
Microbiota Alterations in Lung, Ileum, and Colon of Guinea Pigs with Cough Variant Asthma

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
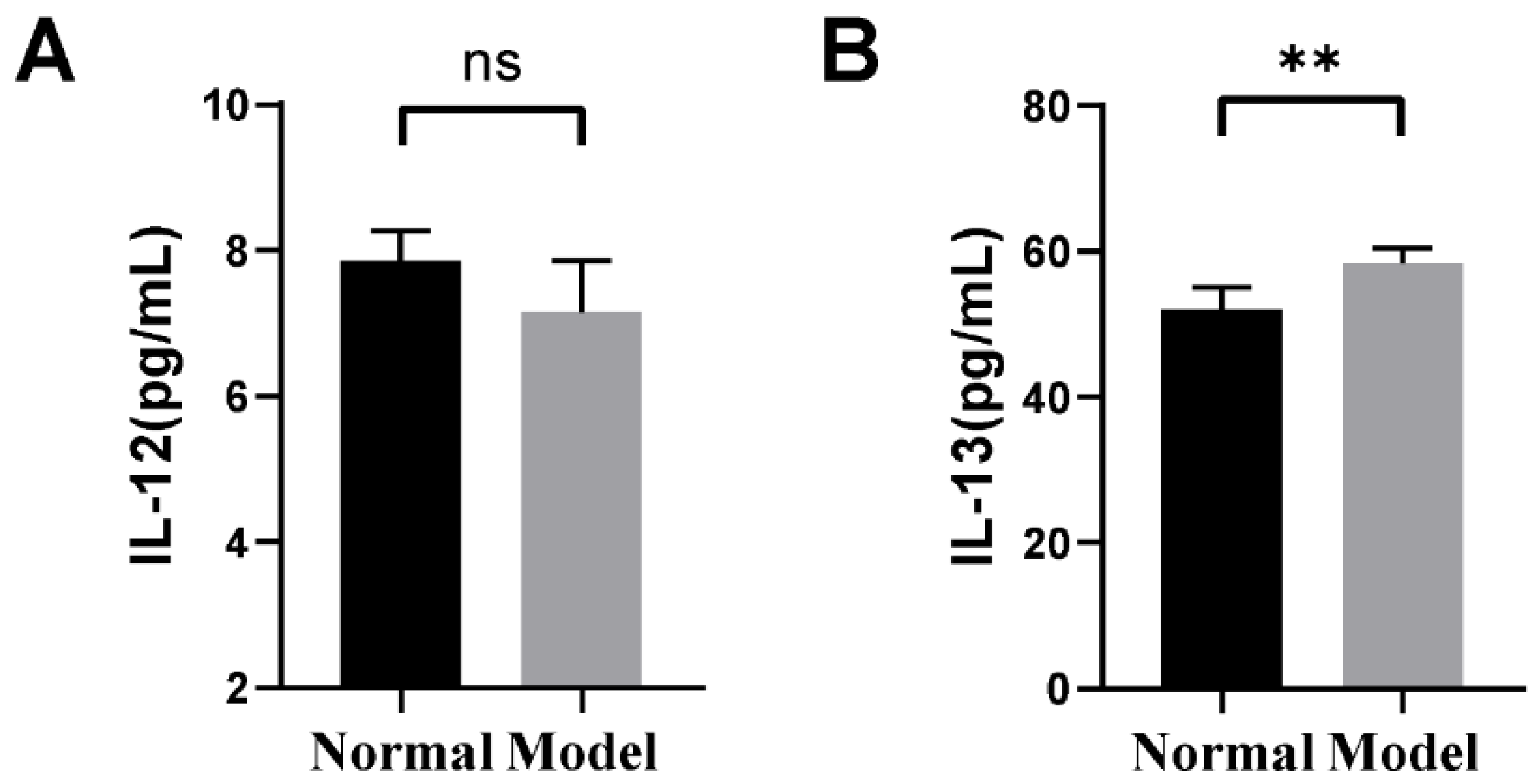
