
Endothelial Mitochondria Transfer to Melanoma Induces M2-Type Macrophage Polarization and Promotes Tumor Growth by the Nrf2/HO-1-Mediated Pathway
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
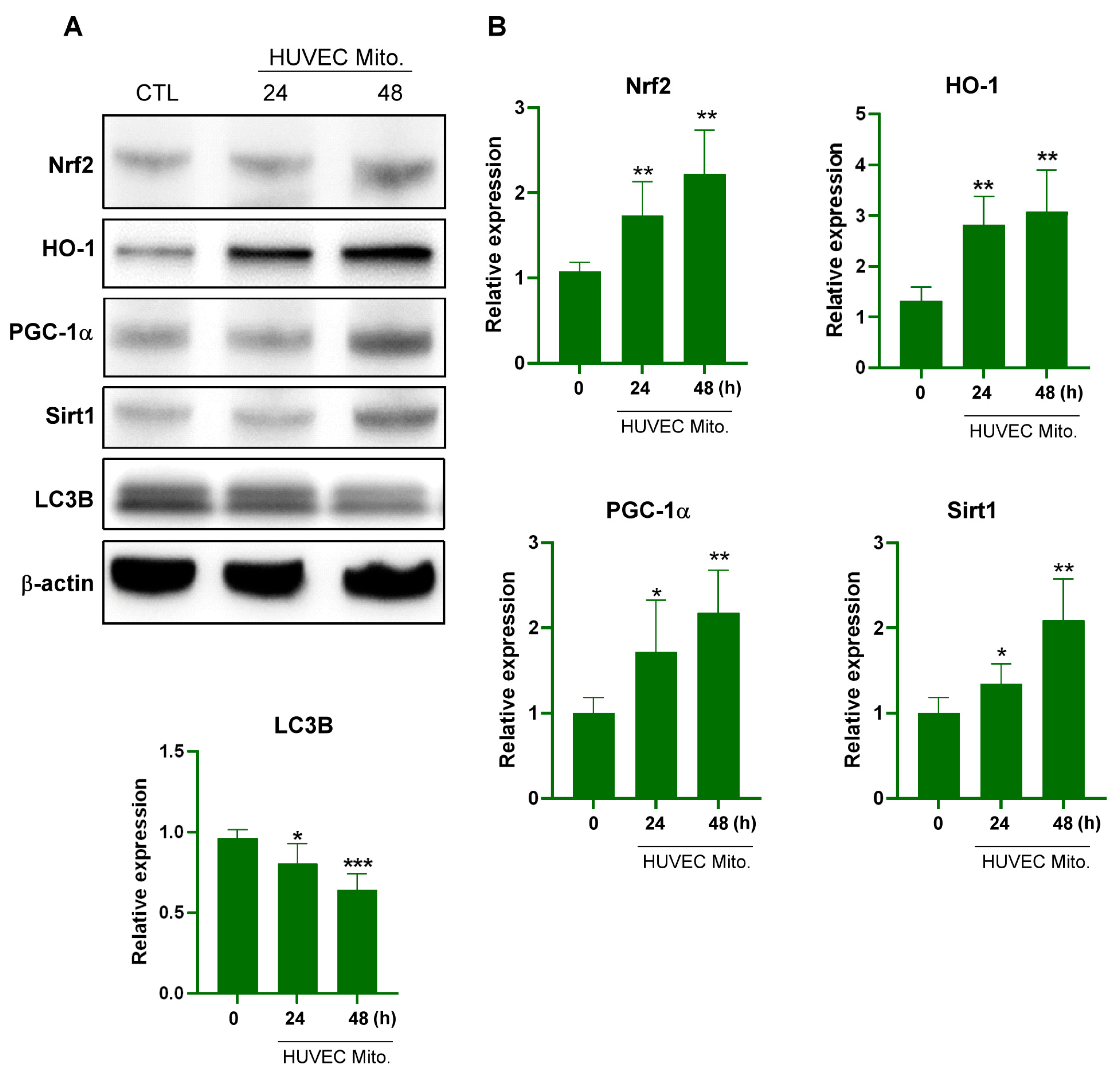
2.1. Transplanted Endothelial Mitochondria Upregulated Mitochondrial Biogenesis with Mediation by the Redox-Sensitive Transcription Factor Nrf2
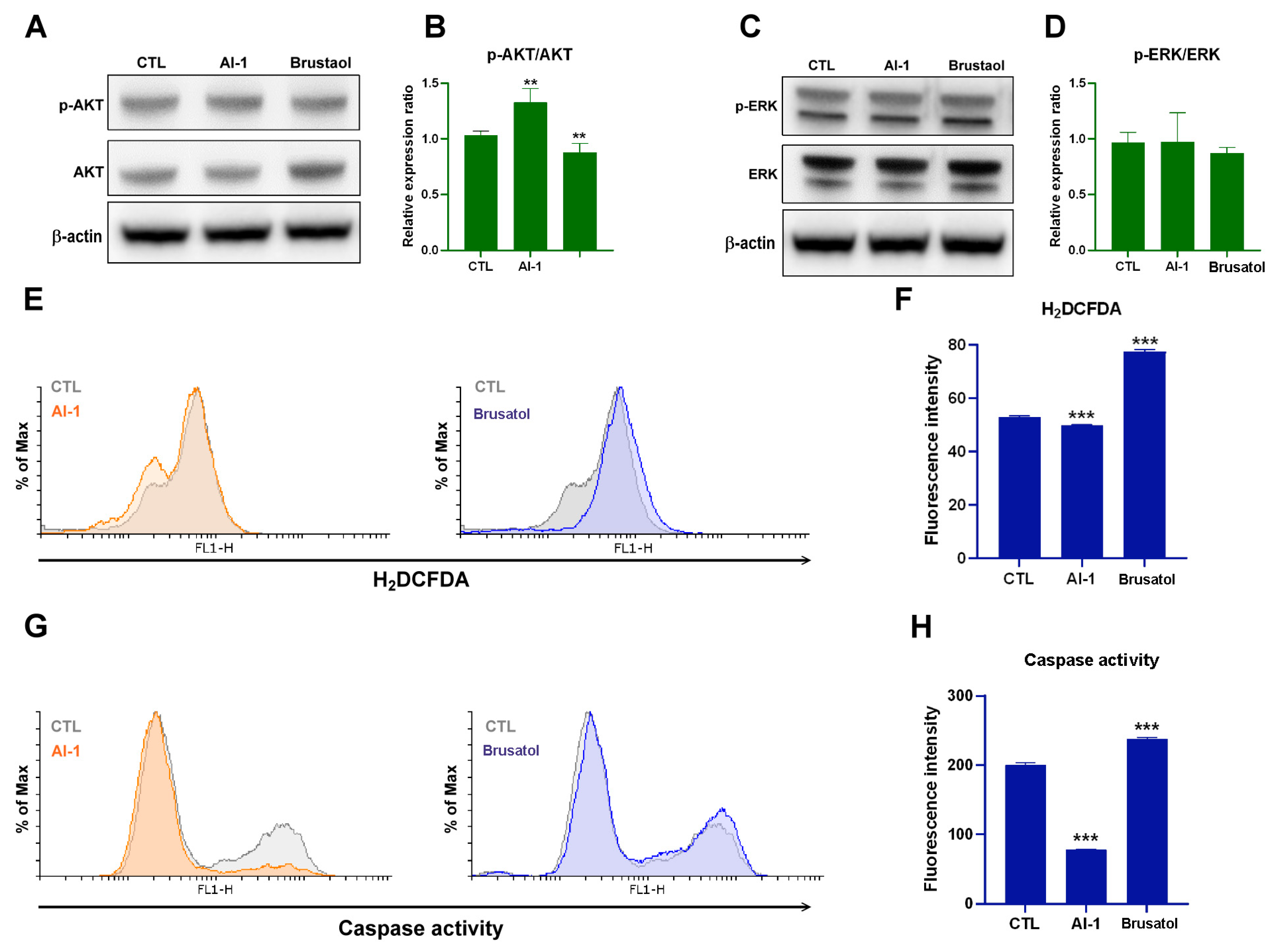
2.2. Transplanted Endothelial Mitochondria Enhanced Melanoma Cells’ Viability through Activation of ERK and AKT Signaling and Suppression of Apoptosis
2.3. Transplanted Endothelial Mitochondria-Suppressed AKT, Oxidative Stress and Apoptosis of Melanoma Cells through Activation of Nrf2-Mediated Signaling
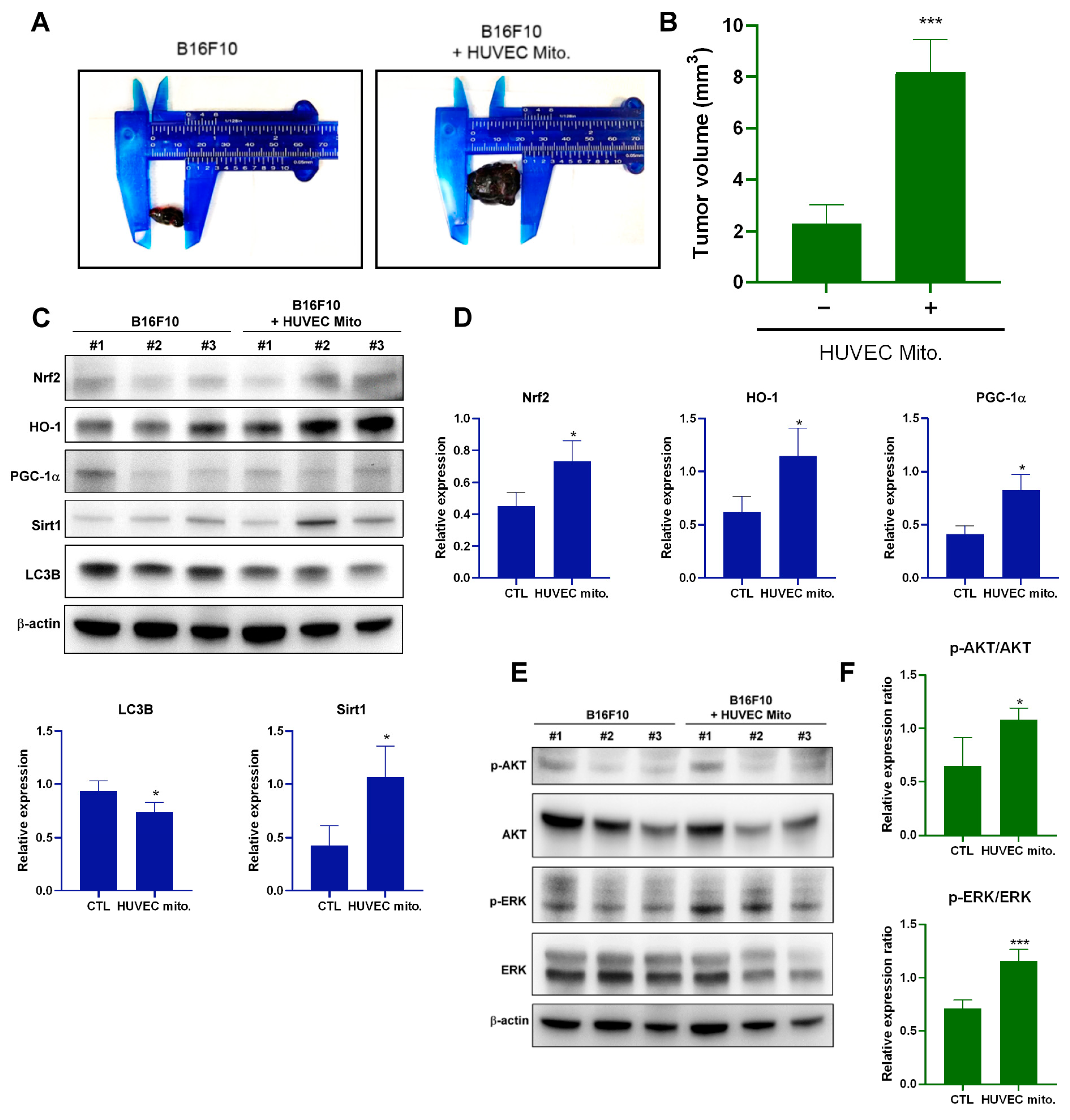
2.4. Transplantation of Endothelial Mitochondria-Promoted Melanoma Tumor Growth through Nrf2-Mediated Pathway in a Tumor Xenograft Animal Model
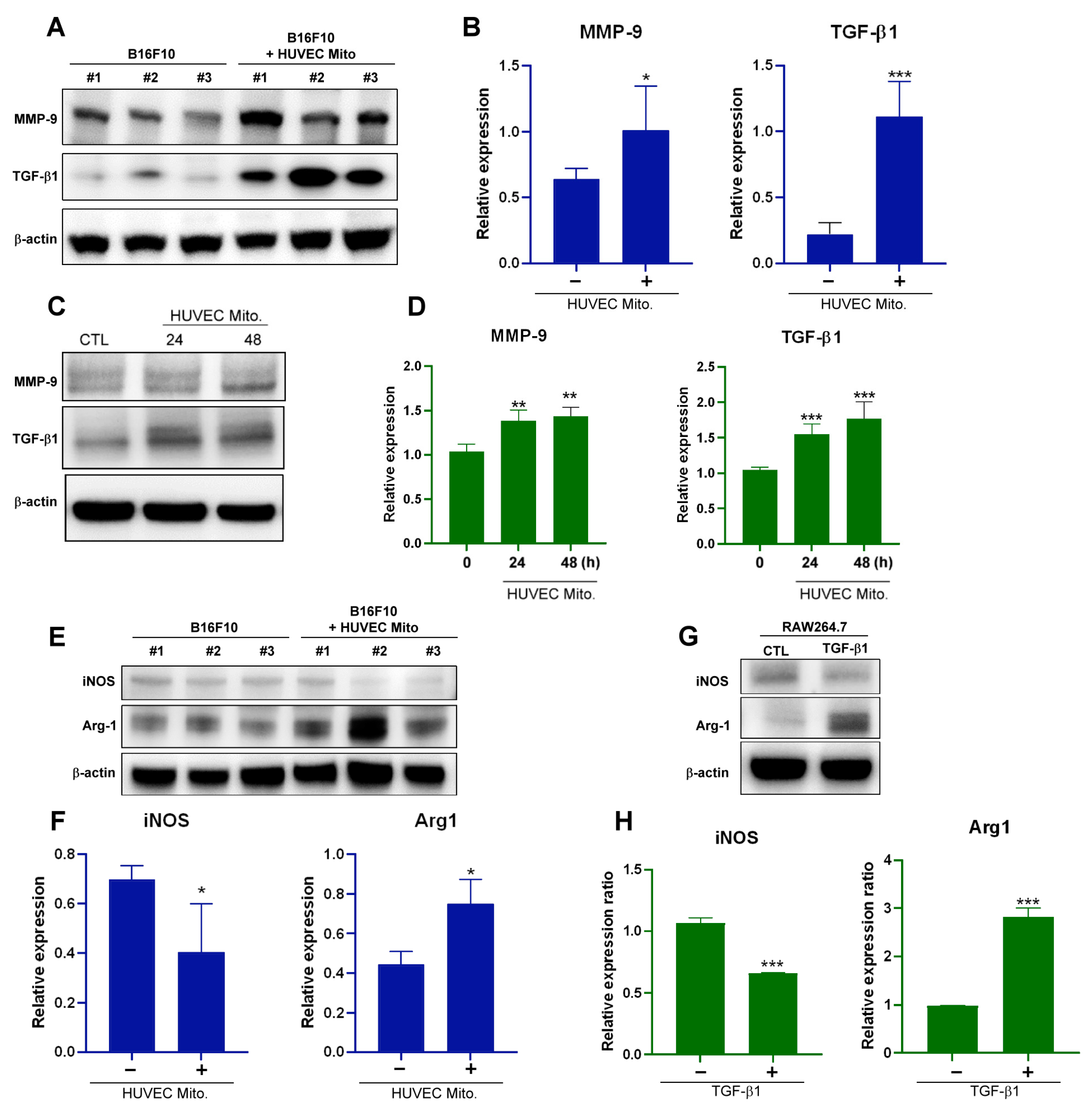
2.5. Transplant of Endothelial Mitochondria to Melanoma Upregulated Matrix Metallopeptidase 9, TGF-β1 and Induced M2-Type TAM in a Tumor Xenograft Animal Model
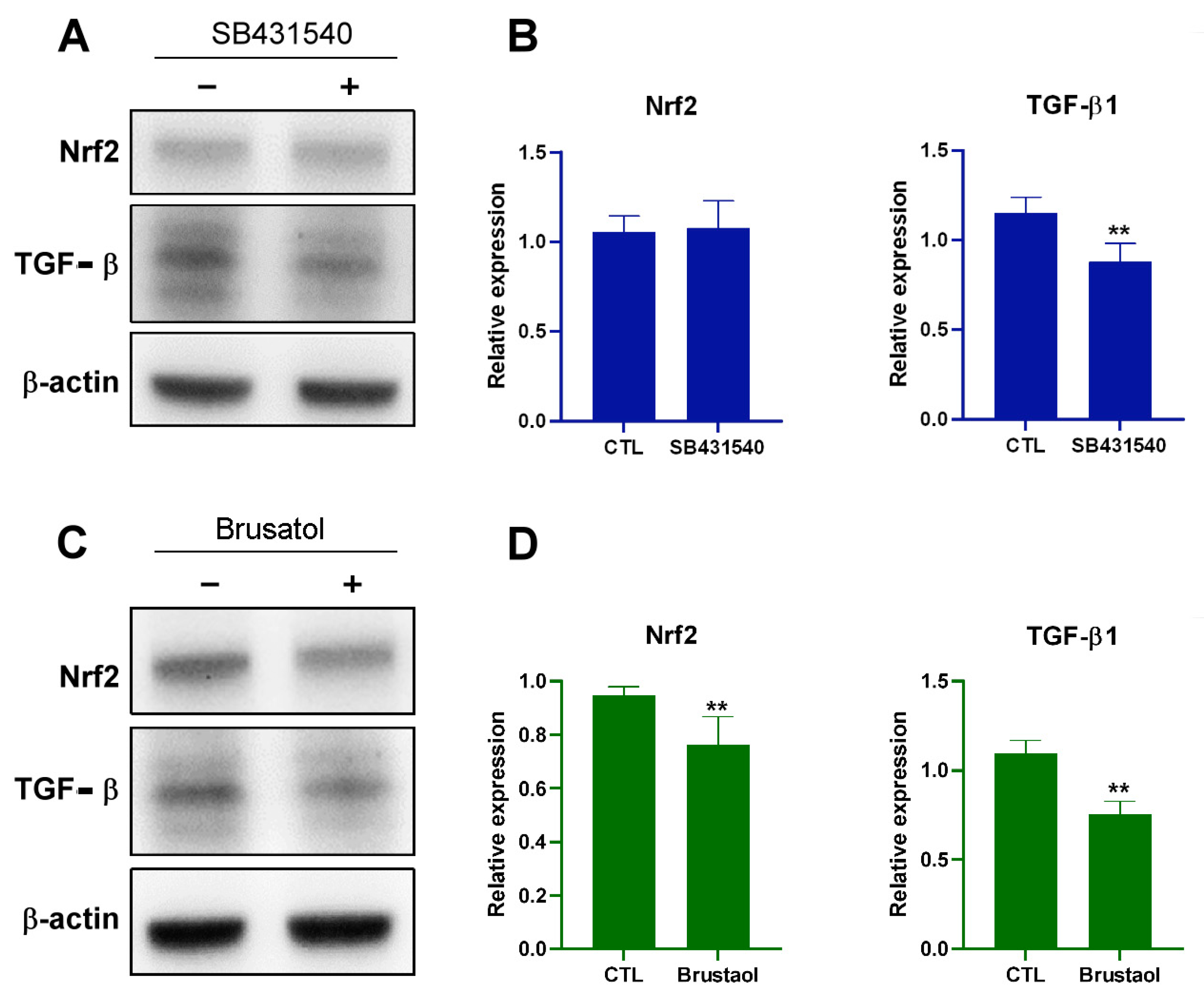
2.6. Transplant of Endothelial Mitochondria into Melanoma Cells Upregulated TGF-β Expression through the Nrf2-Mediated Pathway
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Isolation of Mitochondria and Mitochondrial Transplantation
4.3. Cell Viability Analysis
4.4. Cellular ROS Assay
4.5. Caspase Activity Analysis
4.6. Western Blot
4.7. Xenograft Tumor Experiments
