
Correlation of Experimental and Calculated Inhibition Constants of Protease Inhibitor Complexes



Abstract
:1. Introduction
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| β-Trypsin/SFTI-1 | Measured Ki [nM] | FoldX Ki [nM] | PRODIGY KD [nM] |
|---|---|---|---|
| SFTI-1 | 0.017 | 0.007 | 0.963 |
| SFTI-P14 | - | 0.766 | 1.4 |
| SFTI-R5 | 0.027 | - | 0.99 |
| SFTI-TCTR-N12N14 | 0.7 | 2.86 | - |
| SFTI-RCTR | 4.7 | ||
| SFTI-RCTK | 9.9 | ||
| SFTI-TCTK | 5.5 | ||
| SFTI-TCTK-P14 | 1.6 | ||
| SFTI-TCTR-P14 | 0.43 | ||
| Thrombin/hirudin | Measured Ki [fM] | FoldX Ki [fM] | PRODIGY KD [fM] |
| hirudin-v1/v2 | 22 | 19 | - |
| rhir-v1 | 180 | - | - |
| rhir-v1-Trp3 | 60 | - | - |
| rhir-v1-Phe3 | 30 | - | - |
| hirudin-v2 | 15 | - | |
| v2-Trp3 | 0.077 | - | |
| v2-Arg1-Trp3 | 0.021 | - |
3. Material and Methods
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pérez, S.; Tvaroška, I. Chapter 1-Carbohydrate–Protein Interactions: Molecular Modeling Insights. In Advances in Carbohydrate Chemistry and Biochemistry; Academic Press: Cambridge, MA, USA, 2014; Volume 71, pp. 9–136. ISBN 0065-2318. [Google Scholar]
- Kilburg, D.; Gallicchio, E. Chapter Two—Recent Advances in Computational Models for the Study of Protein–Peptide Interactions. In Insights into Enzyme Mechanisms and Functions from Experimental and Computational Methods; Christov, C.Z.B.T.-A., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 105, pp. 27–57. ISBN 1876-1623. [Google Scholar]
- Papadourakis, M.; Sinenka, H.; Matricon, P.; Hénin, J.; Brannigan, G.; Pérez-Benito, L.; Pande, V.; van Vlijmen, H.; de Graaf, C.; Deflorian, F.; et al. Alchemical Free Energy Calculations on Membrane-Associated Proteins. J. Chem. Theory Comput. 2023, 19, 7437–7458. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, C.L.D.; Completo, G.C.; Nacario, R.C.; Nellas, R.B. Potential Inhibitors of Galactofuranosyltransferase 2 (GlfT2): Molecular Docking, 3D-QSAR, and In Silico ADMETox Studies. Sci. Rep. 2019, 9, 17096. [Google Scholar] [CrossRef] [PubMed]
