
Analysis of the Model of Atherosclerosis Formation in Pig Hearts as a Result of Impaired Activity of DNA Repair Enzymes
 , , ,
, , ,  , , , ,
, , , ,
Abstract
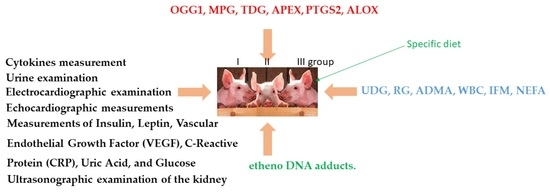
:
1. Introduction
2. Results
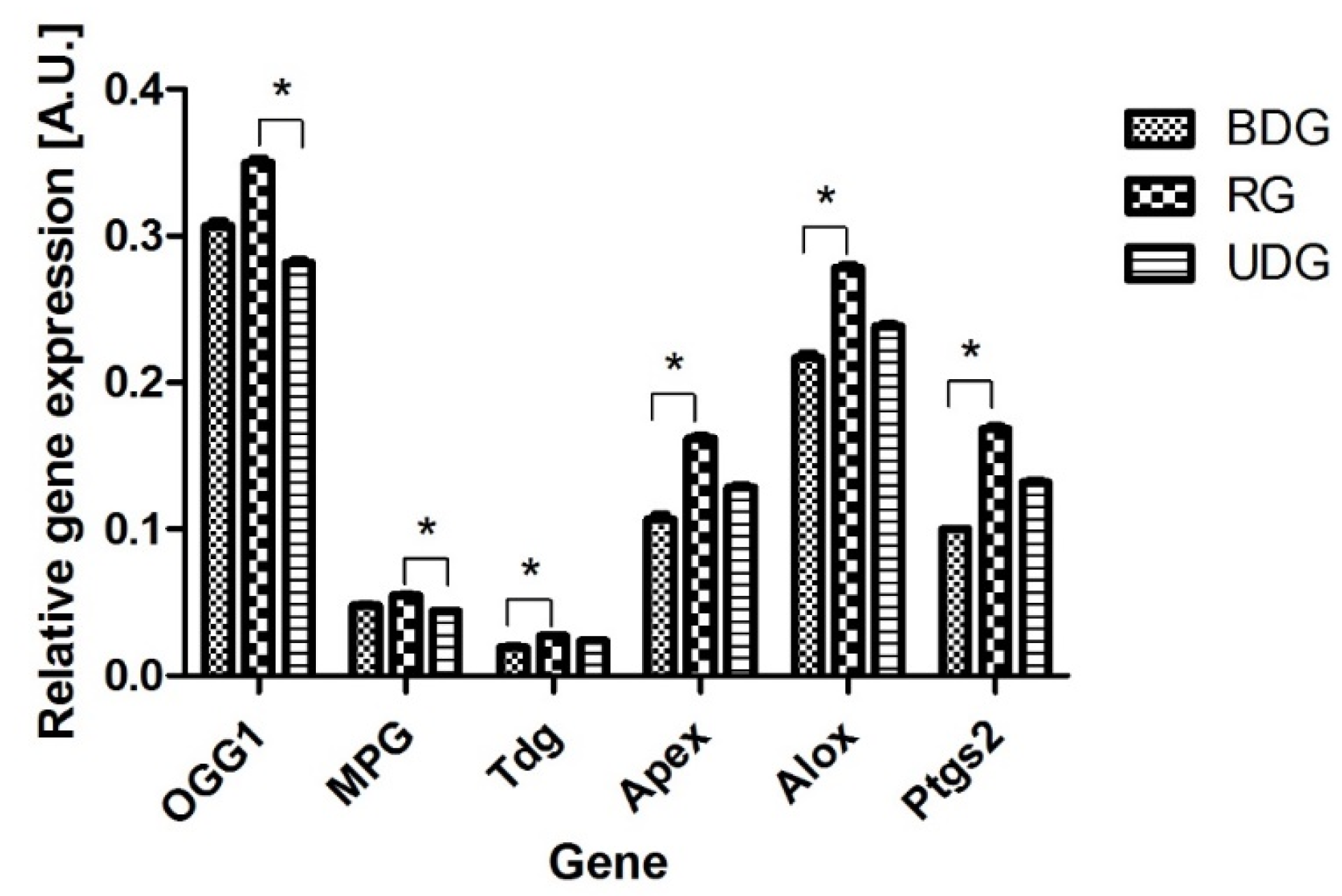
2.1. mRNA Level of Genes Encoding DNA Repair Enzymes
2.1.1. mRNA Abundance of DNA Repair Enzymes
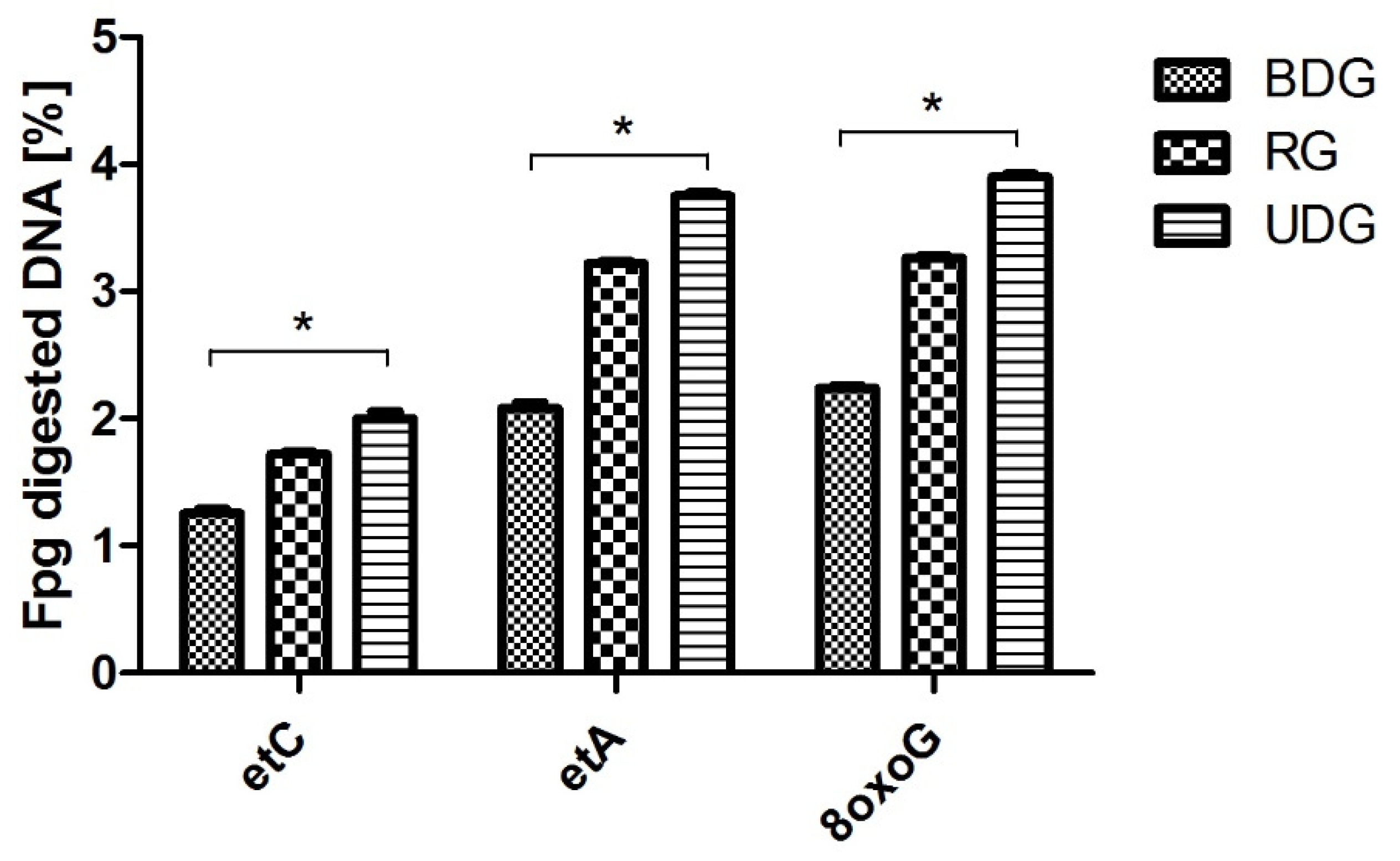
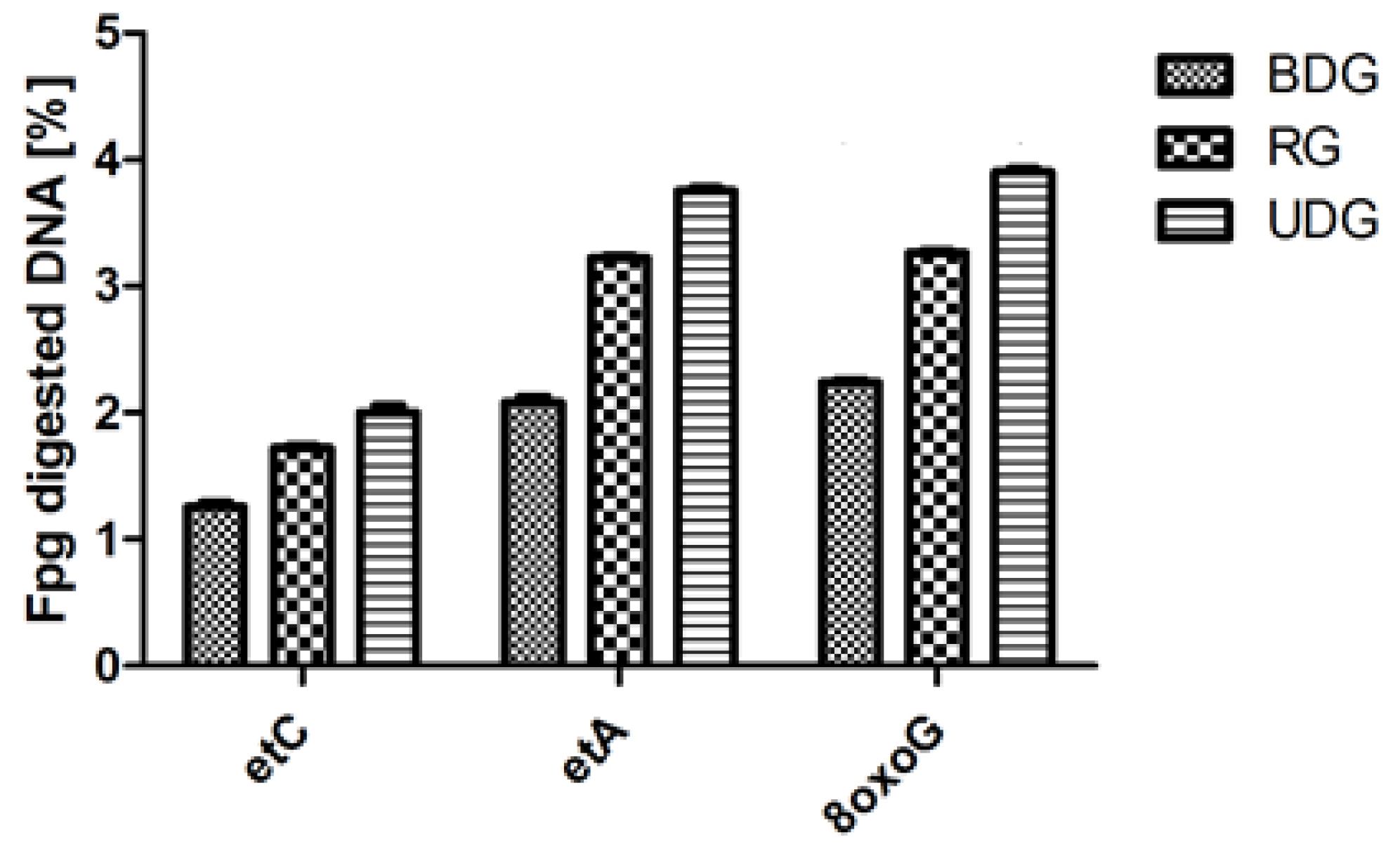
2.1.2. Analysis of DNA Repair after Fpg Cleavage
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Animals and Diets
4.3. Clinical Evaluation
4.4. Measurements of Arterial Blood Pressure
4.5. Blood Examination
4.6. Measurements of Insulin, Leptin, Vascular Endothelial Growth Factor (VEGF), C-Reactive Protein (CRP), Uric Acid, and Glucose
4.7. Cytokines Measurement
4.8. Urine Examination
4.9. Electrocardiographic Examination
4.10. Echocardiographic Measurements
4.11. Ultrasonographic Examination of the Kidney
4.12. The Tissue Collection
4.13. The LC-MS/MS Analysis of DMA, ADMA and SDMA
Analysis of Cholesterol and Triacylglycerol Concentrations in Cardiac Muscle
4.14. Markers of Oxidative Stress
4.14.1. Lipid Peroxidation Assay
4.14.2. Analysis of the Activity of DNA Repair Enzymes
4.14.3. Analysis of Gene Expression of DNA Repair Enzymes
4.14.4. Estimation of Oxidative Damage of Genomic DNA
4.14.5. Statistical Analysis
5. Conclusions
Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Vane, J.R.; Anggard, E.F.; Botting, R.M. Regulatory functions of the vascular endothelium. N. Engl. J. Med. 1990, 323, 27–36. [Google Scholar] [PubMed]
- Heitzer, T.; Schlizig, T.; Krohn, K.; Meinertz, T.; Münzel, T. Endothelial dysfunction, oxidative stress, and risk cardiovascular events in patients with coronary artery disease. Circulation 2001, 104, 2673–2678. [Google Scholar] [CrossRef] [PubMed]
- Torres, N.; Guevara-Cruz, M.; Velázquez-Villegas, L.A.; Tovar, A.R. Nutrition and Atherosclerosis. Arch. Med. Res. 2015, 46, 408–426. [Google Scholar] [CrossRef] [PubMed]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Schaftenaar, F.; Frodermann, V.; Kuiper, J.; Lutgens, E. Atherosclerosis: The interplay between lipids and immune cells. Curr. Opin. Lipidol. 2016, 27, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Falk, E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47 (Suppl. S8), C7–C12. [Google Scholar] [CrossRef]
- Li, B.; Li, W.; Li, X.; Zhou, H. Inflammation: A Novel Therapeutic Target/Direction in Atherosclerosis. Curr. Pharm. Des. 2017, 23, 1216–1227. [Google Scholar] [CrossRef]
- Frostegård, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef]
- Wever, R.; Boer, P.; Hjmering, M.; Stroes, E.; Verhaar, M.; Kastelein, J.; Versluis, K.; van Rijn, H.; Koomans, H.; Rabelink, T. Nitric oxide production is reduced in patients with chronic renal failure. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1168–1172. [Google Scholar] [CrossRef]
- Kehrer, J.P. Free radicals as mediators of tissue injury and disease. Critic. Rev. Toxicol. 1993, 23, 21–48. [Google Scholar] [CrossRef]