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Mitra, D.; Farr, M.; Nagarajan, P.; Ho, J.; Bishop, A.J.; Jhingran, A.; Farooqi, A.S.; Frumovitz, M.; Amaria, R.N.; McQuade, J.L.; et al. Gynecologic tract melanoma in the contemporary therapeutic era: High rates of local and distant disease progression. Gynecol. Oncol. 2022, 167, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Wu, Q.; Song, L.; Wu, W. Treatment of primary vaginal malignant melanoma and review of previous literature: A case report. Medicine 2023, 102, e36128. [Google Scholar]
- Khayyat, A.; Pour, M.A.E.; Mousavi, S.; Khalili-Toosi, A.-R.; Amin, A. Primary Malignant Melanoma of the Genitourinary System: A Systemic Review and Report of Eight Cases. Cureus 2022, 14, e30444. [Google Scholar] [CrossRef]
- Yajima, I.; Kumasaka, M.Y.; Thang, N.D.; Goto, Y.; Takeda, K.; Yamanoshita, O.; Iida, M.; Ohgami, N.; Tamura, H.; Kawamoto, Y.; et al. RAS/RAF/MEK/ERK and PI3K/PTEN/AKT Signaling in Malignant Melanoma Progression and Therapy. Dermatol. Res. Pract. 2011, 2011, 354191. [Google Scholar] [CrossRef]
- Davies, M.A. The Role of the PI3K-AKT Pathway in Melanoma. Cancer J. 2012, 18, 142–147. [Google Scholar] [CrossRef]
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK Signaling Pathway in Melanoma. Int. J. Mol. Sci. 2019, 20, 1483. [Google Scholar] [CrossRef]
- Ramirez, J.A.; Guitart, J.; Rao, M.S.; Diaz, L.K. Cyclin D1 expression in melanocytic lesions of the skin. Ann. Diagn. Pathol. 2005, 9, 185–188. [Google Scholar] [CrossRef]
- Villanueva, J.; Herlyn, M. Melanoma and the tumor microenvironment. Curr. Oncol. Rep. 2008, 10, 439–446. [Google Scholar] [CrossRef]
- Lou, E. A Ticket to Ride: The Implications of Direct Intercellular Communication via Tunneling Nanotubes in Peritoneal and Other Invasive Malignancies. Front. Oncol. 2020, 10, 559548. [Google Scholar] [CrossRef] [PubMed]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Lin, K.; Li, X.; Yuan, X.; Xu, P.; Ni, P.; Xu, D. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front. Immunol. 2020, 11, 1731. [Google Scholar] [CrossRef]
