
Molecular Insights into the Accelerated Sprouting of and Apical Dominance Release in Potato Tubers Subjected to Post-Harvest Heat Stress

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
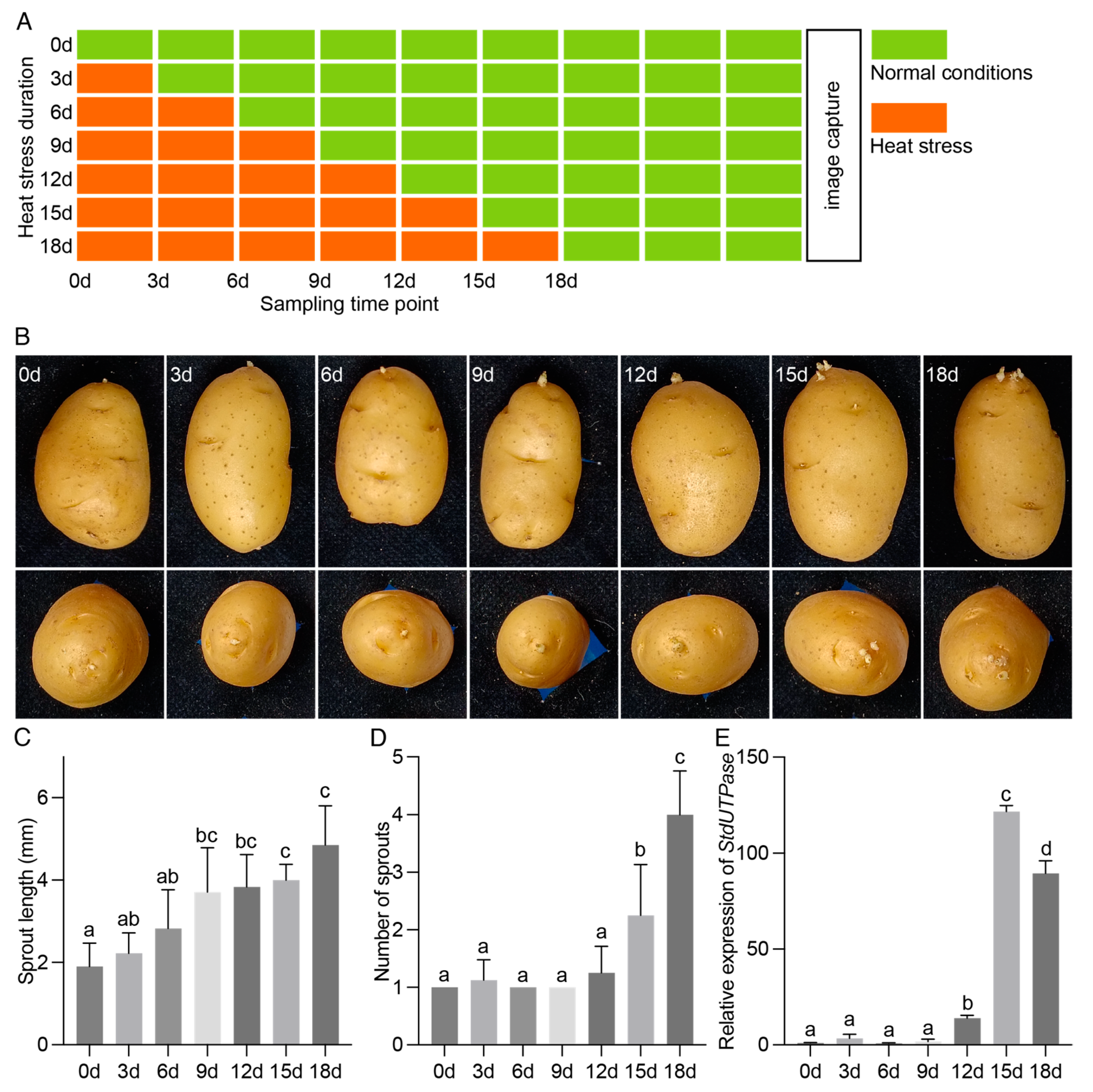
2.1. Post-Harvest HS Promotes Tuber Sprouting and the Release of Apical Dominance
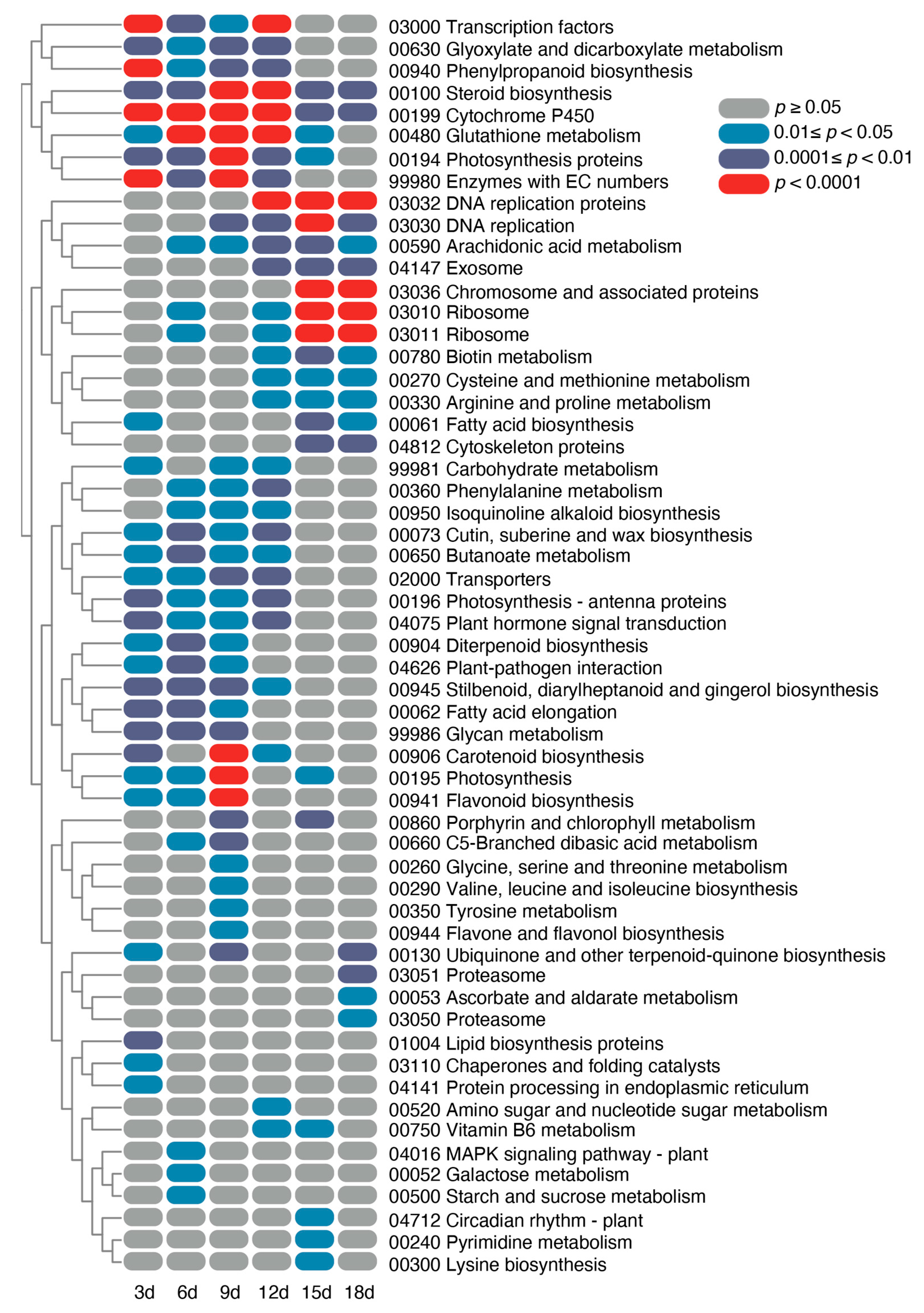
2.2. Dynamic Transcriptome Profiling of Tuber Apical Bud Meristematic Tissue during HS Treatment
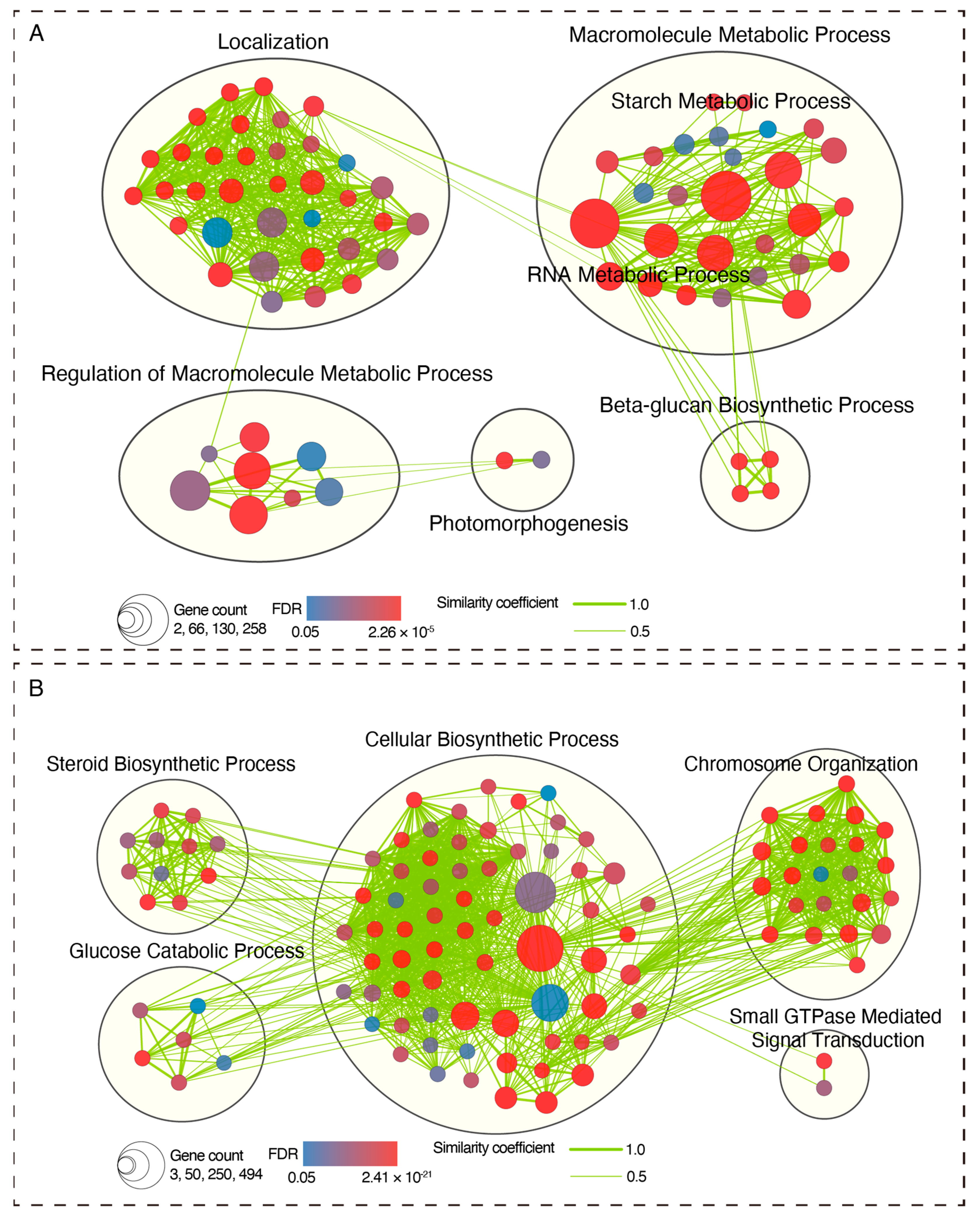
2.3. Identification of DEGs with a Similar Temporal Expression Pattern
2.4. Identification of DEGs with a Similar Temporal Expression Pattern
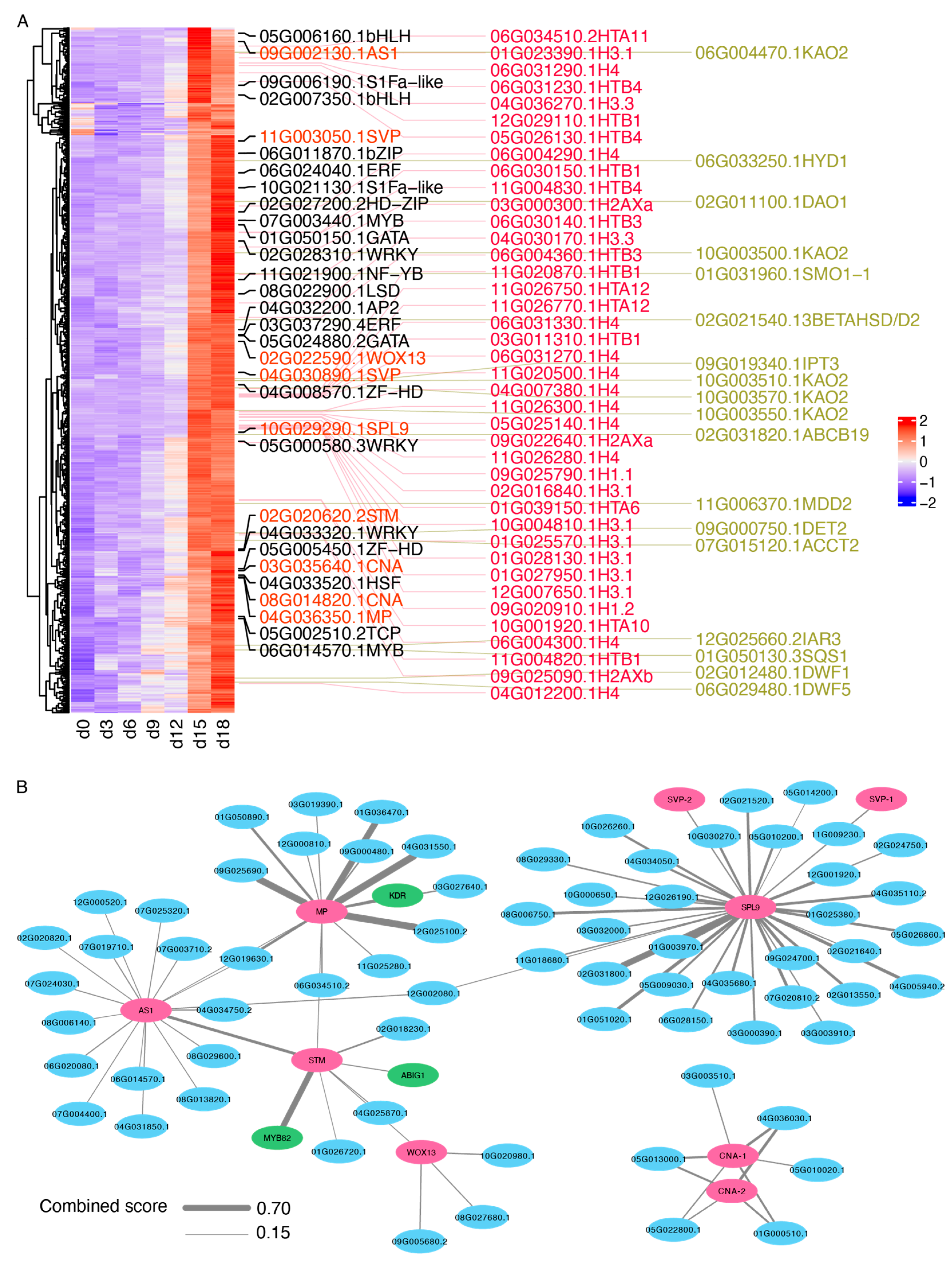
2.5. Identification of the Candidate DEGs Potentially Involved in HS Treatment-Induced Sprouting Variations
3. Discussion
3.1. The Impact of HS Treatment on Post-Harvest Potato Tuber Sprouting
3.2. Transcriptome Unveils Potential Mechanisms of HS Affecting Potato Sprouting
3.3. Limitations of This Study
4. Materials and Methods
4.1. Plant Growth and Tuber Treatments
4.2. RNA Extraction and Quantitative Real-Time PCR
4.3. Library Construction and RNA-Seq
4.4. DEGs Analysis
4.5. KEGG and GO Enrichment Analysis
4.6. Cluster and Heatmap Analysis
4.7. Functional Network Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sonnewald, S.; Sonnewald, U. Regulation of potato tuber sprouting. Planta 2014, 239, 27–38. [Google Scholar] [CrossRef]
- Struik, P.C. Above-ground and below-ground plant development. In Potato Biology and Biotechnology; Elsevier: Amsterdam, The Netherlands, 2007; pp. 219–236. [Google Scholar]
- Pasare, S.A.; Ducreux, L.J.M.; Morris, W.L.; Campbell, R.; Sharma, S.K.; Roumeliotis, E.; Kohlen, W.; van der Krol, S.; Bramley, P.M.; Roberts, A.G.; et al. The role of the potato (Solanum tuberosum) CCD8 gene in stolon and tuber development. N. Phytol. 2013, 198, 1108–1120. [Google Scholar] [CrossRef]