- Mc Cord, J.M. The evolution of free radicals and oxidative stress. Am. J. Med. 2000, 108, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Olinski, R.; Gackowski, D.; Rozalski, R.; Foksinski, M.; Bialkowski, K. Oxidative DNA damage in cancer patients: A cause or a consequence of the disease development? Mutat. Res. 2003, 531, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, H.; Nair, J. Oxidative stress and lipid peroxidation derived DNA-lesions in inflammation driven carcinogenesis. Cancer Detect. Prev. 2004, 28, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Nilsen, H.; Skorpen, F.; Otterlei, M.; Slupphaug, G. Base excision repair of DNA in mammalian cells. Febs Lett. 2000, 476, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H. Analysis of a form of oxidative DNA damage, 8-hydroxy-2’deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutat. Res. 1997, 387, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Salim, S. Oxidative stress and the central nervous system. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Maki, H.; Sekiguchi, M. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 1992, 355, 273–275. [Google Scholar] [CrossRef]
- Gredilla, R. DNA damage and base excision repair in mitochondria and their role in aging. J. Aging Res. 2011, 2011, 257093. [Google Scholar] [CrossRef] [PubMed]
- Olinski, R.; Zastawny, T.; Budzbon, J.; Skokowski, J.; Zegarski, W.; Dizdaroglu, M. DNA base modifications in chromatin of human cancerous tissues. FEBS Lett. 1992, 309, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Prather, E.R.; Stetskiv, M.; Garrison, D.E.; Meade, J.R.; Peace, T.I.; Zhou, T. Inflammaging and oxidative stress in human diseases: From molecular mechanisms to novel treatments. Int. J. Mol. Sci. 2019, 20, 4472. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, M.K.; Gimeno, C.J.; Ames, B.N. Urinary 8-hydroxy-2-deoxyguanosine as a biological marker of in vivo oxidative DNA damage. Proc. Nat. Acad. Sci. USA 1989, 86, 9697–9701. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Can oxidative DNA damage be used as a biomarker of cancer risk in humans? Problems, resolutions, and preliminary results from nutritional supplementation studies. Free Radic. Res. 1998, 29, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Fraga, C.G.; Shigenaga, M.K.; Park, J.W.; Degan, P.; Ames, B.N. Oxidative damage to DNA during aging: 8-hydroxy-2-deoxyguanosine in rat organ DNA and urine. Proc. Nat. Acad. Sci. USA 1990, 87, 4533–4537. [Google Scholar] [CrossRef] [PubMed]
- Sayre, L.M.; Smith, M.A.; Perry, G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Cur. Med. Chem. 2001, 8, 721–738. [Google Scholar] [CrossRef]
- Kasai, H.; Iwamoto-Tanaka, N.; Miyamoto, T.; Kawanami, K.; Kawanami, S.; Kido, R.; Ikeda, M. Life style and urinary 8-hydroxydeoxygua-nosine, a marker of oxidative DNA damage: Effects of exercise, working conditions, meat intake, body mass index, and smoking. Jpn. J. Cancer Res. 2001, 92, 9–15. [Google Scholar] [CrossRef]
- Halliwell, B. Effect of diet on cancer development: Is oxidative DNA damage a biomarker? Free Rad. Biol. Med. 2002, 32, 974–986. [Google Scholar]