- Arlauckas, S.P.; Garren, S.B.; Garris, C.S.; Kohler, R.H.; Oh, J.; Pittet, M.J.; Weissleder, R. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics 2018, 8, 5842–5854. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef] [PubMed]
- Al Amir Dache, Z.; Thierry, A.R. Mitochondria-derived cell-to-cell communication. Cell Rep. 2023, 42, 112728. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.; Camporesi, E.; Peracchia, C. Direct Cell-Cell Communication via Membrane Pores, Gap Junction Channels, and Tunneling Nanotubes: Medical Relevance of Mitochondrial Exchange. Int. J. Mol. Sci. 2022, 23, 6133. [Google Scholar] [CrossRef]
- Spiegelman, B.M. Transcriptional control of mitochondrial energy metabolism through the PGC1 coactivators. Novartis Found Symp. 2007, 287, 60–63. [Google Scholar]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Vallabhaneni, K.C.; Haller, H.; Dumler, I. Vascular Smooth Muscle Cells Initiate Proliferation of Mesenchymal Stem Cells by Mitochondrial Transfer via Tunneling Nanotubes. Stem Cells Dev. 2012, 21, 3104–3113. [Google Scholar] [CrossRef] [PubMed]
- Fahey, M.; Bennett, M.; Thomas, M.; Montney, K.; Vivancos-Koopman, I.; Pugliese, B.; Browning, L.; Bonassar, L.J.; Delco, M. Mesenchymal stromal cells donate mitochondria to articular chondrocytes exposed to mitochondrial, environmental, and mechanical stress. Sci. Rep. 2022, 12, 21525. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Hu, J.; Yan, Q.; Zhu, J.; Zhu, Z.; Chen, Y.; Sun, J.; Zhang, R. Bone marrow-derived mesenchymal stem cells rescue injured H9c2 cells via transferring intact mitochondria through tunneling nanotubes in an in vitro simulated ischemia/reperfusion model. Mol. Med. Rep. 2016, 13, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ning, K.; Wang, D.