
Hematopoietic Prostaglandin D Synthase Is Increased in Mast Cells and Pericytes in Autopsy Myocardial Specimens from Patients with Duchenne Muscular Dystrophy

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
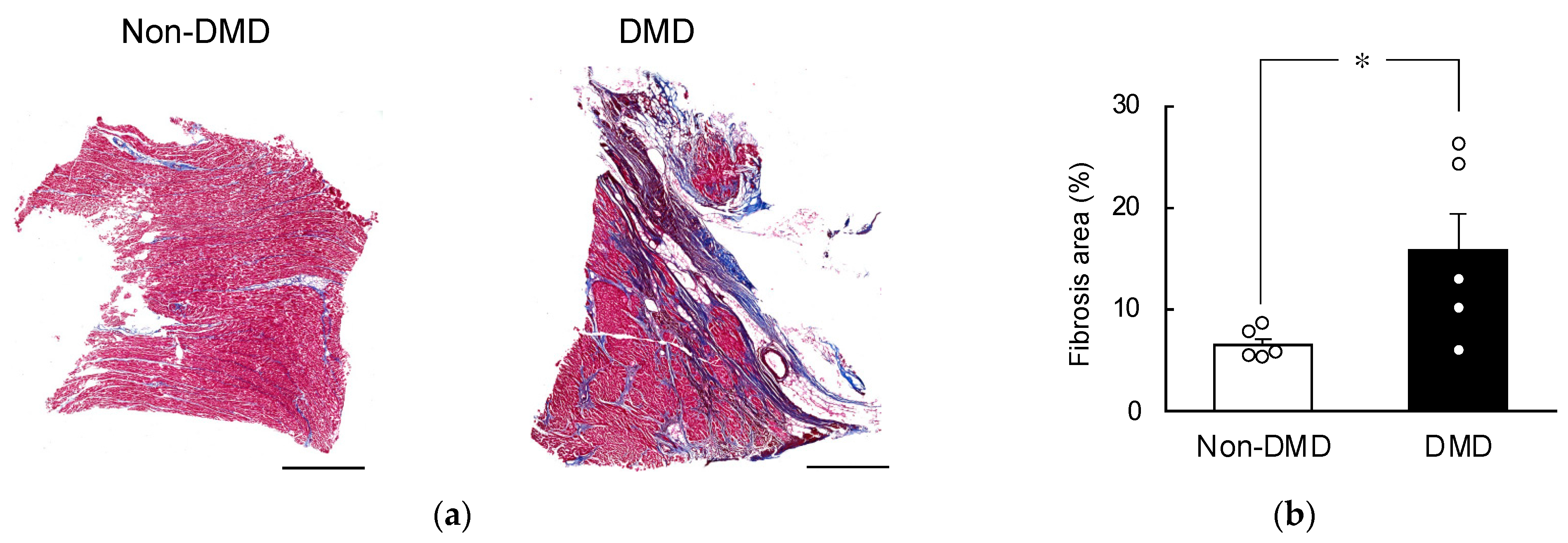
2.1. Fibrotic Progression in DMD Autopsy Myocardial Sections
2.2. Comparison of HPGDS Levels in DMD Autopsy Myocardial Specimens
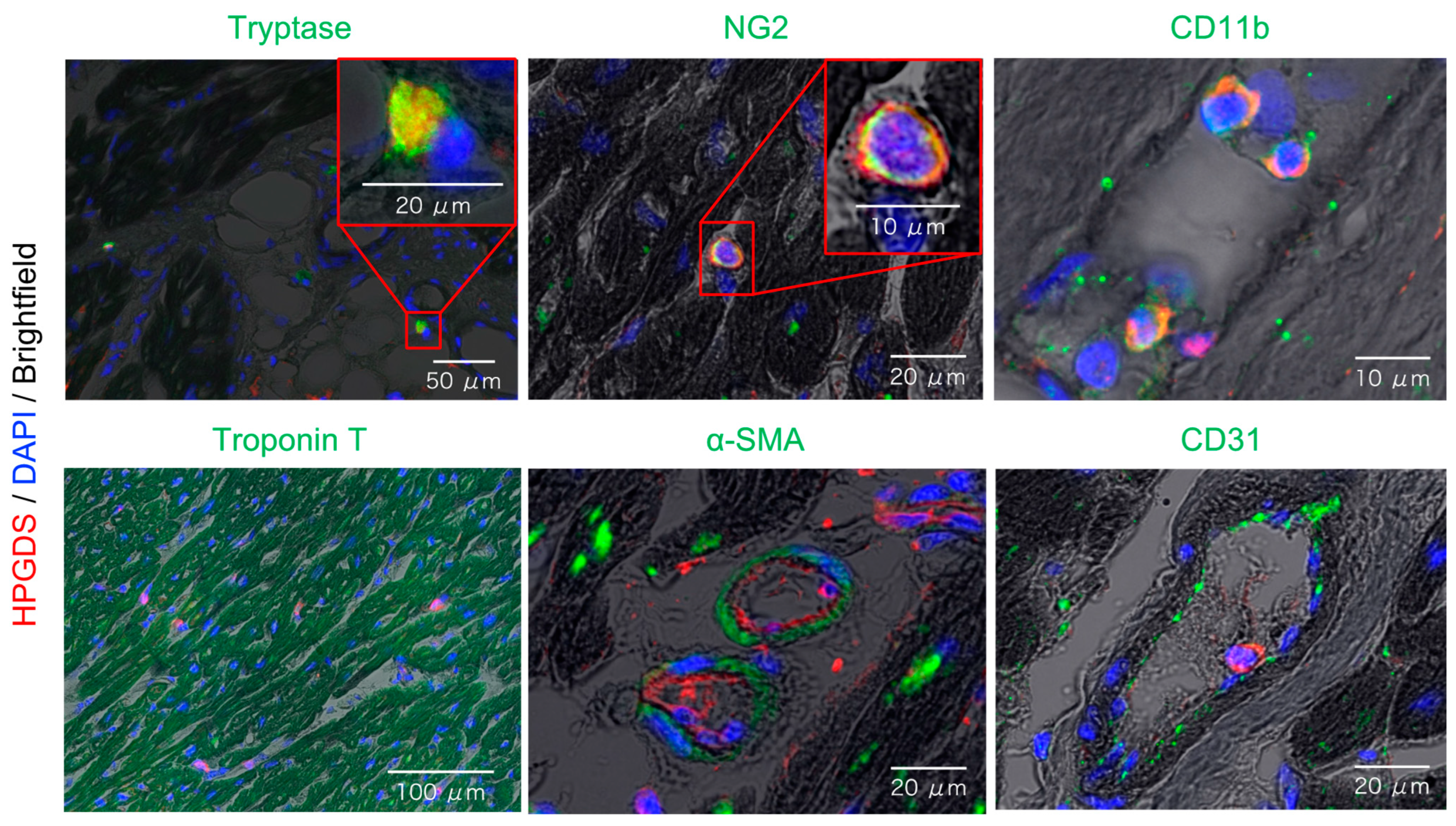
2.3. Localization of HPGDS in Mast Cells, Pericytes, and Myeloid Cells of the Heart
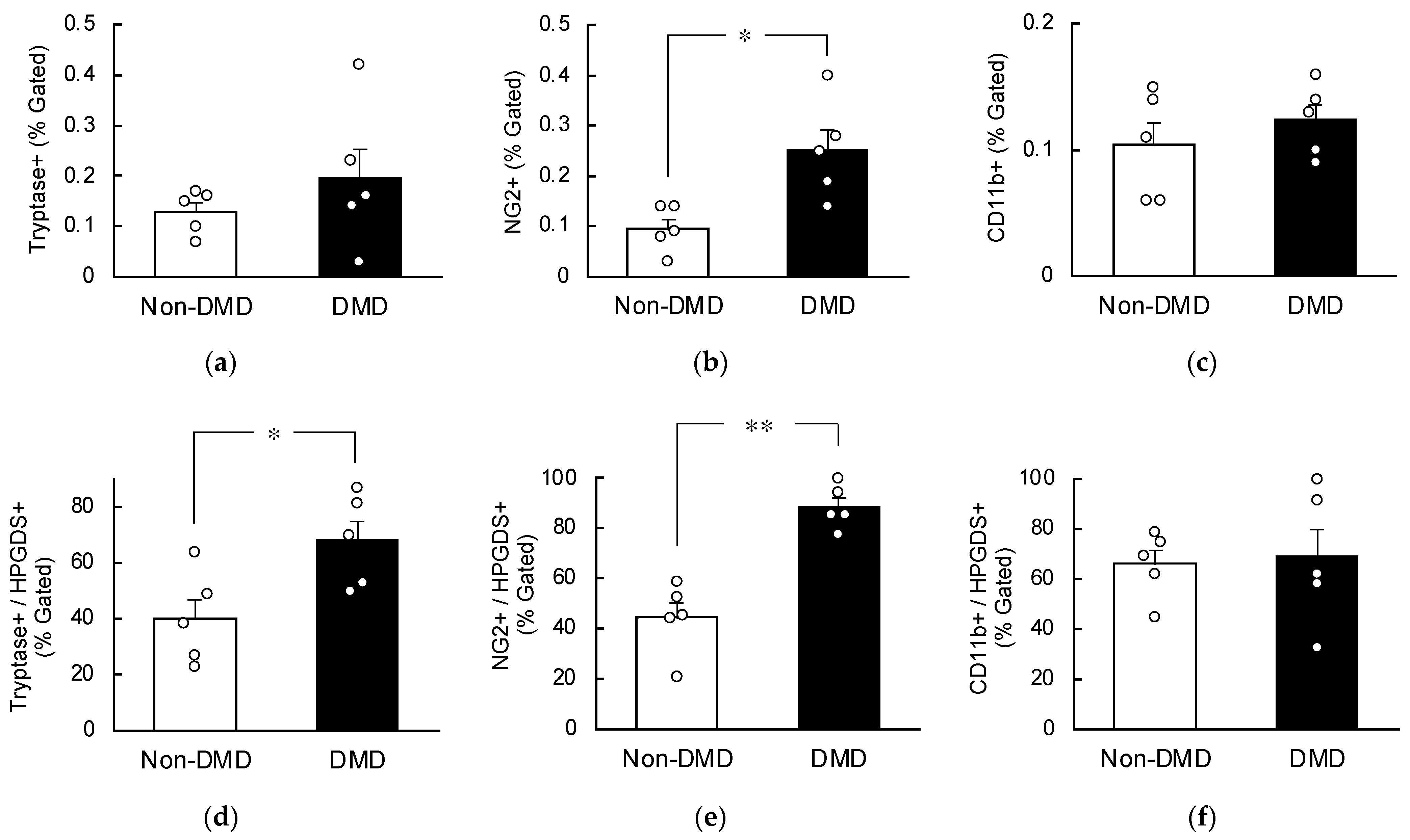
2.4. Increased HPGDS in Mast Cells and Pericytes in DMD Autopsy Myocardial Specimens
3. Discussion
4. Materials and Methods
4.1. Autopsy Myocardial Specimens
4.2. Quantitative Reverse Transcription (RT)-Polymerase Chain Reaction (PCR)
4.3. Western Blotting
4.4. Preparation of Paraffin Sections and Deparaffinization
4.5. Masson Trichrome Staining
4.6. Fluorescent Immunostaining
4.7. Flow Cytometry
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Tyler, K.L. Origins and early descriptions of “Duchenne muscular dystrophy”. Muscle Nerve 2003, 28, 402–422. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Colvin, M.K.; Cripe, L.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: Primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018, 17, 445–455. [Google Scholar] [CrossRef]
- Emery, A.E. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Tsuda, T. Clinical manifestations and overall management strategies for Duchenne muscular dystrophy. Methods Mol. Biol. 2018, 1687, 19–28. [Google Scholar]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Implementation of multidisciplinary care. Lancet Neurol. 2010, 9, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Urade, Y.; Hayaishi, O. Prostaglandin D2 and sleep/wake regulation. Sleep Med. Rev. 2011, 15, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Sang, D.; Lin, K.; Yang, Y.; Ran, G.; Li, B.; Chen, C.; Li, Q.; Ma, Y.; Lu, L.; Cui, X.-Y.; et al. Prolonged sleep deprivation induces a cytokine-storm-like syndrome in mammals. Cell 2023, 186, 5500–5516.e21. [Google Scholar] [CrossRef] [PubMed]