- Ray, S.D.; Lam, L.S.; Rotollo, J.A.; Phadke, S.; Patel, C.; Dontabhaktuni, A.; Mohammad, S.; Lee, H.; Strika, S.; Dobrogowska, A.; et al. Oxidative stress is the master operator of drug and chemically-induced programmed and unprogrammed cell death: Implications of natural antioxidants in vivo. BioFactors 2004, 21, 223–232. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Jaworek, J.; Kot, M.; Sokołowska, B.; Bieleń, A.; Janowska, B.; Cieśla, J.M.; Szparecki, G.; Sadoś, B.; Tudek, B. Inflammation increases oxidative DNA damage repair and stimulates preneoplastic changes in colons of newborn rats. J. Physiol. Pharmacol. 2016, 67, 277–286. [Google Scholar]
- Marnett, L.J.; Plastaras, J.P. Endogenous DNA damage and mutation. Trends Genet. 2001, 17, 214–221. [Google Scholar] [CrossRef]
- Bartsch, H.; Barbin, A.; Marion, M.J.; Nair, J.; Guichard, Y. Formation, detection, and role in carcinogenesis of ethenobases in DNA. Drug Metab. Rev. 1994, 26, 349–371. [Google Scholar] [CrossRef]
- El Ghissassi, F.; Barbin, A.; Nair, J.; Bartsch, H. Formation of 1,N6-ethenoadenine and 3,N4-ethenocytosineby lipid peroxidation products and nucleic acid bases. Chem. Res. Toxicol. 1995, 8, 278–283. [Google Scholar] [CrossRef]
- Winczura, A.; Zdzalik, D.; Tudek, B. Damage of DNA and proteins by major lipid peroxidation products in genome stability. Free Radic. Res. 2012, 46, 442–459. [Google Scholar] [CrossRef] [PubMed]
- Tedgui, A.; Mallat, Z. Cytokines in atherosclerosis: Pathogenic and regulatory pathways. Physiol. Rev. 2006, 86, 515–581. [Google Scholar] [CrossRef] [PubMed]
- Pasławski, R.; Pasławska, U.; Nicpoń, J.; Szuba, A. Swine as a model of experimental atherosclerosis. Adv. Clin. Exp. Med. 2011, 20, 211–215. [Google Scholar]
- Ząbek, A.; Pasławski, R.; Pasławska, U.; Wojtowicz, W.; Drożdż, K.; Polakof, S.; Podhorska, M.; Dzięgiel, P.; Młynarz, P.; Szuba, A. The influence of different diets on metabolism and atherosclerosis processes—A porcine model. Blood serum, urine and tissues 1H NMR metabolomics targeted analysis. PLoS ONE 2017, 12, e0184798. [Google Scholar] [CrossRef] [PubMed]
- Speina, E.; Zielińska, M.; Barbin, A.; Gackowski, D.; Kowalewski, J.; Graziewicz, M.A.; Siedlecki, J.A.; Oliński, R.; Tudek, B. Decreased repair activities of 1, N6 ethenoadeninie and 3, N4-ethenocytosine in lung adenocarcinoma patients. Cancer Res. 2003, 65, 4351–4357. [Google Scholar]
- Wang, Q.; Wen, X.; Kong, J. Recent Progress on Uric Acid Detection: A Review. Crit. Rev. Anal. Chem. 2020, 50, 359–375. [Google Scholar] [CrossRef]
- Wang, T.J.; Nam, B.H.; D’Agostino, R.B.; Wolf, P.A.; Lloyd-Jones, D.M.; MacRae, C.A. Carotid intima-media thickness is associated with premature parental coronary heart disease: The Framingham Heart Study. Circulation 2003, 108, 572–576. [Google Scholar] [CrossRef]
- Böger, R.H.; Bode-Böger, S.M.; Szuba, A.; Tsao, P.S.; Chan, J.R.; Tangphao, O.; Blaschke, T.F.; Cooke, J.P. Asymmetric dimethyloarginine (ADMA): A novel risk factor for endothelial dysfunction: Its role in hypercholesterolemia. Circulation 1998, 98, 1842–1847. [Google Scholar] [CrossRef]