-E.; Xu, H. Progress of Bone Marrow Mesenchymal Stem Cell Mitochondrial Transfer in Organ Injury Repair. Stem Cells Dev. 2023, 32, 379–386. [Google Scholar] [CrossRef]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef]
- Nikoo, A.; Roudkenar, M.H.; Sato, T.; Kuwahara, Y.; Tomita, K.; Pourmohammadi-Bejarpasi, Z.; Najafi-Ghalehlou, N.; Roushandeh, A.M. Mitochondrial transfer in PC-3 cells fingerprinted in ferroptosis sensitivity: A brand new approach targeting cancer metabolism. Hum. Cell 2023, 36, 1441–1450. [Google Scholar] [CrossRef]
- Nakhle, J.; Khattar, K.; Özkan, T.; Boughlita, A.; Moussa, D.A.; Darlix, A.; Lorcy, F.; Rigau, V.; Bauchet, L.; Gerbal-Chaloin, S.; et al. Mitochondria Transfer from Mesenchymal Stem Cells Confers Chemoresistance to Glioblastoma Stem Cells through Metabolic Rewiring. Cancer Res. Commun. 2023, 3, 1041–1056. [Google Scholar] [CrossRef]
- Caicedo, A.; Fritz, V.; Brondello, J.-M.; Ayala, M.; Dennemont, I.; Abdellaoui, N.; de Fraipont, F.; Moisan, A.; Prouteau, C.A.; Boukhaddaoui, H.; et al. MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci. Rep. 2015, 5, 9073. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Martín-Rodríguez, A.; Yáñez-Sepúlveda, R.; Tornero-Aguilera, J.F. Mitochondrial Transfer as a Novel Therapeutic Approach in Disease Diagnosis and Treatment. Int. J. Mol. Sci. 2023, 24, 8848. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, M.; Yang, T.; Wang, X.; Cheng, P.; Zhou, H. What can we do to optimize mitochondrial transplantation therapy for myocardial ischemia-reperfusion injury? Mitochondrion 2023, 72, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, Y.; Qi, Z.; Cao, L.; Ding, S. Mitochondrial transfer/transplantation: An emerging therapeutic approach for multiple diseases. Cell Biosci. 2022, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yuan, S.; Li, Y.; Zhang, Z.; Xiao, W.; Tang, D.; Ye, K.; Liu, Z.; Wang, C.; Zheng, Y.; et al. Regulation on SIRT1-PGC-1α/Nrf2 pathway together with selective inhibition of aldose reductase makes compound hr5F a potential agent for the treatment of diabetic complications. Biochem. Pharmacol. 2018, 150, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef]
- Bales, E.S.; Dietrich, C.; Bandyopadhyay, D.; Schwahn, D.J.; Xu, W.; Didenko, V.; Leiss, P.; Conrad, N.; Pereira-Smith, O.; Orengo, I.; et al. High Levels of Expression of p27KIP1 and Cyclin E in Invasive Primary Malignant Melanomas. J. Investig. Dermatol. 1999, 113, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Napoli, S.; Scuderi, C.; Gattuso, G.; Bella, V.D.; Candido, S.; Basile, M.S.; Libra, M.; Falzone, L. Functional Roles of Matrix Metalloproteinases and Their Inhibitors in Melanoma. Cells 2020, 9, 1151. [Google Scholar] [CrossRef] [PubMed]
- Zimta, A.-A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, R.; Menju, T.; Shimazu, Y.; Toyazaki, T.; Chiba, N.; Miyamoto, H.; Hirayama, Y.; Nishikawa, S.; Tanaka, S.; Yutaka, Y.; et al. M2-like tumor-associated macrophages promote epithelial-mesenchymal transition through the transforming growth factor β/Smad/zinc finger e-box binding homeobox pathway with increased metastatic potential and tumor cell proliferation in lung squamous cell carcinoma. Cancer Sci. 2023, 114, 4521–4534. [Google Scholar] [PubMed]