- Hartmann, A.; Senning, M.; Hedden, P.; Sonnewald, U.; Sonnewald, S. Reactivation of meristem activity and sprout growth in potato tubers require both cytokinin and gibberellin. Plant Physiol. 2011, 155, 776–796. [Google Scholar] [CrossRef] [PubMed]
- Senning, M.; Sonnewald, U.; Sonnewald, S. Deoxyuridine triphosphatase expression defines the transition from dormant to sprouting potato tuber buds. Mol. Breed. 2010, 26, 525–531. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, N.; Wen, Y.; Jin, X.; Yang, J.; Si, H.; Wang, D. Transcriptomic changes during tuber dormancy release process revealed by RNA sequencing in potato. J. Biotechnol. 2015, 198, 17–30. [Google Scholar] [CrossRef]
- Campbell, M.; Suttle, J.; Douches, D.S.; Buell, C.R. Treatment of potato tubers with the synthetic cytokinin 1-(alpha-ethylbenzyl)-3-nitroguanidine results in rapid termination of endodormancy and induction of transcripts associated with cell proliferation and growth. Funct. Integr. Genom. 2014, 14, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Kukreja, S.; Goutam, U. Impact of heat stress on potato (Solanum tuberosum L.): Present scenario and future opportunities. J. Hortic. Sci. Biotechnol. 2019, 95, 407–424. [Google Scholar] [CrossRef]
- Levy, D.; Veilleux, R.E. Adaptation of potato to high temperatures and salinity-a review. Am. J. Potato Res. 2007, 84, 487–506. [Google Scholar] [CrossRef]
- Rosa, J. Relation of tuber maturity and of storage factors to potato dormancy. Hilgardia 1928, 3, 99–124. [Google Scholar] [CrossRef]
- Zhang, G.; Tang, R.; Niu, S.; Si, H.; Yang, Q.; Rajora, O.P.; Li, X.-Q. Heat-stress-induced sprouting and differential gene expression in growing potato tubers: Comparative transcriptomics with that induced by postharvest sprouting. Hortic. Res. 2021, 8, 226. [Google Scholar] [CrossRef]
