mathematical Modelling. PLoS Med. 2016, 13, e1002152. [Google Scholar] [CrossRef]
- Menzies, N.A.; Wolf, E.; Connors, D.; Bellerose, M.; Sbarra, A.N.; Cohen, T.; Hill, A.N.; Yaesoubi, R.; Galer, K.; White, P.J.; et al. Progression from latent infection to active disease in dynamic tuberculosis transmission models: A systematic review of the validity of modelling assumptions. Lancet Infect. Dis. 2018, 18, e228–e238. [Google Scholar] [CrossRef]
- Jarde, A.; Romano, E.; Afaq, S.; Elsony, A.; Lin, Y.; Huque, R.; Elsey, H.; Siddiqi, K.; Stubbs, B.; Siddiqi, N. Prevalence and risks of tuberculosis multimorbidity in low-income and middle-income countries: A meta-review. BMJ Open 2022, 12, e060906. [Google Scholar] [CrossRef]
- Vanino, E.; Granozzi, B.; Akkerman, O.W.; Munoz-Torrico, M.; Palmieri, F.; Seaworth, B.; Tiberi, S.; Tadolini, M. Update of drug-resistant tuberculosis treatment guidelines: A turning point. Int. J. Infect. Dis. 2023, 130 (Suppl. S1), S12–S15. [Google Scholar] [CrossRef]
- Cohen, S.B.; Gern, B.H.; Urdahl, K.B. The Tuberculous Granuloma and Preexisting Immunity. Annu. Rev. Immunol. 2022, 40, 589–614. [Google Scholar] [CrossRef]
- Nisa, A.; Kipper, F.C.; Panigrahy, D.; Tiwari, S.; Kupz, A.; Subbian, S. Different modalities of host cell death and their impact on Mycobacterium tuberculosis infection. Am. J. Physiol. Cell Physiol. 2022, 323, C1444–C1474. [Google Scholar] [CrossRef]
- Hunter, L.; Ruedas-Torres, I.; Agullo-Ros, I.; Rayner, E.; Salguero, F.J. Comparative pathology of experimental pulmonary tuberculosis in animal models. Front. Vet. Sci. 2023, 10, 1264833. [Google Scholar] [CrossRef]
- Klever, A.M.; Alexander, K.A.; Almeida, D.; Anderson, M.Z.; Ball, R.L.; Beamer, G.; Boggiatto, P.; Buikstra, J.E.; Chandler, B.; Claeys, T.A.; et al. The Many Hosts of Mycobacteria 9 (MHM9): A conference report. Tuberculosis 2023, 142, 102377. [Google Scholar] [CrossRef] [PubMed]
- Bucsan, A.N.; Mehra, S.; Khader, S.A.; Kaushal, D. The current state of animal models and genomic approaches towards identifying and validating molecular determinants of Mycobacterium tuberculosis infection and tuberculosis disease. Pathog. Dis. 2019, 77, ftz037. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Wang, D.; Wen, X.; Weiner, D.M.; Via, L.E. One Size Fits All? Not in In Vivo Modeling of Tuberculosis Chemotherapeutics. Front. Cell Infect. Microbiol. 2021, 11, 613149. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Lin, K.; Tiwari, D.; Healy, C.; Trujillo, C.; Liu, Y.; Ioerger, T.R.; Schnappinger, D.; Ehrt, S. Genetic models of latent tuberculosis in mice reveal differential influence of adaptive immunity. J. Exp. Med. 2021, 218, e20210332. [Google Scholar] [CrossRef]
- Subbian, S.; Tsenova, L.; O’Brien, P.; Yang, G.; Kushner, N.L.; Parsons, S.; Peixoto, B.; Fallows, D.; Kaplan, G. Spontaneous latency in a rabbit model of pulmonary tuberculosis. Am. J. Pathol. 2012, 181, 1711–1724. [Google Scholar] [CrossRef]
- Zhan, L.; Tang, J.; Sun, M.; Qin, C. Animal Models for Tuberculosis in Translational and Precision Medicine. Front. Microbiol. 2017, 8, 717. [Google Scholar] [CrossRef] [PubMed]