- Borcherding, N.; Brestoff, J.R. The power and potential of mitochondria transfer. Nature 2023, 623, 283–291. [Google Scholar] [CrossRef]
- Goliwas, K.F.; Libring, S.; Berestesky, E.; Gholizadeh, S.; Schwager, S.C.; Frost, A.R.; Gaborski, T.R.; Zhang, J.; Reinhart-King, C.A. Mitochondrial transfer from cancer-associated fibroblasts increases migration in aggressive breast cancer. J. Cell Sci. 2023, 136, jcs260419. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Rorbach, J.; Downes, K.; Szukszto, M.J.; Pekalski, M.L.; Minczuk, M. Macropinocytic entry of isolated mitochondria in epidermal growth factor-activated human osteosarcoma cells. Sci. Rep. 2017, 7, 12886. [Google Scholar] [CrossRef]
- Jiang, L.; Campagne, C.; Sundström, E.; Sousa, P.; Imran, S.; Seltenhammer, M.; Pielberg, G.; Olsson, M.J.; Egidy, G.; Andersson, L.; et al. Constitutive activation of the ERK pathway in melanoma and skin melanocytes in Grey horses. BMC Cancer 2014, 14, 857. [Google Scholar] [CrossRef]
- Dhawan, P.; Singh, A.B.; Ellis, D.L.; Richmond, A. Constitutive activation of Akt/protein kinase B in melanoma leads to up-regulation of nuclear factor-kappaB and tumor progression. Cancer Res. 2002, 62, 7335–7342. [Google Scholar] [PubMed]
- Ciołczyk-Wierzbicka, D.; Gil, D.; Laidler, P. Treatment of melanoma with selected inhibitors of signaling kinases effectively reduces proliferation and induces expression of cell cycle inhibitors. Med. Oncol. 2017, 35, 7. [Google Scholar] [CrossRef]
- Gammon, B.; Ali, L.; Guitart, J.; Gerami, P. Homogeneous Staining Regions for Cyclin D1, a Marker of Poor Prognosis in Malignant Melanoma. Am. J. Dermatopathol. 2012, 34, 487–490. [Google Scholar] [CrossRef]