- Li, L.-Q.; Zou, X.; Deng, M.-S.; Peng, J.; Huang, X.-L.; Lu, X.; Fang, C.-C.; Wang, X.-Y. Comparative morphology, transcription, and proteomics study revealing the key molecular mechanism of camphor on the potato tuber sprouting effect. Int. J. Mol. Sci. 2017, 18, 2280. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Du, Y.; Liu, X.; Zhang, H.; Liu, Y.; Yan, N.; Zhang, Z. Genome-Wide Analysis of Long Non-Coding RNAs in Potato and Their Potential Role in Tuber Sprouting Process. Int. J. Mol. Sci. 2017, 19, 101. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, N.; Wen, Y.; Si, H.; Wang, D. Identification of differentially expressed genes in potato associated with tuber dormancy release. Mol. Biol. Rep. 2012, 39, 11277–11287. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, L.A.; Kolachevskaya, O.O.; Krivandin, A.V.; Filatova, A.G.; Gradov, O.V.; Plashchina, I.G.; Romanov, G.A. Changes in Structural and Thermodynamic Properties of Starch during Potato Tuber Dormancy. Int. J. Mol. Sci. 2023, 24, 8397. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.J.; Senning, M.; Fischer-Stettler, M.; Streb, S.; Ast, M.; Neuhaus, H.E.; Zeeman, S.C.; Sonnewald, S.; Sonnewald, U. Simultaneous silencing of isoamylases ISA1, ISA2 and ISA3 by multi-target RNAi in potato tubers leads to decreased starch content and an early sprouting phenotype. PLoS ONE 2017, 12, e0181444. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Liu, T.; Reid, S.; Zhang, H.; Peng, X.; Sun, K.; Du, J.; Sonnewald, U.; Song, B. Silencing of α-amylase StAmy23 in potato tuber leads to delayed sprouting. Plant Physiol. Biochem. 2019, 139, 411–418. [Google Scholar] [CrossRef]