- Lima, E.C.; Hosseini-Bandegharaei, A.; Moreno-Piraján, J.C.; Anastopoulos, I. A Critical Review of the Estimation of the Thermodynamic Parameters on Adsorption Equilibria. Wrong Use of Equilibrium Constant in the Van’t Hoff Equation for Calculation of Thermodynamic Parameters of Adsorption. J. Mol. Liq. 2019, 273, 425–434. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics, 4th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2012; ISBN 978-3-527-33074-4. [Google Scholar]
- Masson, P.; Mukhametgalieva, A.R. Partial Reversible Inhibition of Enzymes and Its Metabolic and Pharmaco-Toxicological Implications. Int. J. Mol. Sci. 2023, 24, 12973. [Google Scholar] [CrossRef] [PubMed]
- Szabó, A.; Ludwig, M.; Hegyi, E.; Szépeová, R.; Witt, H.; Sahin-Tóth, M. Mesotrypsin Signature Mutation in a Chymotrypsin C (CTRC) Variant Associated with Chronic Pancreatitis. J. Biol. Chem. 2015, 290, 17282–17292. [Google Scholar] [CrossRef] [PubMed]
- Issa, S.S.; Sokornova, S.V.; Zhidkin, R.R.; Matveeva, T. V The Main Protease of SARS-CoV-2 as a Target for Phytochemicals against Coronavirus. Plants 2022, 11, 1862. [Google Scholar] [CrossRef]
- Gimeno, A.; Cuffaro, D.; Nuti, E.; Ojeda-Montes, M.J.; Beltrán-Debón, R.; Mulero, M.; Rossello, A.; Pujadas, G.; Garcia-Vallvé, S. Identification of Broad-Spectrum MMP Inhibitors by Virtual Screening. Molecules 2021, 26, 4553. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhu, L.; Zhou, W.; Yin, L.; Wang, Y.; Fan, Y.; Chen, Y.; Liu, H. In Silico Prediction of Inhibitory Constant of Thrombin Inhibitors Using Machine Learning. Comb. Chem. High Throughput Screen. 2018, 21, 662–669. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 44130. [Google Scholar] [CrossRef]
- Swedberg, J.E.; de Veer, S.J.; Sit, K.C.; Reboul, C.F.; Buckle, A.M.; Harris, J.M. Mastering the Canonical Loop of Serine Protease Inhibitors: Enhancing Potency by Optimising the Internal Hydrogen Bond Network. PLoS ONE 2011, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Riley, B.T.; de Veer, S.J.; Hoke, D.E.; Van