- Smith, C.L.; Shelagh, A.; Hubank, M.; Leiper, J.M.; Vallance, P. Effects of ADMA upon Gene Expression: An Insight into the Patholphysiological Significance of Raised Plasma ADMA. PLoS Med. 2005, 2, 264. [Google Scholar] [CrossRef]
- Furuki, K.; Adachi, H.; Matsuoka, H.; Enomoto, A.; Satoh, A.; Hino, A.; Hiral, Y.; Imazumi, T. Plasma levels of asymmetric dimethylarginine (ADMA) are related to intima-media thickness of the carotid artery: An epidemiological study. Atherosclerosis 2007, 191, 206–210. [Google Scholar] [CrossRef]
- Montauban van Swijndregt, A.D.; De Lange, E.E.; de Groot, E.; Ackerstaff, R.G. An in vivo evaluation of the reproducibility of intima-media thickness measurements of the carotid artery segments using B-mode ultrasound. Ultrasound Med. Biol. 1999, 25, 323–330. [Google Scholar] [CrossRef]
- Stary, H.C. Natural History and Histological Classification of Atherosclerotic Lesions: An Update. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1177–1178. [Google Scholar] [CrossRef]
- Temelkova-Kurktschev, T.S.; Koehler, C.; Leonhardt, W.; Schaper, F.; Henkel, E.; Siegert, G. Carotid intima-media thickness in newly detected type 2 diabetes: Risk factors. Diabetes Care 1999, 22, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Doroszko, A.; Andrzejak, R.; Szuba, A. Endothelial dysfunction and ADMA in pathogenesis of arterial hypertension. Nadciśnienie Tętnicze 2008, 12, 224–237. [Google Scholar]
- Miriam Font-Nieves, M.; Sans-Fons, G.; Gorina, R.; Bonfill-Teixidor, E.; Salas-Pérdomo, A.; Márquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Induction of COX-2 Enzyme and Down-regulation of COX-1 Expression by Lipopolysaccharide (LPS) Control Prostaglandin E2 Production in Astrocytes. J. Biol. Chem. 2012, 287, 6454–6468. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)). Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Sobczak, A.; Szołtysek-Bołdys, I.; Anczyk, E.; Radek, M.; Prokopowicz, A.; Zaciera, M.; Brewczyński, P.Z. Plasma methylarginine, non-conventional risk factors of coronary-artery disease among subjects exposed and non-exposed to tobacco smoke. Med. Środ 2010, 13, 65–74. [Google Scholar]
- Bode- Böger, S.M.; Scalera, F.; Kielstein, J.T.; Martens-Lobenhoffer, J.; Breithardt, G.; Fobker, M.; Reinecke, H. Symmetrical Dimetyloarginine: A new Combined Parameter for Renal Function and Extent of Coronary Artery Disease. J. Am. Soc. Nephrol. 2006, 17, 1128–1134. [Google Scholar] [PubMed]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [PubMed]
- Bode-Böger, S.M.; Muke, J.; Surdacki, A.; Brabant, R.H.; Böger, R.H.; Frölich, J.C. Oral L-arginine improves endothelial function in healthy individuals older than 70 years. Vasc. Med. 2003, 8, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Tsao, P.S.; Adimoolam, S.; Kimoto, M.; Ogawa, T.; Cooke, J.P. Novel mechanism for endothelial dysfunction: Dysregulation of dimethylarginine dimethylaminohydrolase. Circulation 1999, 99, 3092–3095. [Google Scholar] [CrossRef] [PubMed]