- Chiang, G.C.; Barua, D.; Kramer, E.M.; Amasino, R.M.; Donohue, K. Major flowering time gene, FLOWERING LOCUS C, regulates seed germination in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2009, 106, 11661–11666. [Google Scholar] [CrossRef]
- Michaels, S.D.; Amasino, R.M. FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 1999, 11, 949–956. [Google Scholar] [CrossRef]
- Penfield, S.; Hall, A. A role for multiple circadian clock genes in the response to signals that break seed dormancy in Arabidopsis. Plant Cell 2009, 21, 1722–1732. [Google Scholar] [CrossRef]
- Zhu, Y.; Dong, A.; Shen, W.-H. Histone variants and chromatin assembly in plant abiotic stress responses. Biochim. Biophys. Acta. Gene Regul. Mech. 2012, 1819, 343–348. [Google Scholar] [CrossRef]
- Layat, E.; Bourcy, M.; Cotterell, S.; Zdzieszyńska, J.; Desset, S.; Duc, C.; Tatout, C.; Bailly, C.; Probst, A.V. The Histone chaperone HIRA is a positive regulator of seed germination. Int. J. Mol. Sci. 2021, 22, 4031. [Google Scholar] [CrossRef]
- Regnault, T.; Davière, J.M.; Heintz, D.; Lange, T.; Achard, P. The gibberellin biosynthetic genes At KAO 1 and At KAO 2 have overlapping roles throughout A rabidopsis development. Plant J. 2014, 80, 462–474. [Google Scholar] [CrossRef]
- Choe, S.; Dilkes, B.P.; Gregory, B.D.; Ross, A.S.; Yuan, H.; Noguchi, T.; Fujioka, S.; Takatsuto, S.; Tanaka, A.; Yoshida, S. The Arabidopsis dwarf1 mutant is defective in the conversion of 24-methylenecholesterol to campesterol in brassinosteroid biosynthesis. Plant Physiol. 1999, 119, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Tanaka, A.; Noguchi, T.; Fujioka, S.; Takatsuto, S.; Ross, A.S.; Tax, F.E.; Yoshida, S.; Feldmann, K.A. Lesions in the sterol Δ7 reductase gene of Arabidopsis cause dwarfism due to a block in brassinosteroid biosynthesis. Plant