- Afkhami, S.; D’Agostino, M.R.; Vaseghi-Shanjani, M.; Lepard, M.; Yang, J.X.; Lai, R.; Choi, M.W.Y.; Chacon, A.; Zganiacz, A.; Franken, K.; et al. Intranasal multivalent adenoviral-vectored vaccine protects against replicating and dormant M.tb in conventional and humanized mice. NPJ Vaccines 2023, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Arrey, F.; Lowe, D.; Kuhlmann, S.; Kaiser, P.; Moura-Alves, P.; Krishnamoorthy, G.; Lozza, L.; Maertzdorf, J.; Skrahina, T.; Skrahina, A.; et al. Humanized Mouse Model Mimicking Pathology of Human Tuberculosis for in vivo Evaluation of Drug Regimens. Front. Immunol. 2019, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Endsley, J.J.; Huante, M.B.; Naqvi, K.F.; Gelman, B.B.; Endsley, M.A. Advancing our understanding of HIV co-infections and neurological disease using the humanized mouse. Retrovirology 2021, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Liang, Y.; Wu, X. Animal Models of Tuberculosis Vaccine Research: An Important Component in the Fight against Tuberculosis. Biomed. Res. Int. 2020, 2020, 4263079. [Google Scholar] [CrossRef] [PubMed]
- Lepard, M.; Yang, J.X.; Afkhami, S.; Nazli, A.; Zganiacz, A.; Tang, S.; Choi, M.W.Y.; Vahedi, F.; Deshiere, A.; Tremblay, M.J.; et al. Comparing Current and Next-Generation Humanized Mouse Models for Advancing HIV and HIV/Mtb Co-Infection Studies. Viruses 2022, 14, 1927. [Google Scholar] [CrossRef] [PubMed]
- Morgun, E.; Zhu, J.; Almunif, S.; Bobbala, S.; Aguilar, M.S.; Wang, J.; Conner, K.; Cui, Y.; Cao, L.; Seshadri, C.; et al. Vaccination with mycobacterial lipid loaded nanoparticle leads to lipid antigen persistence and memory differentiation of antigen-specific T cells. eLife 2023, 12, RP87431. [Google Scholar] [CrossRef] [PubMed]
- Calderon, V.E.; Valbuena, G.; Goez, Y.; Judy, B.M.; Huante, M.B.; Sutjita, P.; Johnston, R.K.; Estes, D.M.; Hunter, R.L.; Actor, J.K.; et al. A humanized mouse model of tuberculosis. PLoS ONE 2013, 8, e63331. [Google Scholar] [CrossRef]
- Gong, W.; Liang, Y.; Mi, J.; Jia, Z.; Xue, Y.; Wang, J.; Wang, L.; Zhou, Y.; Sun, S.; Wu, X. Peptides-Based Vaccine MP3RT Induced Protective Immunity Against Mycobacterium tuberculosis Infection in a Humanized Mouse Model. Front. Immunol. 2021, 12, 666290. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Troy, A.; Rowe, J.; Troudt, J.M.; Creissen, E.; McLean, J.; Banerjee, P.; Feuer, G.; Izzo, A.A. Humanized NOG mice as a model for tuberculosis vaccine-induced immunity: A comparative analysis with the mouse and guinea pig models of tuberculosis. Immunology 2017, 152, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, R.; Connelly, T.; Choi, R.; Choi, H.; Samarkina, A.; Li, L.; Gregorio, E.; Chen, Y.; Thakur, R.; Abdel-Mohsen, M.; et al. Tumor-infiltrating mast cells are associated with resistance to anti-PD-1 therapy. Nat. Commun. 2021, 12, 346. [Google Scholar] [CrossRef] [PubMed]
- Altin, J.G.; Sloan, E.K. The role of CD45 and CD45-associated molecules in T cell activation. Immunol. Cell Biol. 1997, 75, 430–445. [Google Scholar] [CrossRef]
- Ramirez-Cardich, M.E.; Kawai, V.; Oberhelman, R.A.; Bautista, C.T.; Castillo, M.E.; Gilman, R.H. Clinical correlates of tuberculosis co-infection in HIV-infected children hospitalized in Peru. Int. J. Infect. Dis. 2006, 10, 278–281. [Google Scholar] [CrossRef]
- Magcwebeba, T.; Dorhoi, A.; du Plessis, N. The Emerging Role of Myeloid-Derived Suppressor Cells in Tuberculosis. Front. Immunol. 2019, 10, 917. [Google Scholar] [CrossRef]
- Subbian, S.; Kumar, R. Immune Correlates of Non-Necrotic and Necrotic Granulomas in Pulmonary Tuberculosis: A Pilot Study. J. Respir. 2021, 1, 248–259. [Google Scholar] [CrossRef]
- Abnousian, A.; Vasquez, J.; Sasaninia, K.; Kelley, M.; Venketaraman, V. Glutathione Modulates Efficacious Changes in the Immune Response against Tuberculosis. Biomedicines 2023, 11, 1340. [Google Scholar] [CrossRef]
