
The Inhibition of Serine Proteases by Serpins Is Augmented by Negatively Charged Heparin: A Concise Review of Some Clinically Relevant Interactions

 , ,
, ,
Abstract
1. Introduction to Serine Proteases and Serine Protease Inhibitors
2. A Primer on Heparin Biology
3. Thrombin, Antithrombin III, and Heparin
4. Plasmin, Anti-Plasmin, and Heparin
5. C1 Esterase/Kallikrein, C1-INH, and Heparin
6. Furin, TMPRSS2, and Their Inhibition by AAT in the Context of HIV, the Hepatitis C Virus, and/or SARS-CoV-2
7. TMPRSS2, Prostate Cancer, and Heparin
8. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Puente, X.S.; Sánchez, L.M.; Overall, C.M.; López-Otín, C. Human and mouse proteases: A comparative genomic approach. Nat. Rev. Genet. 2003, 4, 544–558. [Google Scholar] [CrossRef]
- Neitzel, J.J. Enzyme Catalysis: The Serine Proteases. Nat. Educ. 2010, 3, 21. [Google Scholar]
- Stapels, D.A.C.; Geisbrecht, B.V.; Rooijakkers, S.H.M. Neutrophil serine proteases in antibacterial defense. Curr. Opin. Microbiol. 2015, 23, 42–48. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Tolle, D.P.; Barrett, A.J. Evolutionary families of peptidase inhibitors. Biochem. J. 2004, 378 Pt 3, 705–716. [Google Scholar] [CrossRef]
- Huntington, J.A.; Read, R.J.; Carrell, R.W. Structure of a serpin-protease complex shows inhibition by deformation. Nature 2000, 407, 923–926. [Google Scholar] [CrossRef]
- de Serres, F.J.; Blanco, I.; Fernández-Bustillo, E. Genetic epidemiology of alpha-1 antitrypsin deficiency in North America and Australia/New Zealand: Australia, Canada, New Zealand and the United States of America. Clin. Genet. 2003, 64, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Hazari, Y.M.; Bashir, A.; Habib, M.; Bashir, S.; Habib, H.; Qasim, M.A.; Shah, N.N.; Haq, E.; Teckman, J.; Fazili, K.M. Alpha-1-antitrypsin deficiency: Genetic variations, clinical manifestations and therapeutic interventions. Mutat. Res. 2017, 773, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Kalsheker, N.; Wallis, S.; Speer, A.; Coutelle, C.H.; Woods, D.; Humphries, S.E. The isolation of a clone for human alpha 1-antitrypsin and the detection of alpha 1-antitrypsin in mRNA from liver and leukocytes. Biochem. Biophys. Res. Commun. 1983, 116, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Strnad, P.; McElvaney, N.G.; Lomas, D.A. Alpha1-Antitrypsin Deficiency. N. Engl. J. Med. 2020, 382, 1443–1455. [Google Scholar] [CrossRef] [PubMed]
- Crystal, R.G. The alpha 1-antitrypsin gene and its deficiency states. Trends Genet. 1989, 5, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, J.D.; Haworth, C.S.; Metersky, M.L.; Loebinger, M.R.; Blasi, F.; Sibila, O.; O’Donnell, A.E.; Sullivan, E.J.; Mange, K.C.; Fernandez, C.; et al. WILLOW Investigators. Phase 2 Trial of the DPP-1 Inhibitor Brensocatib in Bronchiectasis. N. Engl. J. Med. 2020, 383, 2127–2137. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057. [Google Scholar] [CrossRef]
- Gozzo, L.; Viale, P.; Longo, L.; Vitale, D.C.; Drago, F. The potential role of heparin in patients with COVID-19: Beyond the anticoagulant effect. A review. Front. Pharmacol. 2020, 11, 1307. [Google Scholar] [CrossRef] [PubMed]
- Hippensteel, J.A.; LaRiviere, W.B.; Colbert, J.F.; Langouet-Astrie, C.J.; Schmidt, E.P. Heparin as a therapy for COVID-19: Current evidence and future possibilities. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 319, L211–L217. [Google Scholar] [CrossRef]