- Mahrouf-Yorgov, M.; Augeul, L.; Da Silva, C.C.; Jourdan, M.; Rigolet, M.; Manin, S.; Ferrera, R.; Ovize, M.; Henry, A.; Guguin, A.; et al. Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differ. 2017, 24, 1224–1238. [Google Scholar] [CrossRef]
- Ohanna, M.; Bonet, C.; Bille, K.; Allegra, M.; Davidson, I.; Bahadoran, P.; Lacour, J.P.; Ballotti, R.; Bertolotto, C. SIRT1 promotes proliferation and inhibits the senescence-like phenotype in human melanoma cells. Oncotarget 2014, 5, 2085–2095. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Pagliei, B.; Cannata, S.M.; Rotilio, G.; Ciriolo, M.R. p53 Orchestrates the PGC-1α-Mediated Antioxidant Response Upon Mild Redox and Metabolic Imbalance. Antioxidants Redox Signal. 2013, 18, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, E.L.; Becker, A.L.; Indra, A.K. NRF2 and Key Transcriptional Targets in Melanoma Redox Manipulation. Cancers 2022, 14, 1531. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.M.; Dafferner, A.J.; Salloom, R.J.; Abdalla, M.Y. Heme Oxygenase-1 Inhibition Modulates Autophagy and Augments Arsenic Trioxide Cytotoxicity in Pancreatic Cancer Cells. Biomedicines 2023, 11, 2580. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Festuccia, C.; Angelucci, A.; Gravina, G.L.; Villanova, I.; Teti, A.; Albini, A.; Bologna, M.; Abini, A. Osteoblast-derived TGF-beta1 modulates matrix degrading protease expression and activity in prostate cancer cells. Int. J. Cancer 2000, 85, 407–415. [Google Scholar] [CrossRef]
- Khalbuss, W.E.; Hossain, M.; Elhosseiny, A. Primary malignant melanoma of the urinary bladder diagnosed by urine cytology: A case report. Acta Cytol. 2001, 45, 631–635. [Google Scholar] [CrossRef]