J. 2000, 21, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Fujioka, S.; Takatsuto, S.; Sakurai, A.; Yoshida, S.; Li, J.; Chory, J. Arabidopsis det2 is defective in the conversion of (24 R)-24-methylcholest-4-en-3-one to (24 R)-24-methyl-5α-cholestan-3-one in brassinosteroid biosynthesis. Plant Physiol. 1999, 120, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Deng, M.; Lyu, C.; Zhang, J.; Peng, J.; Cai, C.; Yang, S.; Lu, L.; Ni, S.; Liu, F. Quantitative phosphoproteomics analysis reveals that protein modification and sugar metabolism contribute to sprouting in potato after BR treatment. Food Chem. 2020, 325, 126875. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Dong, H.; Yin, Y.; Song, X.; Gu, X.; Sang, K.; Zhou, J.; Shi, K.; Zhou, Y.; Foyer, C.H. Brassinosteroid signaling integrates multiple pathways to release apical dominance in tomato. Proc. Natl. Acad. Sci. USA 2021, 118, e2004384118. [Google Scholar] [CrossRef] [PubMed]
- Udvardi, M.K.; Kakar, K.; Wandrey, M.; Montanari, O.; Murray, J.; Andriankaja, A.; Zhang, J.-Y.; Benedito, V.; Hofer, J.M.; Chueng, F. Legume transcription factors: Global regulators of plant development and response to the environment. Plant Physiol. 2007, 144, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; He, H.; Li, Y.; Ai, Q.; Yu, D. MYB82 functions in regulation of trichome development in Arabidopsis. J. Exp. Bot. 2014, 65, 3215–3223. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yoo, S.J.; Park, S.H.; Hwang, I.; Lee, J.S.; Ahn, J.H. Role of SVP in the control of flowering time by ambient temperature in Arabidopsis. Genes Dev. 2007, 21, 397–402. [Google Scholar] [CrossRef]