- Ma, S.-N.; Mao, Z.-X.; Wu, Y.; Liang, M.-X.; Wang, D.-D.; Chen, X.; Chang, P.-A.; Zhang, W.; Tang, J.-H. The anti-cancer properties of heparin and its derivatives: A review and prospect. Cell Adhes. Migr. 2020, 14, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Shute, J.K. Heparin, low molecular weight heparin, and non-anticoagulant derivatives for the treatment of inflammatory lung disease. Pharmaceuticals 2023, 16, 584. [Google Scholar] [CrossRef] [PubMed]
- Dijk, M.; Holkers, J.; Voskamp, P.; Giannetti, B.M.; Waterreus, W.J.; van Veen, H.A.; Pannu, N.S. How Dextran Sulfate Affects C1-inhibitor Activity: A Model for Polysaccharide Potentiation. Structure 2016, 24, 2182–2189. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Buckle, A.M.; Vladar, E.K.; Janoff, E.N.; Khare, R.; Ordway, D.; Beckham, D.; Fornis, L.B.; Majluf-Cruz, A.; Fugit, R.V.; et al. Enoxaparin augments alpha-1-antitrypsin inhibition of TMPRSS2, a promising drug combination against COVID-19. Sci. Rep. 2022, 12, 5207. [Google Scholar] [CrossRef] [PubMed]
- Ersdal-Badju, E.; Lu, A.; Zuo, Y.; Picard, V.; Bock, S.C. Identification of the antithrombin III heparin binding site. J. Biol. Chem. 1997, 272, 19393–19400. [Google Scholar] [CrossRef]
- Chang, G.M.T.; Atkinson, H.M.; Berry, L.R.; Chan, A.K.C. Inhibition of plasmin generation in plasma by heparin, low molecular weight heparin, and a covalent antithrombin-heparin complex. Blood Coagul. Fibrinolysis 2017, 28, 431–437. [Google Scholar] [CrossRef]
- Majluf-Cruz, A.; Nieto-Martinez, S. Long-term follow up analysis of nadroparin for hereditary angioedema: A preliminary report. Int. Immunopharmacol. 2011, 11, 1127–1132. [Google Scholar] [CrossRef]
- Gupta, V.K.; Gowda, L.R. Alpha-1-proteinase inhibitor is a heparin binding serpin: Molecular interactions with the Lys rich cluster of helix-F domain. Biochimie 2008, 90, 749–761. [Google Scholar] [CrossRef]
- Gray, E.; Mulloy, B.; Barrowcliffe, T.W. Heparin and low-molecular-weight heparin. Thromb. Haemost. 2008, 99, 807–818. [Google Scholar]
- Hirsh, J. Low-molecular-weight heparin: A review of the results of recent studies of the treatment of venous thromboembolism and unstable angina. Circulation 1998, 98, 1575–1582. [Google Scholar] [CrossRef] [PubMed]
- Rosett, W.; Hodges, G.R. Antimicrobial Activity of Heparin. J. Clin. Microbiol. 1980, 11, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Abreu, R.; Essler, L.; Loy, A.; Quinn, F.; Giri, P. Heparin inhibits intracellular Mycobacterium tuberculosis bacterial replication by reducing iron levels in human macrophages. Sci. Rep. 2018, 8, 7296. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.A.; Lever, R.; Jones, N.A.; Page, C.P. Effects of heparin and related molecules upon neutrophil aggregation and elastase release in vitro. Br. J. Pharmacol. 2003, 139, 845–853. [Google Scholar] [CrossRef]
- Bae, J.; Desai, U.R.; Pervin, A.; Caldwell, E.E.; Weiler, J.M.; Linhardt, R.J. Interaction of heparin with synthetic antithrombin III peptide analogues. Biochem. J. 1994, 301 Pt 1, 121–129. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; O’Sullivan, J.M.; Preston, R.J.S. Advances in understanding the molecular mechanisms that maintain normal haemostasis. Br. J. Haematol. 2019, 186, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Hsu, E.; Moosavi, L. Biochemistry, Antithrombin III; StatPearls: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK545295/ (accessed on 19 December 2023).
- Pol-Fachin, L.; Franco Becker, C.; Almeida Guimarães, J.; Verli, H. Effects of glycosylation on heparin binding and antithrombin activation by heparin. Proteins 2011, 79, 2735–2745. [Google Scholar] [CrossRef] [PubMed]