- Hermenegildo, C.; Medina, P.; Peiro, M.; Segarra, G.; Vila, J.M.; Ortega, J.; Lluch, S. Plasma concentration of asymmetric dimethylargiine, an endogenous inhibitor of nitric oxide synthase, is elevated in hyperthyroid patients. J. Clin. Endocrinol. Metab. 2002, 87, 5636–5640. [Google Scholar] [CrossRef] [PubMed]
- Closs, E.I.; Mann, G.E. Membrane transport of arginine and cationic aminoacids analogs. In Nitric Oxide in Physiology and Pathology. Ignaro, L.J., Ed.; Academic Press: New York, NY, USA, 2000; pp. 225–241. [Google Scholar]
- Fleszar, M.G.; Wiśniewski, J.; Krzystek-Korpacka, M.; Misiak, B.; Frydecka, D.; Piechowicz, J.; Lorenc-Kukuła, K.; Gamian, A. Quantitative Analysis of l-Arginine, Dimethylated Arginine Derivatives, l-Citrulline, and Dimethylamine in Human Serum Using Liquid Chromatography-Mass Spectrometric Method. Chromatographia 2018, 81, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-J.; Wingerd, B.A.; Arakawa, T.; Smith, W.L. Cyclooxygenase—2gene transcription in a macrophage model of inflammation. J. Immunol. 2006, 177, 8111–8122. [Google Scholar] [CrossRef]
- Paslawska, U.; Paslawski, R.; Janiszewski, A.; Noszczyk-Nowak, A.; Kiczak, L.; Zyśko, D.; Nicpoń, J.; Jankowska, E.A.; Szuba, A.; Ponikowski, P. Normal ECG and echocardiographic (M mode and two dimensional) values in white domestic swine. Acta Vet. Scand. 2014, 56, 54–67. [Google Scholar] [CrossRef]
- Wiśniewski, J.; Fleszar, M.G.; Piechowicz, J.; Krzystek-Korpacka, M.; Chachaj, A.; Szuba, A.; Lorenc-Kukula, K.; Masłowski, L.; Witkiewicz, W.; Gamian, A. A novel mass spectrometry-based method for simultaneous determinantion of asymetric symetric dimethylarginine L-arginine L-cytruline optimized for LC-MS-TOF and LC-MS/MS. Biomed. Chromatogr. 2017, 31, e3994. [Google Scholar] [CrossRef]
- Herosimczyk, A.; Lepczyński, A.; Ożgo, M.; Barszcz, M.; Jaszczuk-Kubiak, E.; Pierzchała, M.; Tuśnio, A.; Skomiał, J. Hepatic proteome changes induced by dietary supplementation with two levels of native chicory inulin in young pigs. Livest. Sci. 2017, 203, 54–62. [Google Scholar] [CrossRef]
- Bochicchio, M.; Latronico, N.; Zani, D.G.; Mariotti, M.; Morandini, L.; Acquarolo, A.M.; Candiani, A. Free radical-induced lipoperoxidation and severe head injury. Intensive Care Med. 1990, 16, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 1993, 15, 532–534, 536–537. [Google Scholar] [PubMed]
- Khan, S.P.; Gul, P.; Ahmed, K.Z.; Ghani, R.; Yaquib, Z. Variation of Carotid Intima-Media Thickness in hypercholesterolemia patients on atorvastatin and rosuvastatin therapy. J. Clin. Exp. Cardiol. 2012, 3, 5. [Google Scholar]
- Lange, H.; Holec, S.; Cognat, V.; Pieuchot, L.; Le Ret, M.; Canaday, J.; Gagliardi, D. Degradation of a polyadenylated rRNA maturation by-product involves one of the three RRP6-like proteins in Arabidopsis thaliana. Mol. Cell Biol. 2008, 28, 3038–3044. [Google Scholar] [CrossRef]
- Wery, M.; Ruidant, S.; Schillewaert, S.; Lepore, N.; Lafontaine, D.L. The nuclear poly(A) polymerase and Exosome cofactor Trf5 is recruited cotranscriptionally to nucleolar surveillance. Rna 2009, 15, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Luu-The, V.; Paquet, N.; Calvo, E.; Cumps, J. Improved real-time RT-PCR method for high-throughput measurements using second derivative calculation and double correction. Biotechniques 2005, 38, 287–293. [Google Scholar] [CrossRef]