- Schwarz, S.; Grande, A.V.; Bujdoso, N.; Saedler, H.; Huijser, P. The microRNA regulated SBP-box genes SPL9 and SPL15 control shoot maturation in Arabidopsis. Plant Mol. Biol. 2008, 67, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Romera-Branchat, M.; Ripoll, J.J.; Yanofsky, M.F.; Pelaz, S. The WOX13 homeobox gene promotes replum formation in the Arabidopsis thaliana fruit. Plant J. 2013, 73, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.E.; Barley, R.; Curtis, M.; Arroyo, J.M.; Dunham, M.; Hudson, A.; Martienssen, R.A. Asymmetric leaves1 mediates leaf patterning and stem cell function in Arabidopsis. Nature 2000, 408, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Long, J.A.; Moan, E.I.; Medford, J.I.; Barton, M.K. A member of the KNOTTED class of homeodomain proteins encoded by the STM gene of Arabidopsis. Nature 1996, 379, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Schlereth, A.; Moller, B.; Liu, W.; Kientz, M.; Flipse, J.; Rademacher, E.H.; Schmid, M.; Jurgens, G.; Weijers, D. MONOPTEROS controls embryonic root initiation by regulating a mobile transcription factor. Nature 2010, 464, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Green, K.A.; Prigge, M.J.; Katzman, R.B.; Clark, S.E. CORONA, a member of the class III homeodomain leucine zipper gene family in Arabidopsis, regulates stem cell specification and organogenesis. Plant Cell 2005, 17, 691–704. [Google Scholar] [CrossRef]
- Liu, T.; Fang, H.; Liu, J.; Reid, S.; Hou, J.; Zhou, T.; Tian, Z.; Song, B.; Xie, C. Cytosolic glyceraldehyde-3-phosphate dehydrogenases play crucial roles in controlling cold-induced sweetening and apical dominance of potato (Solanum tuberosum L.) tubers. Plant Cell Environ. 2017, 40, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhou, T.; Lian, M.; Liu, T.; Hou, J.; Ijaz, R.; Song, B. Genome-Wide Identification and Characterization of the AREB/ABF/ABI5 Subfamily Members from Solanum tuberosum. Int. J. Mol. Sci. 2019, 20, 311. [Google Scholar] [CrossRef]