- Karlaftis, V.; Sritharan, G.; Attard, C.; Corral, J.; Monagle, P.; Ignjatovic, V. Beta (β)-antithrombin activity in children and adults: Implications for heparin therapy in infants and children. J. Thromb. Haemost. 2014, 12, 1141–1144. [Google Scholar] [CrossRef]
- Amiral, J.; Seghatchian, J. Revisiting antithrombin in health and disease, congenital deficiencies and genetic variants, and laboratory studies on α and β forms. Transfus. Apher. Sci. 2018, 57, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Pike, R.N.; Buckle, A.M.; le Bonniec, B.F.; Church, F.C. Control of the coagulation system by serpins. Getting by with a little help from glycosaminoglycans. FEBS J. 2005, 272, 4842–4851. [Google Scholar] [CrossRef] [PubMed]
- Abdelghani, E.; Waller, A.P.; Wolfgang, K.J.; Stanek, J.R.; Parikh, S.V.; Rovin, B.H.; Smoyer, W.E.; Kerlin, B.A. The PNRC Investigators; The NEPTUNE Investigators. Exploring the Role of Antithrombin in Nephrotic Syndrome-Associated Hypercoagulopathy: A Multi-Cohort Study and Meta-Analysis. Clin. J. Am. Soc. Nephrol. 2023, 18, 234–244. [Google Scholar] [CrossRef]
- Chung, W.-S.; Lin, C.-L.; Kao, C.-H. Diabetes increases the risk of deep-vein thrombosis and pulmonary embolism. A population-based cohort study. Thromb. Haemost. 2015, 114, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Busse, P.J.; Christiansen, S.C. Hereditary angioedema. N. Engl. J. Med. 2020, 382, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.M. Angioedema: Etiology, pathophysiology, current and emerging therapies. J. Emerg. Med. 2013, 45, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Katelaris, C.H. Acute Management of Hereditary Angioedema Attacks. Immunol. Allergy Clin. N. Am. 2017, 37, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Cicardi, M.; Zuraw, B.L. Angioedema Due to Bradykinin Dysregulation. J. Allergy Clin. Immunol. Pr. 2018, 6, 1132–1141. [Google Scholar] [CrossRef]
- Aygören-Pürsün, E.; Bygum, A.; Grivcheva-Panovska, V.; Magerl, M.; Graff, J.; Steiner, U.C.; Fain, O.; Huissoon, A.; Kinaciyan, T.; Farkas, H.; et al. Oral Plasma Kallikrein Inhibitor for Prophylaxis in Hereditary Angioedema. N. Engl. J. Med. 2018, 379, 352–362. [Google Scholar] [CrossRef]
- Banerji, A.; Riedl, M.A.; Bernstein, J.A.; Cicardi, M.; Longhurst, H.J.; Zuraw, B.L.; Busse, P.J.; Anderson, J.; Magerl, M.; Martinez-Saguer, I.; et al. HELP Investigators. Effect of Lanadelumab Compared with Placebo on Prevention of Hereditary Angioedema Attacks: A Randomized Clinical Trial. JAMA 2018, 320, 2108–2121. [Google Scholar] [CrossRef]
- Riedl, M.A.; Aygören-Pürsün, E.; Baker, J.; Farkas, H.; Anderson, J.; Bernstein, J.A.; Bouillet, L.; Busse, P.; Manning, M.; Magerl, M.; et al. Evaluation of avoralstat, an oral kallikrein inhibitor, in a Phase 3 hereditary angioedema prophylaxis trial: The OPuS-2 study. Allergy 2018, 73, 1871–1880. [Google Scholar] [CrossRef]
- Maurer, M.; Magerl, M.; Betschel, S.; Aberer, W.; Ansotegui, I.J.; Aygören-Pürsün, E.; Banerji, A.; Bara, N.A.; Boccon-Gibod, I.; Bork, K.; et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2021 revision and update. Allergy 2022, 77, 1961–1990. [Google Scholar] [CrossRef]
- Ecker, E.E.; Gross, P. Anticomplementary power of heparin. J. Infect. Dis. 1929, 44, 287–295. [Google Scholar] [CrossRef]
- Poppelaars, F.; Damman, J.; de Vrij, E.L.; Burgerhof, J.G.; Saye, J.; Daha, M.R.; Leuvenink, H.G.; Uknis, M.E.; Seelen, M.A. New insight into the effects of heparinoids on complement inhibition by C1-inhibitor. Clin. Exp. Immunol. 2016, 184, 378–388. [Google Scholar] [CrossRef]