- Hattenbach, L.O.; Allers, A.; Klais, C.; Koch, F.; Hecker, M. L-Arginine-nitric oxide pathway-related metabolites in the aqueous humor of diabetic patients. Investig. Ophthalmol. Vis. Sci. 2000, 41, 213–217. [Google Scholar]
- Karumanchi, S.A.; Lindheimer, M.D. Preeclampsia and the kidney: Footprints in the urine. Gynecology 2007, 196, 287–288. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Xiong, Y.; Yuan, L.W.; Deng, H.W.; Li, Y.J.; Chen, B.M. Elevated serum endogenous inhibitor of nitric oxide synthase and endothelial dysfunction in aged rats. Clin. Exp. Pharmacol. Physiol. 2001, 28, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Vallance, P.; Leiper, J. Cardiovascular biology of the assymetric dimethyloarginine: Dimethyloarginine dimethylaminohydrolase pathway. Artrioscler. Thromb. Vasc. Biol. 2004, 24, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Janiszewski, A.; Pasławski, R.; Skrzypczak, P.; Pasławska, U.; Szuba, A. The use of a blunt-tipped plastic guide wire improves the safety and reduces the duration of endotracheal intubation in the pig. J. Vet. Med. Sci. 2014, 12, 1260–1262. [Google Scholar]
- Casella, I.B.; Presti, C.; Porta, R.M.P.; Sabbag, C.R.D.; Bosch, M.A.; Yamazaki, Y. A protocol to measure common carotid intima-media thickness. Clinics 2008, 64, 15–20. [Google Scholar] [CrossRef]
- Deanfield, J.; Donald, A.; Ferri, C.; Giannattasio, C.; Halcox, J.; Halligan, S.; Lerman, A.; Mancia, G.; Oliver, J.J.; Pessina, A.C.; et al. Endothelial function and dysfunction. Part I: Methodological issues for assessment In the different vascular beds: A statement by the Working Group on Endothelin and Endothelial Factors of the European Society of Hypertension. J. Hypert. 2005, 23, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, D.; Paslawski, R.; Gomulkiewicz, A.; Gladine, C.; Janczak, D.; Grzegorek, I.; Jablonska, K.; Drozdz, K.; Chmielewska, M.; Piotrowska, A.; et al. Alterations of aorta intima and media transcriptome in swine fed high-fat diet over one-year follow-up period and of the reversal to normal diet. Nutr. Metab. Cardiovas. 2020, 30, 1201–1215. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; Dragoni, S.; Lisi, M.; Di Stolfo, G.; Sonnati, S.; Fineschi, M.; Parker, J.D. Conduit artery constriction mediated by low flow: A novel noninvasive method for the assessment of vascular function. J. Am. Coll. Cardiol. 2008, 51, 1953–1958. [Google Scholar] [CrossRef]
- Maruhashi, T.; Hisatome, I.; Kihara, Y.; Higashi, Y. Hyperuricemia and endothelial function: From molecular background to clinical perspectives. Atherosclerosis 2018, 278, 226–231. [Google Scholar] [CrossRef]