- Kanaoka, Y.; Urade, Y. Hematopoietic prostaglandin D synthase. Prostaglandins Leukot. Essent. Fatty Acids 2003, 69, 163–167. [Google Scholar] [CrossRef]
- Rittchen, S.; Heinemann, A. Therapeutic potential of hematopoietic prostaglandin D(2) synthase in allergic inflammation. Cells 2019, 8, 619. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; Urade, Y.; Jakobsson, P.-J. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef] [PubMed]
- Okinaga, T.; Mohri, I.; Fujimura, H.; Imai, K.; Ono, J.; Urade, Y.; Taniike, M. Induction of hematopoietic prostaglandin D synthase in hyalinated necrotic muscle fibers: Its implication in grouped necrosis. Acta Neuropathol. 2002, 104, 377–384. [Google Scholar] [CrossRef]
- Nakagawa, T.; Takeuchi, A.; Kakiuchi, R.; Lee, T.; Yagi, M.; Awano, H.; Iijima, K.; Takeshima, Y.; Urade, Y.; Matsuo, M. A prostaglandin D2 metabolite is elevated in the urine of Duchenne muscular dystrophy patients and increases further from 8 years old. Clin. Chim. Acta 2013, 423, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, E.; Komaki, H.; Tachimori, H.; Miyoshi, K.; Yamamiya, I.; Shimizu-Motohashi, Y.; Ishiyama, A.; Saito, T.; Nakagawa, E.; Sugai, K.; et al. Urinary prostaglandin metabolites as Duchenne muscular dystrophy progression markers. Brain Dev. 2018, 40, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Mohri, I.; Aritake, K.; Taniguchi, H.; Sato, Y.; Kamauchi, S.; Nagata, N.; Maruyama, T.; Taniike, M.; Urade, Y. Inhibition of prostaglandin D synthase suppresses muscular necrosis. Am. J. Pathol. 2009, 174, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, H.; Shibata, N.; Endo, A.; Ito, T.; Yanase, Y.; Murakami, Y.; Fujii, K.; Hamamura, K.; Saeki, Y.; Naito, M.; et al. Discovery of a highly potent and selective degrader targeting hematopoietic prostaglandin D synthase via in silico design. J. Med. Chem. 2021, 64, 15868–15882. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.; Yasuda, S.; McNally, E.; Metzger, J.M. Distinct pathophysiological mechanisms of cardiomyopathy in hearts lacking dystrophin or the sarcoglycan complex. FASEB J. 2011, 25, 3106–3114. [Google Scholar] [CrossRef]