- Wuillemin, W.A.; te Velthuis, H.; Lubbers, Y.T.; de Ruig, C.P.; Eldering, E.; Hack, C.E. Potentiation of C1 inhibitor by glycosaminoglycans: Dextran sulfate species are effective inhibitors of in vitro complement activation in plasma. J. Immunol. 1997, 159, 1953–1960. [Google Scholar] [CrossRef]
- Wijeyewickrema, L.C.; Lameignere, E.; Hor, L.; Duncan, R.C.; Shiba, T.; Travers, R.J.; Kapopara, P.R.; Lei, V.; Smith, S.A.; Kim, H.; et al. Polyphosphate is a novel cofactor for regulation of complement by the serpin, C1-inhibitor. Blood 2016, 128, 1766–1776. [Google Scholar] [CrossRef]
- Weiler, J.M.; Stechschulte, D.J.; Levine, H.T.; Edens, R.E.; Maves, K.K. Inhaled heparin in the treatment of hereditary angioedema. Complement. Inflamm. 1991, 8, 240–241. [Google Scholar]
- Levine, H.T.; Stechschulte, D.J. Possible efficacy nebulized heparin therapy in hereditary angioedema. Immunol. Allergy Pract. 1992, 14, 31–36. [Google Scholar]
- Weiler, J.M.; Quinn, S.A.; Woodworth, G.G.; Brown, D.D.; Layton, T.A.; Maves, K.K. Does heparin prophylaxis prevent exacerbations of hereditary angioedema? J. Allergy Clin. Immunol. 2002, 109, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.D.; Thomas, L.; Hayflick, J.S.; Thomas, G. Inhibition of HIV-1 gp160-dependent membrane fusion by a furin directed alpha 1-antitrypsin variant. J. Biol. Chem. 1993, 268, 24887–24891. [Google Scholar] [CrossRef] [PubMed]
- Jean, F.; Stella, K.; Thomas, L.; Liu, G.; Xiang, Y.; Reason, A.J.; Thomas, G. alpha1-Antitrypsin Portland, a bioengineered serpin highly selective for furin: Application as an antipathogenic agent. Proc. Natl. Acad. Sci. USA 1998, 95, 7293–7298. [Google Scholar] [CrossRef] [PubMed]
- Hada, K.; Isshiki, K.; Matsuda, S.; Yuasa, K.; Tsuji, A. Engineering of α1-antitrypsin variants with improved specificity for the proprotein convertase furin using site-directed random mutagenesis. Protein Eng. Des. Sel. 2013, 26, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Scott, B.M.; Sheffield, W.P. Engineering the serpin α1-antitrypsin: A diversity of goals and techniques. Protein Sci. 2020, 29, 856–871. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, L.; Pott, G.B.; Ralston, A.H. Alpha-1-antitrypsin inhibits human immunodeficiency virus type 1. FASEB J. 2001, 15, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Pasquato, A.; Dettin, M.; Basak, A.; Gambaretto, R.; Tonin, L.; Seidah, N.G.; Di Bello, C.D. Heparin enhances the furin cleavage of HIV-1 gp160 peptides. FEBS Lett. 2007, 581, 5807–5813. [Google Scholar] [CrossRef]
- Esumi, M.; Ishibashi, M.; Yamaguchi, H.; Nakajima, S.; Tai, Y.; Kikuta, S.; Sugitani, M.; Takayama, T.; Tahara, M.; Takeda, M.; et al. Transmembrane serine protease TMPRSS2 activates hepatitis C virus infection. Hepatology 2015, 61, 437–446. [Google Scholar] [CrossRef]
- Propst, T.; Propst, A.; Dietze, O.; Judmaier, G.; Braunsteiner, H.; Vogel, W. High prevalence of viral infection in adults with homozygous and heterozygous alpha 1-antitrypsin deficiency and chronic liver disease. Ann. Intern. Med. 1992, 117, 641–645. [Google Scholar] [CrossRef]
- Fuentes-Prior, P. Priming of SARS-CoV-2 S protein by several membrane-bound serine proteinases could explain enhanced viral infectivity and systemic COVID-19 infection. J. Biol. Chem. 2020, 296, 100135. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Ko, C.J.; Hsu, T.W.; Wu, S.R.; Lan, S.W.; Hsiao, T.F.; Lin, H.Y.; Lin, H.H.; Tu, H.F.; Lee, C.F.; Huang, C.C.; et al. Inhibition of TMPRSS2 by HAI-2 reduces prostate cancer cell invasion and metastasis. Oncogene 2020, 39, 5950–5963. [Google Scholar] [CrossRef]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef]