2.1. The Changes in Inflammatory Factors of Guinea Pigs
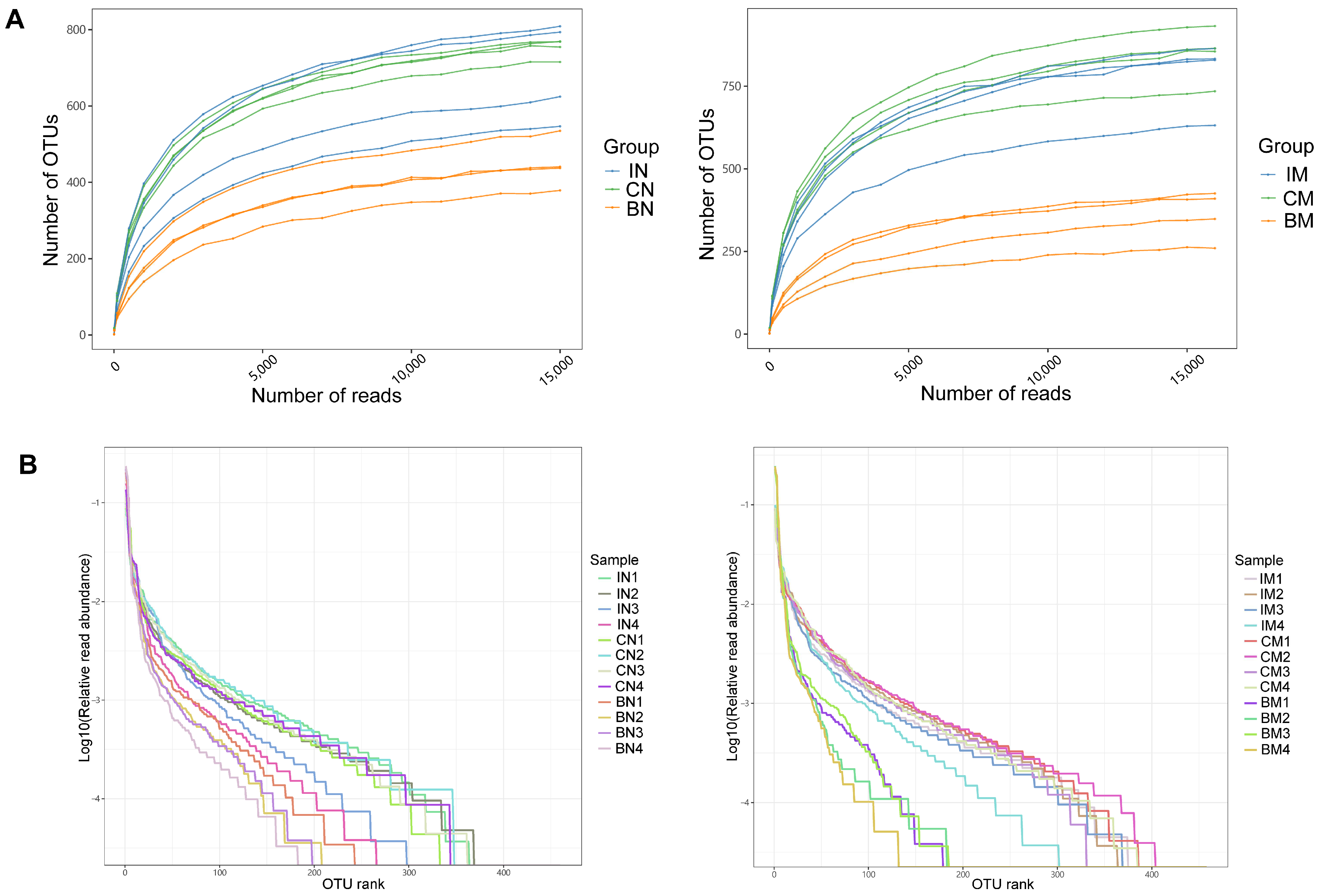
2.2. Metadata and Sequencing of Microbiota
2.3. Evaluation of Microbial Composition