- Houang, E.M.; Sham, Y.Y.; Bates, F.S.; Metzger, J.M. Muscle membrane integrity in Duchenne muscular dystrophy: Recent advances in copolymer-based muscle membrane stabilizers. Skelet. Muscle 2018, 8, 31. [Google Scholar] [CrossRef]
- Pettipher, R. The roles of the prostaglandin D(2) receptors DP(1) and CRTH2 in promoting allergic responses. Br. J. Pharmacol. 2008, 153, S191–S199. [Google Scholar] [CrossRef] [PubMed]
- Boie, Y.; Sawyer, N.; Slipetz, D.M.; Metters, K.M.; Abramovitz, M. Molecular cloning and characterization of the human prostanoid DP receptor. J. Biol. Chem. 1995, 270, 18910–18916. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Shirakabe, A.; Ikeda, Y.; Sciarretta, S.; Zablocki, D.K.; Sadoshima, J. Aging and autophagy in the heart. Circ. Res. 2016, 118, 1563–1576. [Google Scholar] [CrossRef]
- Zissler, A.; Stoiber, W.; Steinbacher, P.; Geissenberger, J.; Monticelli, F.C.; Pittner, S. Postmortem protein degradation as a tool to estimate the PMI: A systematic review. Diagnostics 2020, 10, 1014. [Google Scholar] [CrossRef]
- Lee, L.L.; Chintalgattu, V. Pericytes in the heart. Adv. Exp. Med. Biol. 2019, 1122, 187–210. [Google Scholar] [PubMed]
- Loperfido, M.; Steele-Stallard, H.B.; Tedesco, F.S.; VandenDriessche, T. Pluripotent stem cells for gene therapy of degenerative muscle diseases. Curr. Gene Ther. 2015, 15, 364–380. [Google Scholar] [CrossRef]
- Tedesco, F.S.; Moyle, L.A.; Perdiguero, E. Muscle interstitial cells: A brief field guide to non-satellite cell populations in skeletal muscle. Methods Mol. Biol. 2017, 1556, 129–147. [Google Scholar]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Dobie, R.; Wilson-Kanamori, J.R.; Henderson, B.E.P.; Smith, J.R.; Matchett, K.P.; Portman, J.R.; Wallenborg, K.; Picelli, S.; Zagorska, A.; Pendem, S.V.; et al. Single-cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis. Cell Rep. 2019, 29, 1832–1847.e8. [Google Scholar] [CrossRef]