- Asai, Y.; Yanagawa, N.; Osakabe, M.; Yamada, N.; Sugimoto, R.; Sato, A.; Ito, K.; Koike, Y.; Tanji, T.; Sakuraba, M.; et al. The clinicopathological impact of tumor-associated macrophages in patients with cutaneous malignant melanoma. J. Surg. Oncol. 2024, 129, 381–391. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, F.-C.; Tsai, H.-Y.; Cheng, B.-L.; Tsai, K.-J.; Chen, P.-C.; Huang, Y.-B.; Liu, C.-J.; Wu, D.-C.; Wu, M.-C.; Huang, B.; et al. Endothelial Mitochondria Transfer to Melanoma Induces M2-Type Macrophage Polarization and Promotes Tumor Growth by the Nrf2/HO-1-Mediated Pathway. Int. J. Mol. Sci. 2024, 25, 1857. https://doi.org/10.3390/ijms25031857
Kuo F-C, Tsai H-Y, Cheng B-L, Tsai K-J, Chen P-C, Huang Y-B, Liu C-J, Wu D-C, Wu M-C, Huang B, et al. Endothelial Mitochondria Transfer to Melanoma Induces M2-Type Macrophage Polarization and Promotes Tumor Growth by the Nrf2/HO-1-Mediated Pathway. International Journal of Molecular Sciences. 2024; 25(3):1857. https://doi.org/10.3390/ijms25031857
Chicago/Turabian StyleKuo, Fu-Chen, Hsin-Yi Tsai, Bi-Ling Cheng, Kuen-Jang Tsai, Ping-Chen Chen, Yaw-Bin Huang, Chung-Jung Liu, Deng-Chyang Wu, Meng-Chieh Wu, Bin Huang, and et al. 2024. "Endothelial Mitochondria Transfer to Melanoma Induces M2-Type Macrophage Polarization and Promotes Tumor Growth by the Nrf2/HO-1-Mediated Pathway" International Journal of Molecular Sciences 25, no. 3: 1857. https://doi.org/10.3390/ijms25031857
APA StyleKuo, F.-C., Tsai, H.-Y., Cheng, B.-L., Tsai, K.-J., Chen, P.-C., Huang, Y.-B., Liu, C.-J., Wu, D.-C., Wu, M.-C., Huang, B., & Lin, M.-W. (2024). Endothelial Mitochondria Transfer to Melanoma Induces M2-Type Macrophage Polarization and Promotes Tumor Growth by the Nrf2/HO-1-Mediated Pathway. International Journal of Molecular Sciences, 25(3), 1857. https://doi.org/10.3390/ijms25031857