- Baskin, D.G.; Blevins, J.E.; Schwartz, M.W. How the brain regulates food intake and body weight: The role of leptin. J. Pediatr. Endocrinol. Metab. 2001, 14 (Suppl. S6), 1417–1429. [Google Scholar]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Shimomura, I.; Hammer, R.E.; Ikemoto, S.; Brown, M.S.; Goldstein, J.L. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature 1999, 401, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Oda, N.; Imamura, S.; Fujita, T.; Uchida, Y.; Inagaki, K.; Kakizawa, H.; Hayakawa, N.; Suzuki, A.; Takeda, J.; Horikawa, Y.; et al. The ratio of leptin to adiponectin can be used as an index of insulin resistance. Metabolism 2008, 57, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Milasan, A.; Smaani, A.; Martel, C. Early Rescue of Lymphatic Function Limits Atherosclerosis Progression in Ldlr Mice. Atherosclerosis 2019, 283, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Houssari, M.; Dumesnil, A.; Tardif, V.; Kivelä, R.; Pizzinat, N.; Boukhalfa, I.; Godefroy, D.; Schapman, D.; Hemanthakumar, K.A.; Bizou, M.; et al. Lymphatic and Immune Cell Cross-Talk Regulates Cardiac Recovery After Experimental Myocardial Infarction. Arter. Thromb. Vasc. Biol. 2020, 40, 1722–1737. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BDG | UDG | RG | ||||
|---|---|---|---|---|---|---|
| Start | End | Start | End | Start | End | |
| Body mass (kg) | 40 | 246 ± 27.5 | 40 | 260 ± 21 | 40 | 245 ± 16.5 |
| Blood pressure (mmHg) | ||||||
| Systolic/ | 148 ± 10 | 153 ± 16 | 143 ± 14 | 161 ± 17 | 143 ± 14 | 159 ± 15 |
| Diastolic | 87 ± 11 | 98 ± 13 | 93 ± 13 | 103 ± 13 | 93 ± 13 | 109 ± 11 |
| IVSd (mm) | 0.78 ± 0.06 | 10.5 ± 0.9 | 0.78 ± 0.06 | 10.4 ± 0.9 | 0.78 ± 0.06 | 10.4 ± 0.9 |
| LWd (mm) | 0.7 ± 0.06 | 10.4 ± 0.7 | 0.7 ± 0.06 | 10.4 ± 0.5 | 0.7 ± 0.06 | 10.4 ± 10.6 |
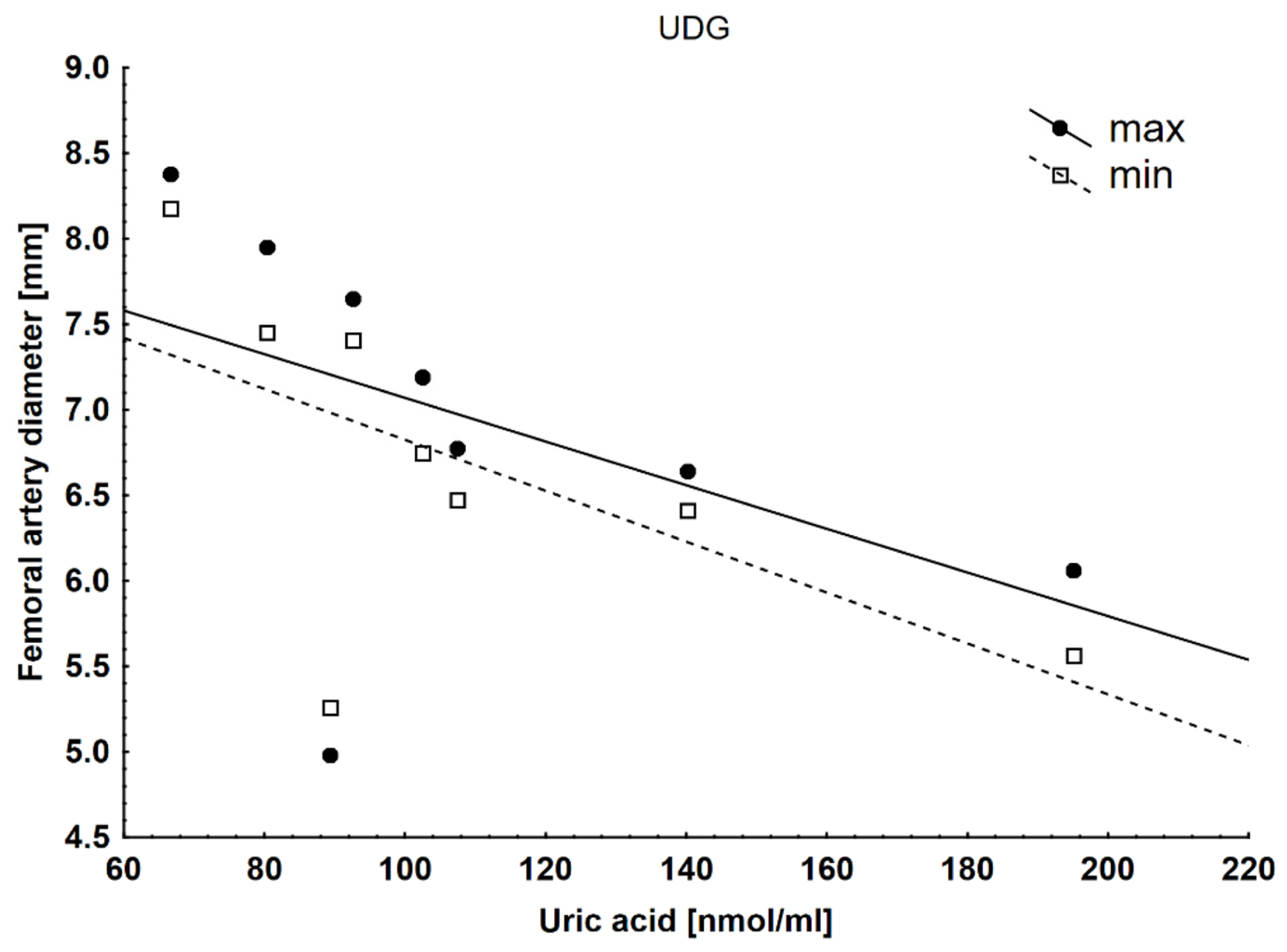
| Dmax FA (mm) | 5.07 ± 0.64 | 7.43 ± 1.18 | 5.18 ± 0.53 | 7.3 ± 1.05 | 5.18 ± 0.53 | 6.80 ± 0.88 |
| WBC (G/L) | 12.5 ± 2.96 | 10.82 ± 3.82 | 11.6 ± 2.77 | 9.47 ± 1.76 | 14.1 ± 2.62 | 9.87 ± 2.14 |
| Urea (mmol/L) | 3.35 ± 0.66 | 2.9 ± 0.78 | 6.62 ± 10.7 | 3.04 ± 1.04 | 3.76 ± 1.75 | 4.43 ± 1.37 ^ |
| Creatinine (µmol/L) | 182 ± 44 | 149 ± 24 | 165 ± 20 | 181 ± 44 | 180 ± 33 | 194 ± 30 ^ |
| HOMA serum (Units) | 0.21 ± 0.05 | 0.21 ± 0.06 | 0.25 ± 0.07 | 0.25 ± 0.07 | 0.35 ± 0.26 | 0.34 ± 0.25 |
| NEFA plasma (mmol/L) | - | 0.3 ± 0.15 | - | 0.29 ± 0.14 | - | 0.64 ± 0.44 |
| CRP (ng/mL) | 114 ± 46 | 60 ± 36 | 87 ± 43 | 52 ± 53 | 99 ± 33 | 41 ± 21 |
| IL-1β (pg/L) | - | 5.59 ± 6.08 | - | 3.89 ± 3.32 | - | 0.98 ± 1.59 |
| IL-6 (pg/mL | - | 4.52 ± 3.10 | - | 9.03 ± 3.03 | - | 9.70 ± 12.25 |
| TNFα (pg/L) | - | 59.0 ± 99.47 | - | 55.65 ± 65.86 | - | 36.50 ± 10.20 |
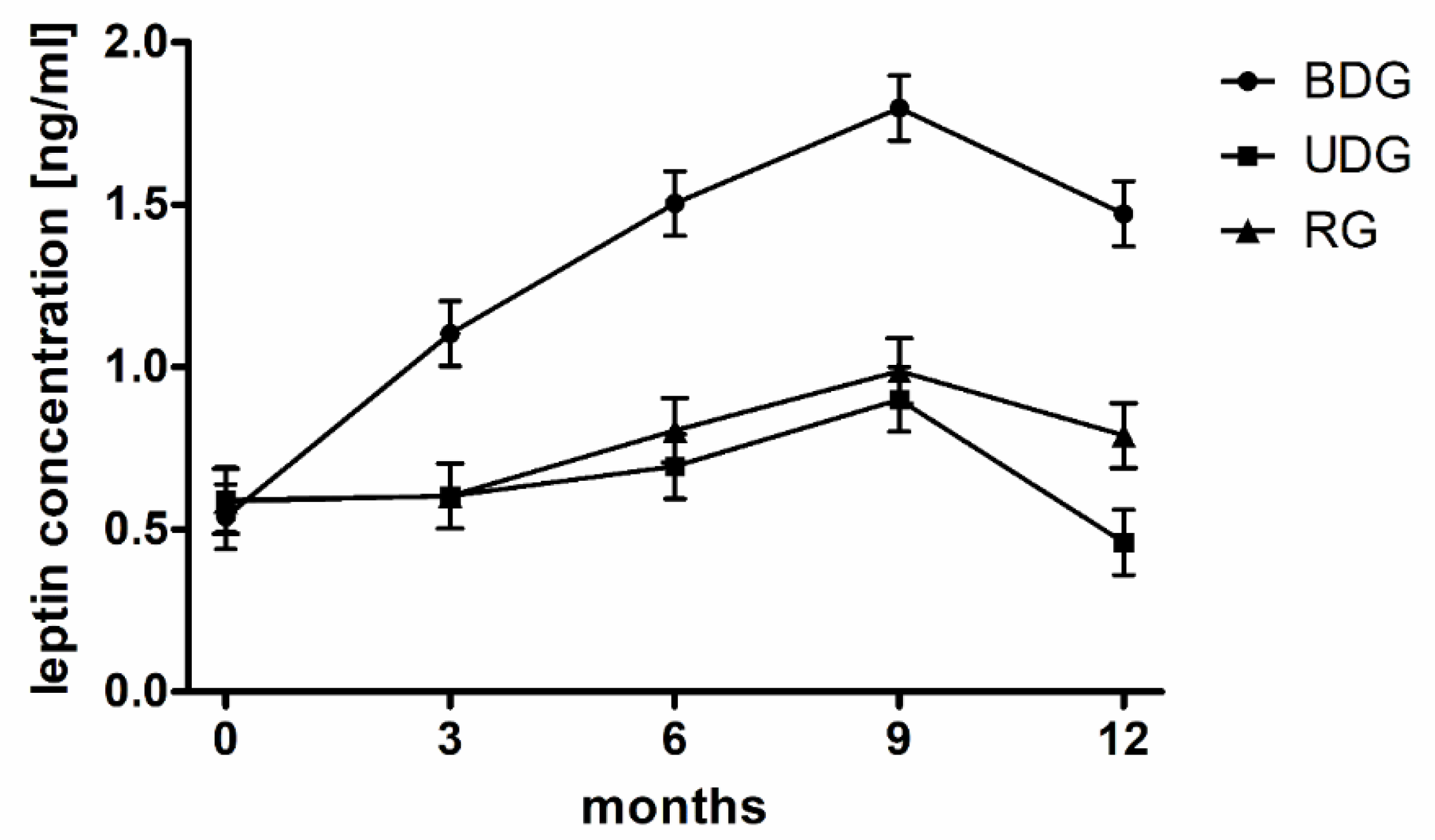
| Leptin serum (ng/mL) | 0.59 ± 1.38 | 1.48 ± 1.6 | 0.55 ± 0.47 | 0.47 ± 0.32 * | 0.59 ± 0.45 | 0.83 ± 0.46 |
| Uric acid serum (µmol/L) | 111 ± 24 | 110 ± 24 | 103 ± 30 | 109 ± 41 | 111 ± 17 | 130 ± 24 □ |
| L-arginine—heart tissue (µmol/L) | 74 ± 14 | 81 ± 10 | 76 ± 11 | 78 ± 13 | 76 ± 11 | 80 ± 12 |
| ADMA heart tissue (µmol/L) | 2.04 ± 0.25 | 1.37 ± 0.16 # | 2.15 ± 0.1 | 1.46 ± 0.19 # | 2.15 ± 0.1 | 1.5 ± 0.1 ^# |
| SDMA heart tissue (µmol/L) | 0.73 ± 0.1 | 0.47 ± 0.07 | 0.8 ± 0.1 | 0.53 ± 0.07 * | 0.7 ± 0.1 | 0.5 ± 0.08 |
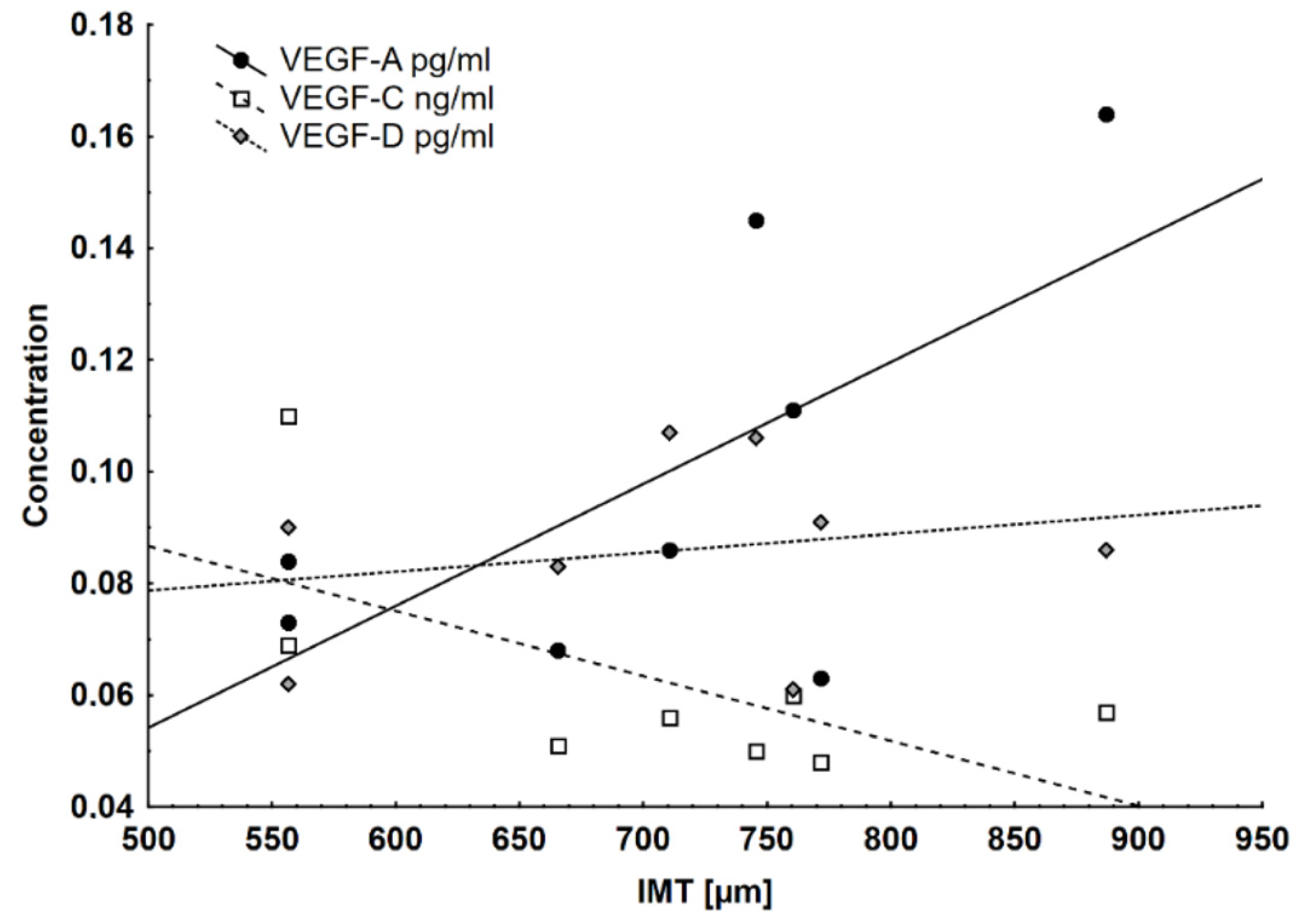