- Kuppe, C.; Ibrahim, M.M.; Kranz, J.; Zhang, X.; Ziegler, S.; Perales-Patón, J.; Jansen, J.; Reimer, K.C.; Smith, J.R.; Dobie, R.; et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 2021, 589, 281–286. [Google Scholar] [CrossRef]
- Quijada, P.; Park, S.; Zhao, P.; Kolluri, K.S.; Wong, D.; Shih, K.D.; Fang, K.; Pezhouman, A.; Wang, L.; Daraei, A.; et al. Cardiac pericytes mediate the remodeling response to myocardial infarction. J. Clin. Investig. 2023, 133, e162188. [Google Scholar] [CrossRef]
- Takeshita, E.; Komaki, H.; Shimizu-Motohashi, Y.; Ishiyama, A.; Sasaki, M.; Takeda, S. A phase I study of TAS-205 in patients with Duchenne muscular dystrophy. Ann. Clin. Transl. Neurol. 2018, 5, 1338–1349. [Google Scholar] [CrossRef]
- Komaki, H.; Maegaki, Y.; Matsumura, T.; Shiraishi, K.; Awano, H.; Nakamura, A.; Kinoshita, S.; Ogata, K.; Ishigaki, K.; Saitoh, S.; et al. Early phase 2 trial of TAS-205 in patients with Duchenne muscular dystrophy. Ann. Clin. Transl. Neurol. 2020, 7, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Kanaoka, Y.; Fujimori, K.; Kikuno, R.; Sakaguchi, Y.; Urade, Y.; Hayaishi, O. Structure and chromosomal localization of human and mouse genes for hematopoietic prostaglandin D synthase. Conservation of the ancestral genomic structure of sigma-class glutathione S-transferase. Eur. J. Biochem. 2000, 267, 3315–3322. [Google Scholar] [CrossRef] [PubMed]
- Mohri, I.; Eguchi, N.; Suzuki, K.; Urade, Y.; Taniike, M. Hematopoietic prostaglandin D synthase is expressed in microglia in the developing postnatal mouse brain. Glia 2003, 42, 263–274. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-DMD | |||||
|---|---|---|---|---|---|
| Number | Age | Gender | Postmortem Time at Autopsy | Diagnosis | Gene Search |
| #1 | 53 | M | 2 h 24 min | Amyotrophic lateral sclerosis | - |
| #2 | 79 | F | 1 h 20 min | Neurofibrillary dementia | - |
| #3 | 66 | M | 14 h 22 min | Distal myopathy | - |
| #4 | 61 | M | 4 h 10 min | Spinal muscular atrophy | SMN1 exon 7 deletion |
| #5 | 61 | M | 18 h 00 min | Anti-SRP antibody positivenecrotizing myositis | - |