- Gunst, J.D.; Staerke, N.B.; Pahus, M.H.; Kristensen, L.H.; Bodilsen, J.; Lohse, N.; Dalgaard, L.S.; Brønnum, D.; Fröbert, O.; Hønge, B.; et al. Efficacy of the TMPRSS2 inhibitor camostat mesilate in patients hospitalized with Covid-19-a double-blind randomized controlled trial. EClinicalMedicine 2021, 35, 100849. [Google Scholar] [CrossRef]
- Azouz, N.P.; Klingler, A.M.; Callahan, V.; Akhrymuk, I.V.; Elez, K.; Raich, L.; Henry, B.M.; Benoit, J.L.; Benoit, S.W.; Noé, F.; et al. Alpha 1 antitrypsin is an inhibitor of the SARS-CoV2-priming protease TMPRSS2. Pathog. Immun. 2021, 6, 55–74. [Google Scholar] [CrossRef]
- Ritzmann, F.; Chitirala, P.; Krüger, N.; Hoffmann, M.; Zuo, W.; Lammert, F.; Smola, S.; Tov, N.; Alagem, N.; Lepper, P.M.; et al. AAT-in-COVID-19 study group. Therapeutic Application of Alpha-1-Antitrypsin in COVID-19. Am. J. Respir. Crit. Care Med. 2021, 204, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Wettstein, L.; Weil, T.; Conzelmann, C.; Müller, J.A.; Groß, R.; Hirschenberger, M.; Seidel, A.; Klute, S.; Zech, F.; Bozzo, C.P.; et al. Alpha-1 antitrypsin inhibits TMPRSS2 protease activity and SARS-CoV-2 infection. Nat. Commun. 2021, 12, 1726. [Google Scholar] [CrossRef] [PubMed]
- Oguntuyo, K.Y.; Stevens, C.S.; Siddiquey, M.N.; Schilke, R.M.; Woolard, M.D.; Zhang, H.; Acklin, J.A.; Ikegame, S.; Hung, C.T.; Lim, J.K.; et al. In plain sight: The role of alpha-1-antitrypsin in COVID-19 pathogenesis and therapeutics. bioRxiv 2020, preprint. [Google Scholar]
- Bhattacharyya, C.; Das, C.; Ghosh, A.; Singh, A.K.; Mukherjee, S.; Majumder, P.P.; Basu, A.; Biswas, N.K. SARS-CoV-2 mutation 614G creates an elastase cleavage site enhancing its spread in high AAT-deficient regions. Infect. Genet. Evol. 2021, 90, 104760. [Google Scholar] [CrossRef] [PubMed]
- Rosendal, E.; Mihai, I.S.; Becker, M.; Das, D.; Frängsmyr, L.; Persson, B.D.; Rankin, G.D.; Gröning, R.; Trygg, J.; Forsell, M.; et al. Serine Protease Inhibitors Restrict Host Susceptibility to SARS-CoV-2 Infections. mBio 2022, 13, e0089222. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; O’Reilly, S.; Gewaid, H.; Bowie, A.G.; Gautier, V.; Worrall, D.M. Reactive Centre Loop Mutagenesis of SerpinB3 to Target TMPRSS2 and Furin: Inhibition of SARS-CoV-2 Cell Entry and Replication. Int. J. Mol. Sci. 2022, 23, 12522. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Schountz, T.; Buckle, A.M.; Talbert, J.L.; Sandhaus, R.A.; Chan, E.D. Alpha-1-antitrypsin antagonizes COVID-19: A review of the epidemiology, molecular mechanisms, and clinical evidence. Biochem. Soc. Trans. 2023, 51, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Blanco, I.; de Serres, F.J.; Fernandez-Bustillo, E.; Lara, B.; Miravitlles, M. Estimated numbers and prevalence of PI*S and PI*Z alleles of alpha1-antitrypsin deficiency in European countries. Eur. Respir. J. 2006, 27, 77–84. [Google Scholar] [CrossRef] [PubMed]
- McElvaney, O.J.; McEvoy, N.L.; McElvaney, O.F.; Carroll, T.P.; Murphy, M.P.; Dunlea, D.M.; Choileain, O.N.; Clarke, J.; O’Connor, E.; Hogan, G.; et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am. J. Respir. Crit. Care Med. 2020, 202, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Shapira, G.; Shomron, N.; Gurwitz, D. Ethnic differences in alpha-1 antitrypsin deficiency allele frequencies may partially explain national differences in COVID-19 fatality rates. FASEB J. 2020, 34, 14160–14165. [Google Scholar] [CrossRef] [PubMed]