Haeften, J.; Leahy, D.; Swedberg, J.E.; Brattsand, M.; Hartfield, P.J.; Buckle, A.M.; et al. Potent, Multi-Target Serine Protease Inhibition Achieved by a Simplified β-Sheet Motif. PLoS ONE 2019, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New Ways to Boost Molecular Dynamics Simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Van Durme, J.; Delgado, J.; Stricher, F.; Serrano, L.; Schymkowitz, J.; Rousseau, F. A Graphical Interface for the FoldX Forcefield. Bioinformatics 2011, 27, 1711–1712. [Google Scholar] [CrossRef]
- Delgado, J.; Radusky, L.G.; Cianferoni, D.; Serrano, L. FoldX 5.0: Working with RNA, Small Molecules and a New Graphical Interface. Bioinformatics 2019, 35, 4168–4169. [Google Scholar] [CrossRef]
- Vangone, A.; Bonvin, A.M.J.J. Contacts-Based Prediction of Binding Affinity in Protein–Protein Complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A Web Server for Predicting the Binding Affinity of Protein–Protein Complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- de Veer, S.J.; White, A.M.; Craik, D.J. Sunflower Trypsin Inhibitor-1 (SFTI-1): Sowing Seeds in the Fields of Chemistry and Biology. Angew. Chem. Int. Ed. Engl. 2021, 60, 8050–8071. [Google Scholar] [CrossRef]
- Krowarsch, D.; Cierpicki, T.; Jelen, F.; Otlewski, J. Canonical Protein Inhibitors of Serine Proteases. Cell. Mol. Life Sci. C. 2003, 60, 2427–2444. [Google Scholar] [CrossRef]
- Schechter, I.; Berger, A. On the Size of the Active Site in Proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Evans, S.A.; Olson, S.T.; Shore, J.D. P-Aminobenzamidine as a Fluorescent Probe for the Active Site of Serine Proteases. J. Biol. Chem. 1982, 257, 3014–3017. [Google Scholar] [CrossRef] [PubMed]
- Perilo, C.S.; Pereira, M.T.; Santoro, M.M.; Nagem, R.A.P. Structural Binding Evidence of the Trypanocidal Drugs Berenil® and Pentacarinate® Active Principles to a Serine Protease Model. Int. J. Biol. Macromol. 2010, 46, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Pendlebury, D.; Wang, R.; Henin, R.D.; Hockla, A.; Soares, A.S.; Madden, B.J.; Kazanov, M.D.; Radisky, E.S. Sequence and Conformational Specificity in Substrate Recognition: Several Human Kunitz Protease Inhibitor Domains Are Specific Substrates of Mesotrypsin. J. Biol. Chem. 2014, 289, 32783–32797. [Google Scholar] [CrossRef]