| DMD | |||||
| Number | Age | Gender | Postmortem Time at Autopsy | Diagnosis | Gene Search |
| #1 | 29 | M | 12 h 01 min | DMD | exon 5–16 deletion (in-frame) |
| #2 | 28 | M | 17 h 07 min | DMD | exon 52-deletion |
| #3 | 41 | M | 1 h 33 min | DMD | No data |
| #4 | 25 | M | 3 h 26 min | DMD | exon 52-deletion |
| #5 | 39 | M | 2 h 24 min | DMD | Deletion/duplication (−) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamamura, K.; Yoshida, Y.; Oyama, K.; Li, J.; Kawano, S.; Inoue, K.; Toyooka, K.; Yamadera, M.; Matsunaga, N.; Matsumura, T.; et al. Hematopoietic Prostaglandin D Synthase Is Increased in Mast Cells and Pericytes in Autopsy Myocardial Specimens from Patients with Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2024, 25, 1846. https://doi.org/10.3390/ijms25031846
Hamamura K, Yoshida Y, Oyama K, Li J, Kawano S, Inoue K, Toyooka K, Yamadera M, Matsunaga N, Matsumura T, et al. Hematopoietic Prostaglandin D Synthase Is Increased in Mast Cells and Pericytes in Autopsy Myocardial Specimens from Patients with Duchenne Muscular Dystrophy. International Journal of Molecular Sciences. 2024; 25(3):1846. https://doi.org/10.3390/ijms25031846
Chicago/Turabian StyleHamamura, Kengo, Yuya Yoshida, Kosuke Oyama, Junhao Li, Shimpei Kawano, Kimiko Inoue, Keiko Toyooka, Misaki Yamadera, Naoya Matsunaga, Tsuyoshi Matsumura, and et al. 2024. "Hematopoietic Prostaglandin D Synthase Is Increased in Mast Cells and Pericytes in Autopsy Myocardial Specimens from Patients with Duchenne Muscular Dystrophy" International Journal of Molecular Sciences 25, no. 3: 1846. https://doi.org/10.3390/ijms25031846
APA StyleHamamura, K., Yoshida, Y., Oyama, K., Li, J., Kawano, S., Inoue, K., Toyooka, K., Yamadera, M., Matsunaga, N., Matsumura, T., & Aritake, K. (2024). Hematopoietic Prostaglandin D Synthase Is Increased in Mast Cells and Pericytes in Autopsy Myocardial Specimens from Patients with Duchenne Muscular Dystrophy. International Journal of Molecular Sciences, 25(3), 1846. https://doi.org/10.3390/ijms25031846