- Vianello, A.; Braccioni, F. Geographical overlap between alpha-1 antitrypsin deficiency and COVID-19 infection in Italy: Casual or causal? Arch. Bronconeumol. 2020, 56, 609–610. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.K.; Goswami, K. Host genomics of COVID-19: Evidence point towards Alpha 1 antitrypsin deficiency as a putative risk factor for higher mortality rate. Med. Hypotheses 2021, 147, 110485. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.; Inês Costa, M.; Gomes, J.; Sucena, M. Alpha-1 antitrypsin deficiency severity and the risk of COVID-19: A Portuguese cohort. Respir. Med. 2021, 181, 106387. [Google Scholar] [CrossRef] [PubMed]
- Ferrarotti, I.; Ottaviani, S.; Balderacchi, A.M.; Barzon, V.; De Silvestri, A.; Piloni, D.; Mariani, F.; Corsico, A.G. COVID-19 infection in severe alpha 1-antitrypsin deficiency: Looking for a rationale. Respir. Med. 2021, 183, 106440. [Google Scholar] [CrossRef]
- Murgia, N.; Corsico, A.G.; D’Amato, G.; Maesano, C.N.; Tozzi, A.; Annesi-Maesano, I. Do gene-environment interactions play a role in COVID-19 distribution? The case of Alpha-1 Antitrypsin, air pollution and COVID-19. Multidiscip. Respir. Med. 2021, 16, 741. [Google Scholar] [CrossRef]
- Yang, C.; Chapman, K.R.; Wong, A.; Liu, M. α1-Antitrypsin deficiency and the risk of COVID-19: An urgent call to action. Lancet Respir. Med. 2021, 9, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Yoshikura, H. Epidemiological correlation between COVID-19 epidemic and prevalence of alpha-1 antitrypsin deficiency in the world. Glob. Health Med. 2021, 3, 73–81. [Google Scholar] [CrossRef]
- Parr, D.G.; Chorostowska-Wynimko, J.; Corsico, A.; Esquinas, C.; McElvaney, G.N.; Sark, A.D.; Sucena, M.; Tanash, H.; Turner, A.M.; Miravitlles, M. Impact of COVID-19 in Patients With Severe Alpha-1 Antitrypsin Deficiency: The IMCA1 Study of the EARCO Clinical Research Collaboration. Arch. Bronconeumol. 2022, 58, 840–842. [Google Scholar] [CrossRef]
- Pertzov, B.; Shapira, G.; Abushkara, S.; Cohen, S.; Turjeman, A.; Kramer, M.R.; Gurwitz, D.; Shomron, N. Lower serum alpha 1 antitrypsin levels in patients with severe COVID-19 compared with patients hospitalized due to non-COVID-19 pneumonia. Infect. Dis. 2022, 54, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Philippe, A.; Puel, M.; Narjoz, C.; Gendron, N.; Durey-Dragon, M.A.; Vedie, B.; Balduyck, M.; Chocron, R.; Hauw-Berlemont, C.; Sanchez, O.; et al. Imbalance between alpha-1-antitrypsin and interleukin 6 is associated with in-hospital mortality and thrombosis during COVID-19. Biochimie 2022, 202, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Hermosa, J.L.; Vargas Centanaro, G.; González Castro, M.E.; Miravitlles, M.; Lázaro-Asegurado, L.; Jiménez-Rodríguez, B.M.; Rodríguez, R.A.; Moreno Méndez, R.; Torres-Duran, M.; Hernández-Pérez, J.M.; et al. Severe COVID-19 Illness and α1-Antitrypsin Deficiency: COVID-AATD Study. Biomedicines 2023, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- Shimi, G.; Sohrab, G.; Pourvali, K.; Ghorbani, A.; Balam, F.H.; Rostami, K.; Zand, H. Correlation of Low Levels of alpha-1 Antitrypsin and Elevation of Neutrophil to Lymphocyte Ratio with Higher Mortality in Severe COVID-19 Patients. Mediat. Inflamm. 2021, 2021, 5555619. [Google Scholar] [CrossRef]