| VEGF-A serum (pg/mL) | 0.11 ± 0.04 | 0.09 ± 0.02 | 0.09 ± 0.02 | 0.1 ± 0.04 | 0.11 ± 0.05 | 0.08 ± 0.02 |
| VEGF-C serum (pg/mL) | 0.07 ± 0.02 | 0.05 ± 0.01 | 0.08 ± 0.02 | 0.07 ± 0.02 | 0.07 ± 0.02 | 0.07 ± 0.02 ^ |
| VEGF-D serum (pg/mL) | 0.08 ± 0.02 | 0.09 ± 0.02 | 0.07 ± 0.01 | 0.08 ± 0.02 | 0.08 ± 0.02 | 0.08 ± 0.03 |
| Cholesterol heart tissue (µmol/g) | - | 0.57 ± 0.27 | - | 0.62 ± 0.16 | - | 0.71 ± 0.18 |
| TG heart tissue (µmol/g) | - | 6.68 ± 1.17 | - | 6.07 ± 0.2 * | - | 5.63 ± 0.96 ^ |
| TBARS heart (nmol/g) | - | 0.89 ± 0.22 | - | 1.12 ± 0.48 | - | 1.19 ± 0.5 |
| etC heart (fmol/μg protein/h) | - | 4.81 ± 1.59 | - | 4.28 ± 1.25 | - | 5.94 ± 1.5^ |
| etA heart (fmol/μg protein/h) | - | 6.55 ± 2.84 | - | 6.16 ± 2.78 | - | 8.45 ± 2.2 |
| 8 oxoG heart (fmol/μg protein/h) | - | 11.75 ± 2.67 | - | 11.05 ± 3.38 | - | 13.7 ± 2.5 |
| Gene | Primer | Sequence (5′ to 3′) |
|---|---|---|
| MPG | MpgF | GTCCTAGTCCGGCGACTTCC |
| MpgR | CTT GTCTGGGCAGGCCCTTTG C | |
| OGG1 | OGG1F | CTCAGAAATTCCAAGGTGTTC |
| OGG1R | CCGCTCCACCAT-GCCAGTG. | |
| ALOX12 | Alox12F | TTGCATACTTTGTAGACAGTCTCC |
| Alox12R | CTGAGTTTCATCCATTTTGGTCATG | |
| Ptgs2 | Ptgs2F | AAGGAGATGGCAGCAGAGTT |
| Ptgs2R | GTGGCCGTCTTGACAATGTT | |
| 18S rRNA | 18SpfF | ATCCTTCGATGTCGGCTCTT |
| 18SpfR | ACTAACCTGTCTCACGACGGTC | |
| ANPG | ANPGpeF | CGCAGCATCTATTTCTCAAGC |
| ANPGpeR | GTGCCATTAGGAAGTCGCC | |
| TDG | TDGpeF | TAATGGGCAGTGGATGACCC |
| TDGpeR | TGCAGCATTTAAGCAGAGCTGA | |
| APEX1 | APE1peF | GAATGCTGGCTTCACTCCACA |
| APE1peR | AAAGGTGTAGGCATACGCCGT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paslawski, R.; Kowalczyk, P.; Paslawska, U.; Wiśniewski, J.; Dzięgiel, P.; Janiszewski, A.; Kiczak, L.; Zacharski, M.; Gawdzik, B.; Kramkowski, K.; et al. Analysis of the Model of Atherosclerosis Formation in Pig Hearts as a Result of Impaired Activity of DNA Repair Enzymes. Int. J. Mol. Sci. 2024, 25, 2282. https://doi.org/10.3390/ijms25042282
Paslawski R, Kowalczyk P, Paslawska U, Wiśniewski J, Dzięgiel P, Janiszewski A, Kiczak L, Zacharski M, Gawdzik B, Kramkowski K, et al. Analysis of the Model of Atherosclerosis Formation in Pig Hearts as a Result of Impaired Activity of DNA Repair Enzymes. International Journal of Molecular Sciences. 2024; 25(4):2282. https://doi.org/10.3390/ijms25042282
Chicago/Turabian StylePaslawski, Robert, Paweł Kowalczyk, Urszula Paslawska, Jerzy Wiśniewski, Piotr Dzięgiel, Adrian Janiszewski, Liliana Kiczak, Maciej Zacharski, Barbara Gawdzik, Karol Kramkowski, and et al. 2024. "Analysis of the Model of Atherosclerosis Formation in Pig Hearts as a Result of Impaired Activity of DNA Repair Enzymes" International Journal of Molecular Sciences 25, no. 4: 2282. https://doi.org/10.3390/ijms25042282
APA StylePaslawski, R., Kowalczyk, P., Paslawska, U., Wiśniewski, J., Dzięgiel, P., Janiszewski, A., Kiczak, L., Zacharski, M., Gawdzik, B., Kramkowski, K., & Szuba, A. (2024). Analysis of the Model of Atherosclerosis Formation in Pig Hearts as a Result of Impaired Activity of DNA Repair Enzymes. International Journal of Molecular Sciences, 25(4), 2282. https://doi.org/10.3390/ijms25042282