- Liu, T.; Kawochar, M.A.; Begum, S.; Wang, E.; Zhou, T.; Jing, S.; Liu, T.; Yu, L.; Nie, B.; Song, B. Potato tonoplast sugar transporter 1 controls tuber sugar accumulation during postharvest cold storage. Hortic. Res. 2023, 10, uhad035. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [PubMed]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5. [Google Scholar] [CrossRef]
- Gu, Z. Complex heatmap visualization. IMETA 2022, 1, e43. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Wu, Q.; Zhou, S.; Xia, J.; Yin, W.; Deng, L.; Song, B.; He, T. Molecular Insights into the Accelerated Sprouting of and Apical Dominance Release in Potato Tubers Subjected to Post-Harvest Heat Stress. Int. J. Mol. Sci. 2024, 25, 1699. https://doi.org/10.3390/ijms25031699
Liu T, Wu Q, Zhou S, Xia J, Yin W, Deng L, Song B, He T. Molecular Insights into the Accelerated Sprouting of and Apical Dominance Release in Potato Tubers Subjected to Post-Harvest Heat Stress. International Journal of Molecular Sciences. 2024; 25(3):1699. https://doi.org/10.3390/ijms25031699
Chicago/Turabian StyleLiu, Tengfei, Qiaoyu Wu, Shuai Zhou, Junhui Xia, Wang Yin, Lujun Deng, Botao Song, and Tianjiu He. 2024. "Molecular Insights into the Accelerated Sprouting of and Apical Dominance Release in Potato Tubers Subjected to Post-Harvest Heat Stress" International Journal of Molecular Sciences 25, no. 3: 1699. https://doi.org/10.3390/ijms25031699
APA StyleLiu, T., Wu, Q., Zhou, S., Xia, J., Yin, W., Deng, L., Song, B., & He, T. (2024). Molecular Insights into the Accelerated Sprouting of and Apical Dominance Release in Potato Tubers Subjected to Post-Harvest Heat Stress. International Journal of Molecular Sciences, 25(3), 1699. https://doi.org/10.3390/ijms25031699