- Abd, H.A.; Kasim, A.A.; Shareef, L.G. Serum levels of a1-antitrypsin, interleukin-1b and interleukin-6 in Iraqi COVID-19 patients: A cross-sectional study. F1000Research 2022, 11, 921. [Google Scholar] [CrossRef]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J. Biol. Chem. 2000, 275, 27258–27265. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Li, Z.; Janciauskiene, S.; Mahadeva, R. Oxidation of Z α1-antitrypsin by cigarette smoke induces polymerization: A novel mechanism of early-onset emphysema. Am. J. Respir. Cell Mol. Biol. 2011, 45, 261–269. [Google Scholar] [CrossRef] [PubMed]
- McElvaney, O.F.; Asakura, T.; Meinig, S.L.; Torres-Castillo, J.L.; Hagan, R.S.; Gabillard-Lefort, C.; Murphy, M.P.; Thorne, L.B.; Borczuk, A.; Reeves, E.P.; et al. Protease-anti-protease compartmentalization in SARS-CoV-2 ARDS: Therapeutic implications. EBioMedicine 2022, 77, 103894. [Google Scholar] [CrossRef]
- Cai, Q.; Kim, M.; Harada, A.; Idowu, M.O.; Sundaresan, G.; Zweit, J.; Oh, Y. Alpha-1 Antitrypsin Inhibits Tumorigenesis and Progression of Colitis-Associated Colon Cancer through Suppression of Inflammatory Neutrophil-Activated Serine Proteases and IGFBP-3 Proteolysis. Int. J. Mol. Sci. 2022, 23, 13737. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yu, J.; Mani, R.S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Y.; Zhang, J.; Hu, Q.; Zhi, F.; Zhang, S.; Mao, D.; Zhang, Y.; Liang, H. Significance of the TMPRSS2:ERG gene fusion in prostate cancer. Mol. Med. Rep. 2017, 16, 5450–5458. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.C. Expanding the clinical indications for α(1)-antitrypsin therapy. Mol. Med. 2012, 18, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Phelps, M.K.; Olson, L.M.; Van Berkel Patel, M.A.; Thompson, M.J.; Murphy, C.V. Nebulized Heparin for Adult Patients With Smoke Inhalation Injury: A Review of the Literature. J. Pharm. Technol. 2020, 36, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, B.; Bagdasarian, A.; Mitchell, B.; Talamo, R.C.; Colman, R.W.; Rosenberg, R.D. Antithrombin-heparin cofactor: An inhibitor of plasma kallikrein. Arch. Biochem. Biophys. 1976, 175, 737–747. [Google Scholar] [CrossRef]
- Rezaie, A.R.; Giri, H. Anticoagulant and signaling functions of antithrombin. J. Thromb. Haemost. 2020, 18, 3142–3153. [Google Scholar] [CrossRef]
- Davis, A.E., III; Mejia, P.; Lu, F. Biological activities of C1 inhibitor. Mol. Immunol. 2008, 45, 4057–4063. [Google Scholar] [CrossRef]
- Frenzel, E.; Wrenger, S.; Brügger, B.; Salipalli, S.; Immenschuh, S.; Aggarwal, N.; Lichtinghagen, R.; Mahadeva, R.; Marcondes, A.M.; Dinarello, C.A.; et al. α1-Antitrypsin Combines with Plasma Fatty Acids and Induces Angiopoietin-like Protein 4 Expression. J. Immunol. 2015, 195, 3605–3616. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.E.; Murray, G.; Gogoi, D.; Yusuf, A.; McCarthy, C.; Wormald, M.R.; Casey, M.; Gabillard-Lefort, C.; McElvaney, N.G.; Reeves, E.P. A Review of Alpha-1 Antitrypsin Binding Partners for Immune Regulation and Potential Therapeutic Application. Int. J. Mol. Sci. 2022, 23, 2441. [Google Scholar] [CrossRef] [PubMed]

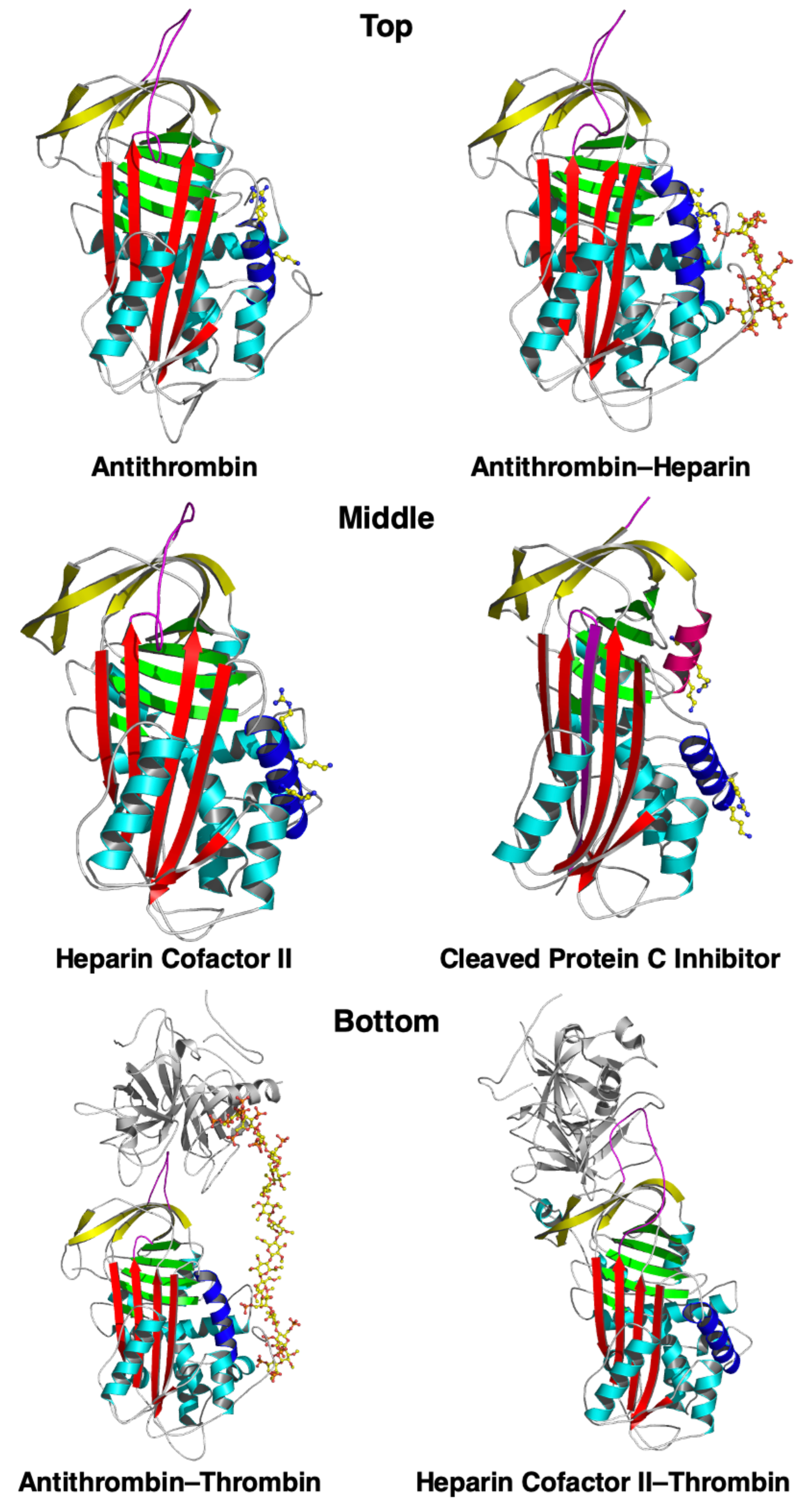
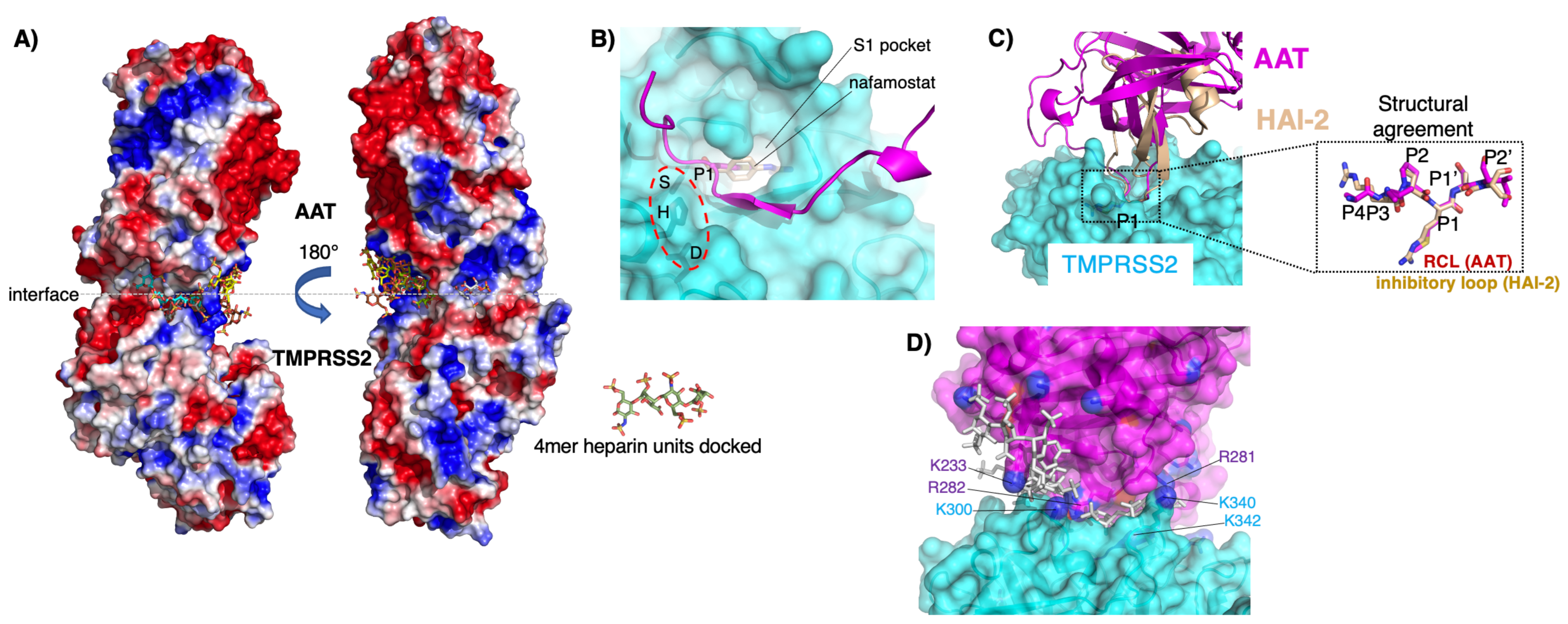

{kind=link}
{kind=link}
{kind=link}
| Serine Protease and Serpin Pair | Augmentation by Negatively Charged Polysaccharide |
|---|---|
| Thrombin and antithrombin III | Heparin [19] |
| Plasmin and anti-plasmin | Heparin [20] |
| C1-esterase and C1-INH | Heparin (nadroparin and enoxaparin)Dextran [17] |
| C1-esterase and kallikrein | Heparin, nadroparin [21] |
| Furin and AAT Portland | Unknown |
| TMPRSS2 and AAT | Enoxaparin, UFH [18] |
| Neutrophil elastase and AAT | Heparin [22] |
| Trypsin and AAT | Heparin [22] |