- Long, Y.-Q.; Lee, S.-L.; Lin, C.-Y.; Enyedy, I.J.; Wang, S.; Li, P.; Dickson, R.B.; Roller, P.P. Synthesis and Evaluation of the Sunflower Derived Trypsin Inhibitor as a Potent Inhibitor of the Type II Transmembrane Serine Protease, Matriptase. Bioorg. Med. Chem. Lett. 2001, 11, 2515–2519. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Chen, L.; Meehan, E.J.; Daly, N.; Craik, D.J.; Huang, M.; Ngo, J.C. Structure of Catalytic Domain of Matriptase in Complex with Sunflower Trypsin Inhibitor-1. BMC Struct. Biol. 2011, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, J.E.; Wu, G.; Mahatmanto, T.; Durek, T.; Caradoc-Davies, T.T.; Whisstock, J.C.; Law, R.H.P.; Craik, D.J. Highly Potent and Selective Plasmin Inhibitors Based on the Sunflower Trypsin Inhibitor-1 Scaffold Attenuate Fibrinolysis in Plasma. J. Med. Chem. 2019, 62, 552–560. [Google Scholar] [CrossRef]
- Riley, B.T.; Ilyichova, O.; de Veer, S.J.; Swedberg, J.E.; Wilson, E.; Hoke, D.E.; Harris, J.M.; Buckle, A.M. KLK4 Inhibition by Cyclic and Acyclic Peptides: Structural and Dynamical Insights into Standard-Mechanism Protease Inhibitors. Biochemistry 2019, 58, 2524–2533. [Google Scholar] [CrossRef]
- Quimbar, P.; Malik, U.; Sommerhoff, C.P.; Kaas, Q.; Chan, L.Y.; Huang, Y.-H.; Grundhuber, M.; Dunse, K.; Craik, D.J.; Anderson, M.A.; et al. High-Affinity Cyclic Peptide Matriptase Inhibitors. J. Biol. Chem. 2013, 288, 13885–13896. [Google Scholar] [CrossRef]
- Luckett, S.; Garcia, R.S.; Barker, J.J.; Konarev, A.; Shewry, P.R.; Clarke, A.R.; Brady, R.L. High-Resolution Structure of a Potent, Cyclic Proteinase Inhibitor from Sunflower Seeds1 1Edited by I. A. Wilson. J. Mol. Biol. 1999, 290, 525–533. [Google Scholar] [CrossRef]