- Barbosa Bomfim, C.C.; Pinheiro Amaral, E.; Santiago-Carvalho, I.; Almeida Santos, G.; Machado Salles, E.; Hastreiter, A.A.; Silva do Nascimento, R.; Almeida, F.M.; Lopes Bia Ventura Simao, T.; Linhares Rezende, A.; et al. Harmful Effects of Granulocytic Myeloid-Derived Suppressor Cells on Tuberculosis Caused by Hypervirulent Mycobacteria. J. Infect. Dis. 2021, 223, 494–507. [Google Scholar] [CrossRef]
- Jontvedt Jorgensen, M.; Jenum, S.; Tonby, K.; Mortensen, R.; Walzl, G.; Du Plessis, N.; Dyrhol-Riise, A.M. Monocytic myeloid-derived suppressor cells reflect tuberculosis severity and are influenced by cyclooxygenase-2 inhibitors. J. Leukoc. Biol. 2021, 110, 177–186. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Knaul, J.K.; Jorg, S.; Oberbeck-Mueller, D.; Heinemann, E.; Scheuermann, L.; Brinkmann, V.; Mollenkopf, H.J.; Yeremeev, V.; Kaufmann, S.H.; Dorhoi, A. Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1053–1066. [Google Scholar] [CrossRef]
- Orr, M.T.; Windish, H.P.; Beebe, E.A.; Argilla, D.; Huang, P.W.; Reese, V.A.; Reed, S.G.; Coler, R.N. Interferon gamma and Tumor Necrosis Factor Are Not Essential Parameters of CD4+ T-Cell Responses for Vaccine Control of Tuberculosis. J. Infect. Dis. 2015, 212, 495–504. [Google Scholar] [CrossRef]
- Robinson, C.M.; Jung, J.Y.; Nau, G.J. Interferon-gamma, tumor necrosis factor, and interleukin-18 cooperate to control growth of Mycobacterium tuberculosis in human macrophages. Cytokine 2012, 60, 233–241. [Google Scholar] [CrossRef]
- Bolajoko, E.B.; Arinola, O.G.; Odaibo, G.N.; Maiga, M. Plasma levels of tumor necrosis factor-alpha, interferon-gamma, inducible nitric oxide synthase, and 3-nitrotyrosine in drug-resistant and drug-sensitive pulmonary tuberculosis patients, Ibadan, Nigeria. Int. J. Mycobacteriol. 2020, 9, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Vijayamalini, M.; Manoharan, S. Lipid peroxidation, vitamins C, E and reduced glutathione levels in patients with pulmonary tuberculosis. Cell Biochem. Funct. 2004, 22, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Safe, I.P.; Amaral, E.P.; Araújo-Pereira, M.; Lacerda, M.V.G.; Printes, V.S.; Souza, A.B.; Beraldi-Magalhães, F.; Monteiro, W.M.; Sampaio, V.S.; Barreto-Duarte, B.; et al. Adjunct N-Acetylcysteine Treatment in Hospitalized Patients With HIV-Associated Tuberculosis Dampens the Oxidative Stress in Peripheral Blood: Results From the RIPENACTB Study Trial. Front. Immunol. 2021, 11, 602589. [Google Scholar] [CrossRef] [PubMed]
- Kachour, N.; Beever, A.; Owens, J.; Cao, R.; Kolloli, A.; Kumar, R.; Sasaninia, K.; Vaughn, C.; Singh, M.; Truong, E.; et al. Liposomal Glutathione Helps to Mitigate Mycobacterium tuberculosis Infection in the Lungs. Antioxidants 2022, 11, 673. [Google Scholar] [CrossRef] [PubMed]
- Kolloli, A.; Ramasamy, S.; Kumar, R.; Nisa, A.; Kaplan, G.; Subbian, S. A phosphodiesterase-4 inhibitor reduces lung inflammation and fibrosis in a hamster model of SARS-CoV-2 infection. Front. Immunol. 2023, 14, 1270414. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolloli, A.; Kumar, R.; Venketaraman, V.; Subbian, S. Immunopathology of Pulmonary Mycobacterium tuberculosis Infection in a Humanized Mouse Model. Int. J. Mol. Sci. 2024, 25, 1656. https://doi.org/10.3390/ijms25031656
Kolloli A, Kumar R, Venketaraman V, Subbian S. Immunopathology of Pulmonary Mycobacterium tuberculosis Infection in a Humanized Mouse Model. International Journal of Molecular Sciences. 2024; 25(3):1656. https://doi.org/10.3390/ijms25031656
Chicago/Turabian StyleKolloli, Afsal, Ranjeet Kumar, Vishwanath Venketaraman, and Selvakumar Subbian. 2024. "Immunopathology of Pulmonary Mycobacterium tuberculosis Infection in a Humanized Mouse Model" International Journal of Molecular Sciences 25, no. 3: 1656. https://doi.org/10.3390/ijms25031656