| Biologic Effect | Example(s) |
|---|---|
| Anticoagulation | Inhibits coagulation through inactivation of thrombin and factor X. |
| Antiviral | Competitively inhibits cell surface heparan sulfate, a co-receptor SARS-CoV-2. |
| Antibacterial | Possibly inhibits growth of certain bacteria, including Staphylococcus aureus [25]. |
| Antimycobacterial | Reduces hepcidin expression (an iron-sequestering protein) in macrophages infected with Mycobacterium tuberculosis (MTB), increases ferroportin expression (an iron exporter protein), and decreases the intracellular burden of MTB [26]. |
| Anti-elastase and antitrypsin | Inhibits the enzymatic activity of neutrophil elastase on its release from neutrophils [27] and enhances the inhibitory effect of AAT on elastase and trypsin [22]. |
| Anti-inflammatory | Inhibits inflammation by inhibiting nuclear factor-kappa B (NFκB) activation, binding to pro-inflammatory molecules (IL-8, major basic protein, and complement components), and reducing the release and activity of IL-6. |
| Anticomplement and contact system | Augments C1-esterase inhibitor (C1-INH) inhibition of kallikrein ⟶ a decrease in capillary leak. |
| Cell protection | Binds to extracellular histones released from dead cells, mitigating histone-mediated endothelial and organ dysfunction. Interacts with endothelial cells to maintain vascular integrity. |
| Anticancer | Blocks angiogenesis and adhesion of cancer cells to platelets, potentially interrupting the metastases of cancer cells. |
| Serpin | Serine Proteases Inhibited |
|---|---|
| Antithrombin III | Thrombin, factor Xa, kallikrein, and to a lesser extent IXa and XIIa [97,98]. |
| Anti-plasmin | Plasmin and plasminogen. |
| C1-esterase inhibitor | C1 esterase (C1r, C1s), MASP2, plasmin, tissue plasminogen activator, factor XI, factor XIIa, and kallikrein [99]. |
| Alpha-1-antitrypsin | Proteinase-3, trypsin, chymotrypsin, myeloperoxidase, cathepsins, a-defensins, tryptase, plasmin, thrombin, factor Xa, urokinase, a disintegrin and metalloprotease 17 (ADAM17, aka tumor necrosis factor converting enzyme), and Transmembrane Protease 2 (TMPRSS2) [18,72,100,101]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, E.D.; King, P.T.; Bai, X.; Schoffstall, A.M.; Sandhaus, R.A.; Buckle, A.M. The Inhibition of Serine Proteases by Serpins Is Augmented by Negatively Charged Heparin: A Concise Review of Some Clinically Relevant Interactions. Int. J. Mol. Sci. 2024, 25, 1804. https://doi.org/10.3390/ijms25031804
Chan ED, King PT, Bai X, Schoffstall AM, Sandhaus RA, Buckle AM. The Inhibition of Serine Proteases by Serpins Is Augmented by Negatively Charged Heparin: A Concise Review of Some Clinically Relevant Interactions. International Journal of Molecular Sciences. 2024; 25(3):1804. https://doi.org/10.3390/ijms25031804
Chicago/Turabian StyleChan, Edward D., Paul T. King, Xiyuan Bai, Allen M. Schoffstall, Robert A. Sandhaus, and Ashley M. Buckle. 2024. "The Inhibition of Serine Proteases by Serpins Is Augmented by Negatively Charged Heparin: A Concise Review of Some Clinically Relevant Interactions" International Journal of Molecular Sciences 25, no. 3: 1804. https://doi.org/10.3390/ijms25031804
APA StyleChan, E. D., King, P. T., Bai, X., Schoffstall, A. M., Sandhaus, R. A., & Buckle, A. M. (2024). The Inhibition of Serine Proteases by Serpins Is Augmented by Negatively Charged Heparin: A Concise Review of Some Clinically Relevant Interactions. International Journal of Molecular Sciences, 25(3), 1804. https://doi.org/10.3390/ijms25031804