- Vincent, J.P.; Lazdunski, M. Trypsin-Pancreatic Trypsin Inhibitor Association. Dynamics of the Interaction and Role of Disulfide Bridges. Biochemistry 1972, 11, 2967–2977. [Google Scholar] [CrossRef]
- Marquart, M.; Walter, J.; Deisenhofer, J.; Bode, W.; Huber, R. The Geometry of the Reactive Site and of the Peptide Groups in Trypsin, Trypsinogen and Its Complexes with Inhibitors. Acta Crystallogr. Sect. B 1983, 39, 480–490. [Google Scholar] [CrossRef]
- Stone, S.R.; Hofsteenge, J. Kinetics of the Inhibition of Thrombin by Hirudin. Biochemistry 1986, 25, 4622–4628. [Google Scholar] [CrossRef] [PubMed]
- Rydel, T.J.; Tulinsky, A.; Bode, W.; Huber, R. Refined Structure of the Hirudin-Thrombin Complex. J. Mol. Biol. 1991, 221, 583–601. [Google Scholar] [CrossRef] [PubMed]
- Dall, E.; Hollerweger, J.C.; Dahms, S.O.; Cui, H.; Häussermann, K.; Brandstetter, H. Structural and Functional Analysis of Cystatin E Reveals Enzymologically Relevant Dimer and Amyloid Fibril States. J. Biol. Chem. 2018, 293, 13151–13165. [Google Scholar] [CrossRef] [PubMed]
- Dall, E.; Fegg, J.C.; Briza, P.; Brandstetter, H. Structure and Mechanism of an Aspartimide-Dependent Peptide Ligase in Human Legumain. Angew. Chem. Int. Ed. 2015, 54, 2917–2921. [Google Scholar] [CrossRef] [PubMed]
- Johansen-Leete, J.; Ullrich, S.; Fry, S.E.; Frkic, R.; Bedding, M.J.; Aggarwal, A.; Ashhurst, A.S.; Ekanayake, K.B.; Mahawaththa, M.C.; Sasi, V.M.; et al. Antiviral Cyclic Peptides Targeting the Main Protease of SARS-CoV-2. Chem. Sci. 2022, 13, 3826–3836. [Google Scholar] [CrossRef] [PubMed]
- Ruderisch, N.; Schlatter, D.; Kuglstatter, A.; Guba, W.; Huber, S.; Cusulin, C.; Benz, J.; Rufer, A.C.; Hoernschemeyer, J.; Schweitzer, C.; et al. Potent and Selective BACE-1 Peptide Inhibitors Lower Brain Aβ Levels Mediated by Brain Shuttle Transport. EBioMedicine 2017, 24, 76–92. [Google Scholar] [CrossRef]
- Kusumoto, Y.; Hayashi, K.; Sato, S.; Yamada, T.; Kozono, I.; Nakata, Z.; Asada, N.; Mitsuki, S.; Watanabe, A.; Wakasa-Morimoto, C.; et al. Highly Potent and Oral Macrocyclic Peptides as a HIV-1 Protease Inhibitor: MRNA Display-Derived Hit-to-Lead Optimization. ACS Med. Chem. Lett. 2022, 13, 1634–1641. [Google Scholar] [CrossRef]
- Lichte, A.; Kolkenbrock, H.; Tschesche, H. The Recombinant Catalytic Domain of Membrane-Type Matrix Metalloproteinase-1 (MT1-MMP) Induces Activation of Progelatinase A and Progelatinase A Complexed with TIMP-2. FEBS Lett. 1996, 397, 277–282. [Google Scholar] [CrossRef]
- Fernandez-Catalan, C.; Bode, W.; Huber, R.; Turk, D.; Calvete, J.J.; Lichte, A.; Tschesche, H.; Maskos, K. Crystal Structure of the Complex Formed by the Membrane Type 1-Matrix Metalloproteinase with the Tissue Inhibitor of Metalloproteinases-2, the Soluble Progelatinase A Receptor. EMBO J. 1998, 17, 5238–5248. [Google Scholar] [CrossRef] [PubMed]
- Batra, J.; Robinson, J.; Soares, A.S.; Fields, A.P.; Radisky, D.C.; Radisky, E.S. Matrix Metalloproteinase-10 (MMP-10) Interaction with Tissue Inhibitors of Metalloproteinases TIMP-1 and TIMP-2. J. Biol. Chem. 2012, 287, 15935–15946. [Google Scholar] [CrossRef] [PubMed]
- Gomis-Rüth, F.-X.; Maskos, K.; Betz, M.; Bergner, A.; Huber, R.; Suzuki, K.; Yoshida, N.; Nagase, H.; Brew, K.; Bourenkov, G.P.; et al. Mechanism of Inhibition of the Human Matrix Metalloproteinase Stromelysin-1 by TIMP-1. Nature 1997, 389, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Elamin, T.; Brandstetter, H.; Dall, E. Legumain Activity Is Controlled by Extended Active Site Residues and Substrate Conformation. Int. J. Mol. Sci. 2022, 23, 12548. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Tewari, R.; Raghava, G.P.S. Ki DoQ: Using Docking Based Energy Scores to Develop Ligand Based Model for Predicting Antibacterials. BMC Bioinform. 2010, 11, 125. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Cerutti, D.S.; Cisneros, G.A.; Cruzeiro, V.W.D.; Forouzesh, N.; Giese, T.J.; Götz, A.W.; Gohlke, H.; et al. AmberTools. J. Chem. Inf. Model. 2023, 63, 6183–6191. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. Sect. D 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Agirre, J.; Atanasova, M.; Bagdonas, H.; Ballard, C.B.; Baslé, A.; Beilsten-Edmands, J.; Borges, R.J.; Brown, D.G.; Burgos-Mármol, J.J.; Berrisford, J.M.; et al. The CCP4 Suite: Integrative Software for Macromolecular Crystallography. Acta Crystallogr. Sect. D 2023, 79, 449–461. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Junren, C.; Xiaofang, X.; Huiqiong, Z.; Gangmin, L.; Yanpeng, Y.; Xiaoyu, C.; Yuqing, G.; Yanan, L.; Yue, Z.; Fu, P.; et al. Pharmacological Activities and Mechanisms of Hirudin and Its Derivatives—A Review. Front. Pharmacol. 2021, 12, 1–23. [Google Scholar] [CrossRef]
- Lazar, J.B.; Winant, R.C.; Johnson, P.H. Hirudin: Amino-Terminal Residues Play a Major Role in the Interaction with Thrombin. J. Biol. Chem. 1991, 266, 685–688. [Google Scholar] [CrossRef]
- Bloch, J.S.; Mukherjee, S.; Kowal, J.; Filippova, E.V.; Niederer, M.; Pardon, E.; Steyaert, J.; Kossiakoff, A.A.; Locher, K.P. Development of a Universal Nanobody-Binding Fab Module for Fiducial-Assisted Cryo-EM Studies of Membrane Proteins. Proc. Natl. Acad. Sci. USA 2021, 118, e2115435118. [Google Scholar] [CrossRef]
- Jiang, H.; Deng, R.; Yang, X.; Shang, J.; Lu, S.; Zhao, Y.; Song, K.; Liu, X.; Zhang, Q.; Chen, Y.; et al. Peptidomimetic Inhibitors of APC–Asef Interaction Block Colorectal Cancer Migration. Nat. Chem. Biol. 2017, 13, 994–1001. [Google Scholar] [CrossRef]
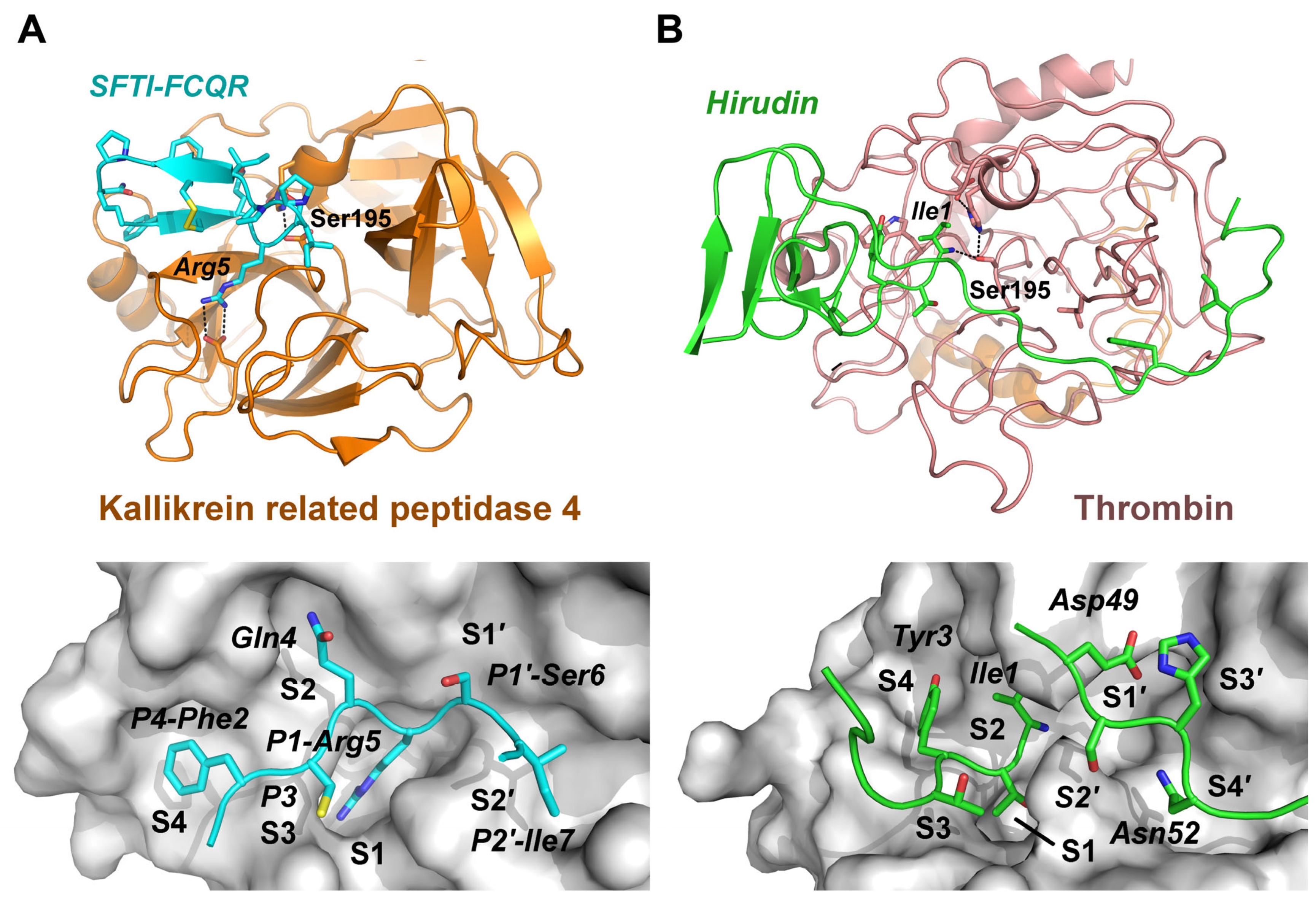
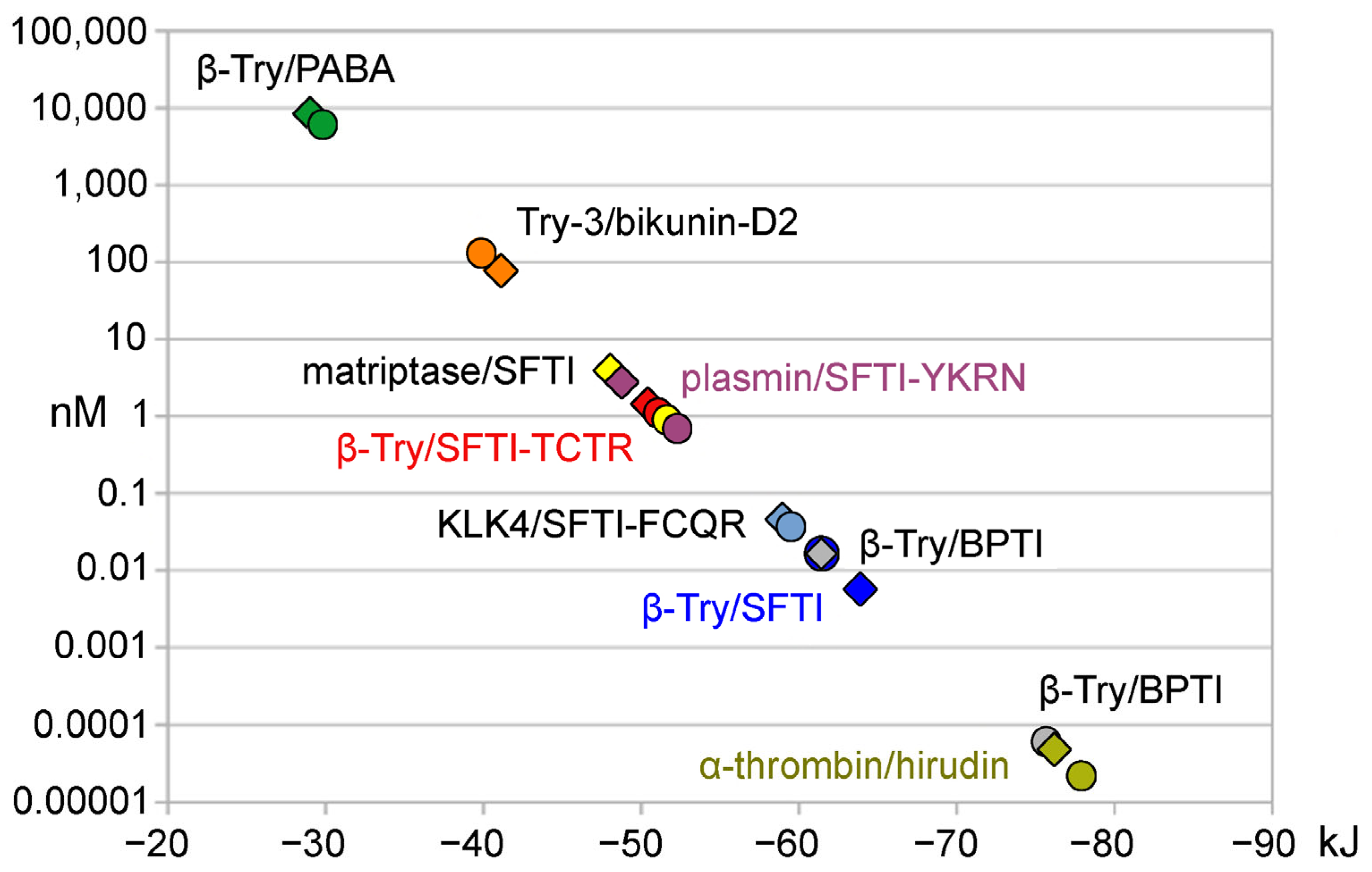
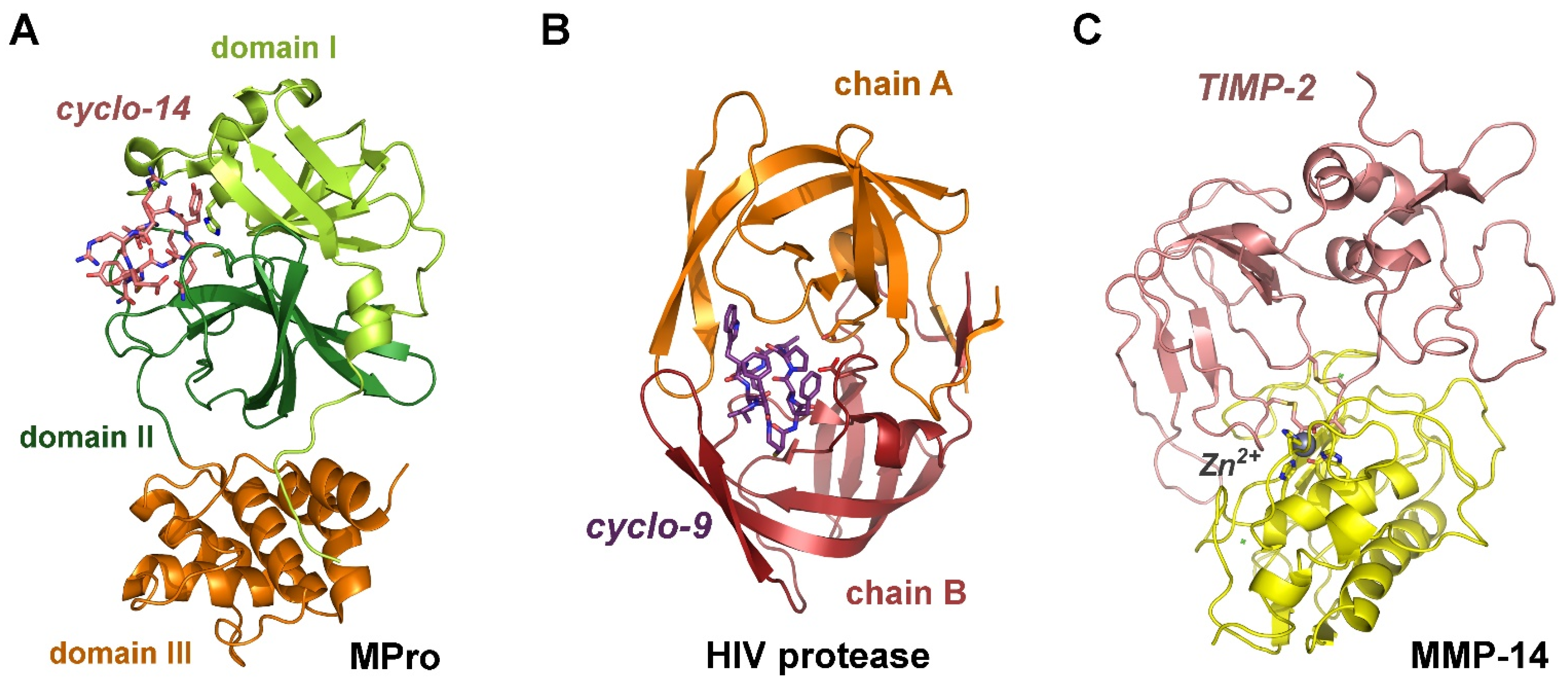

| Complex | FoldX ΔG | Ki (exp) | ΔG (exp) | ΔG Prodigy | Structure | ||
|---|---|---|---|---|---|---|---|
| Protease/Inhibitor | kJ/mol | kcal/mol | nM | nM | kJ/mol | kJ/mol | PDB |
| β-Try/PABA | −29.02 | −6.93 | 8220 | 6100 [25] | −29.79 | −23.93 1 | 3GY4 [26] |
| Try-3/bikunin-D2 | −40.96 | −9.79 | 78 | 138 [27] | −39.87 | −43.10 | 4U30 [27] |
| matriptase/SFTI-1 | −48.02 | −11.47 | 3.83 | 0.92 [28] | −51.55 | −39.75 | 3P8F [29] |
| plasmin/SFTI-Y4K5R7N14 | −50.41 | −12.04 | 1.46 | 1.20 [30] | −50.98 | −54.03 | 6D3Z [30] |
| β-Try/SFTI-T2R5N12N14 | −48.74 | −11.65 | 2.86 | 0.70 [15] | −52.23 | −48.53 | 6BVH [15] |
| KLK4/SFTI-F2Q4R5N14 | −58.95 | −14.09 | 0.046 | 0.039 [31] | −59.40 | (−41.84) | 4KEL [31] |
| β-Try/SFTI-1 | −63.18 | −15.10 | 0.0066 | 0.017 [32] | −61.47 | (−51.46) | 1SFI [33] |
| β-Try/BPTI | −61.42 | −14.68 | 0.017 | 0.00006 [34] | −75.43 | (−51.46) | 2PTC [35] |
| α-thrombin/hirudin-v2 | −78.95 | −18.87 | 0.000015 | 0.000022 [36] | −77.91 | (−49.37) | 4HTC [37] |
| legumain/cystatin E | (−31.88) | −7.62 | 46.4/19.8 2 | 0.0107 [38] | −62.59/43.95 2 | −41.84 | 4N6O [39] |
| MPro/cyclo-14-mer | (−30.46) | −7.28 | 17 | 14 [40] | −44.81 | −44.33 | 7RNW [40] |
| BACE-1/22-mer | −53.68 | −12.83 | 0.39/10.0 | 3.2 3 [41] | −48.46 | −45.60/47.28 | 5MCQ [41] |
| HIV/cyclo-9-mer | −51.97 | −12.42 | 0.779 | 4.02 3 [42] | −47.90 | (−36.00) | 7YF6 [42] |
| MMP-14/TIMP-2 | −52.09 | −12.45 | 0.740/0.149 | 0.104 [43] | −56.95 | −56.07 | 1BUV [44] |
| MMP-3/TIMP-1 | (−65.90) | −15.75 | 0.003/0.087 4 | 0.130 [45] | −56.40 | −57.32 4 | 1UEA [46] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goettig, P.; Chen, X.; Harris, J.M. Correlation of Experimental and Calculated Inhibition Constants of Protease Inhibitor Complexes. Int. J. Mol. Sci. 2024, 25, 2429. https://doi.org/10.3390/ijms25042429
Goettig P, Chen X, Harris JM. Correlation of Experimental and Calculated Inhibition Constants of Protease Inhibitor Complexes. International Journal of Molecular Sciences. 2024; 25(4):2429. https://doi.org/10.3390/ijms25042429
Chicago/Turabian StyleGoettig, Peter, Xingchen Chen, and Jonathan M. Harris. 2024. "Correlation of Experimental and Calculated Inhibition Constants of Protease Inhibitor Complexes" International Journal of Molecular Sciences 25, no. 4: 2429. https://doi.org/10.3390/ijms25042429
APA StyleGoettig, P., Chen, X., & Harris, J. M. (2024). Correlation of Experimental and Calculated Inhibition Constants of Protease Inhibitor Complexes. International Journal of Molecular Sciences, 25(4), 2429. https://doi.org/10.3390/ijms25042429