in Three Different Ecological Niches of Normal Guinea Pigs
2.4. Microbial Composition of the Ileum of Guinea Pigs Varied Maximally in the Three Different Ecological Niches
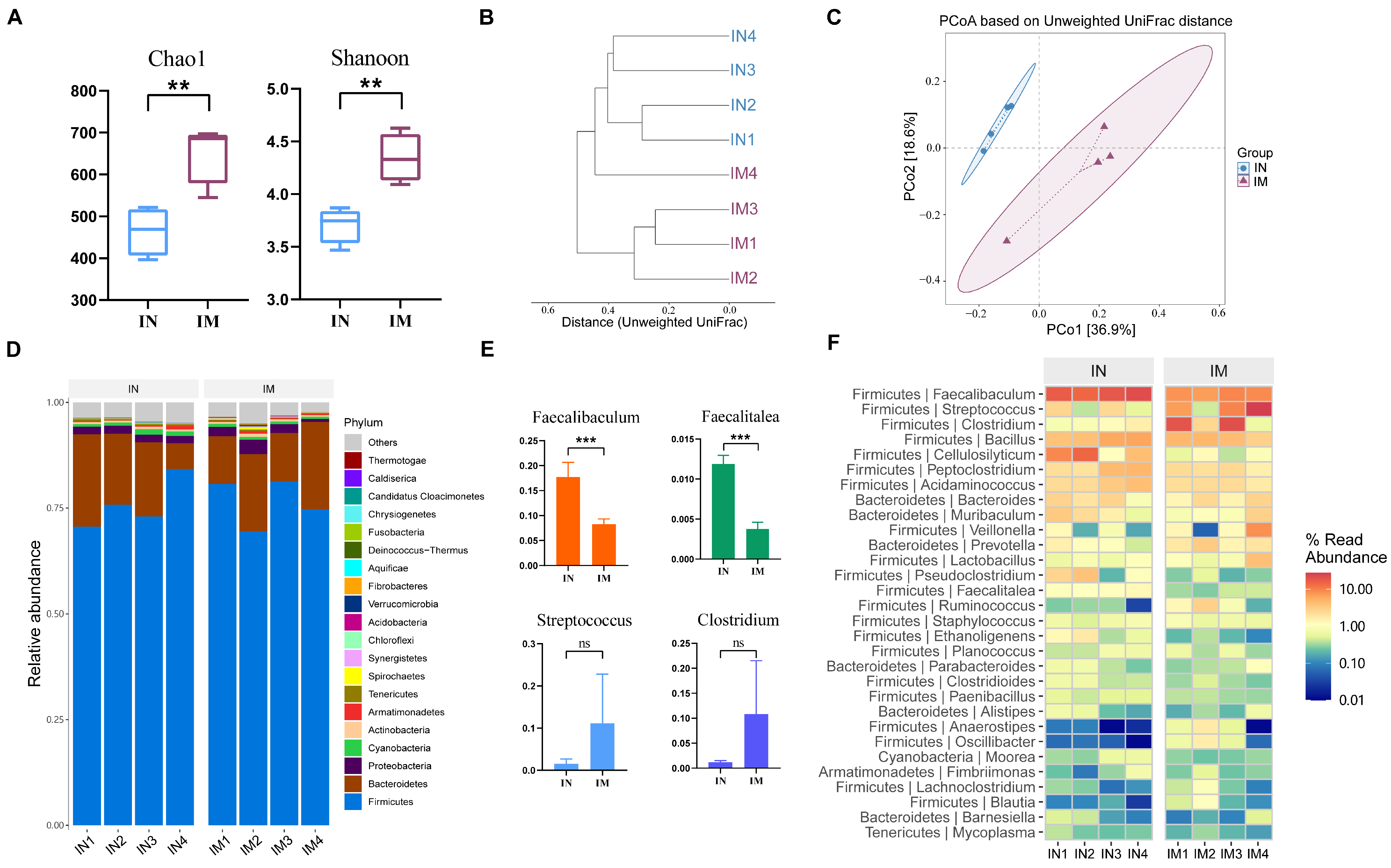
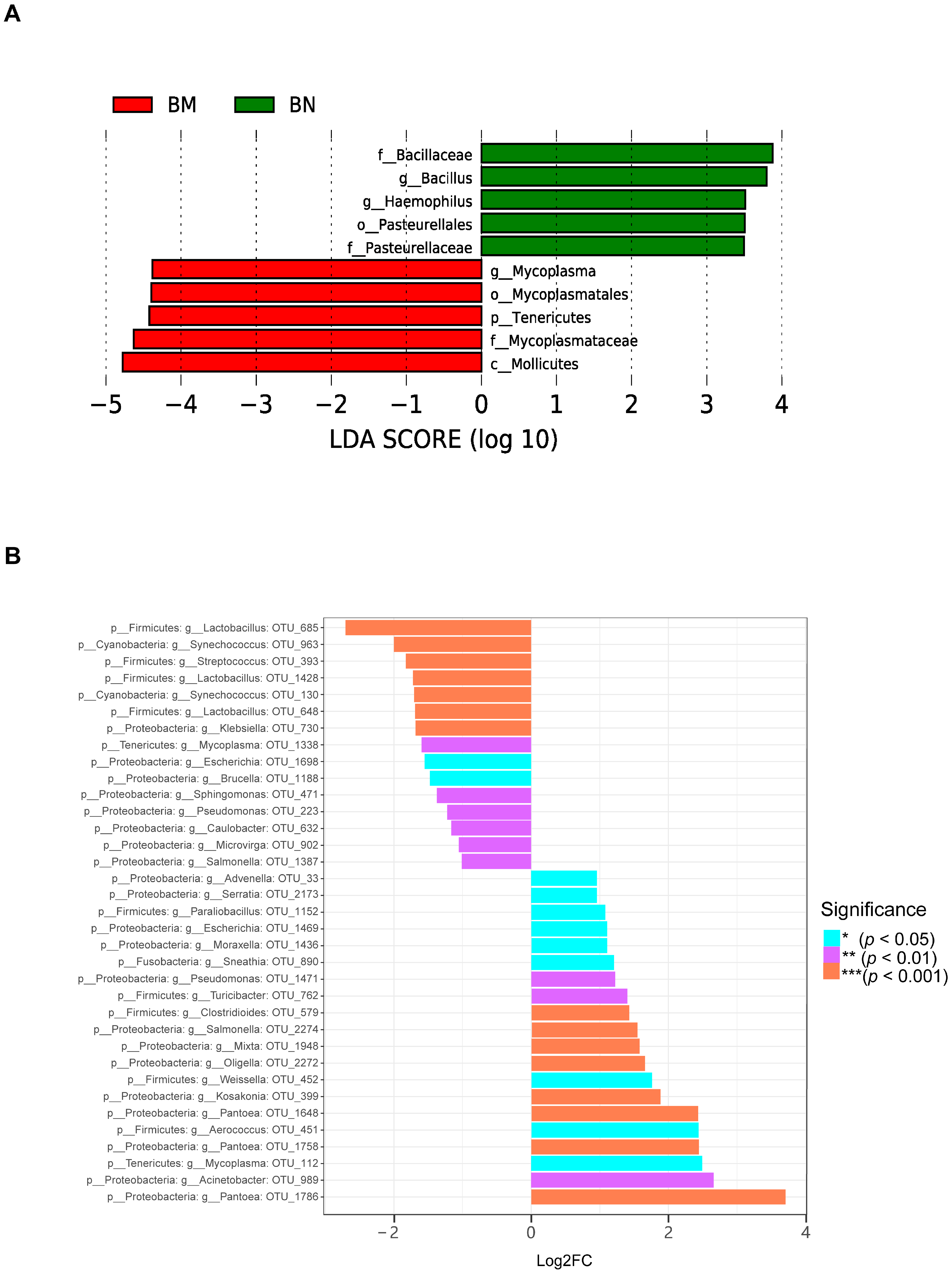
2.5. Microbial Composition of the Lung of Guinea Pigs Varied Secondarily in the Three Different Ecological Niches
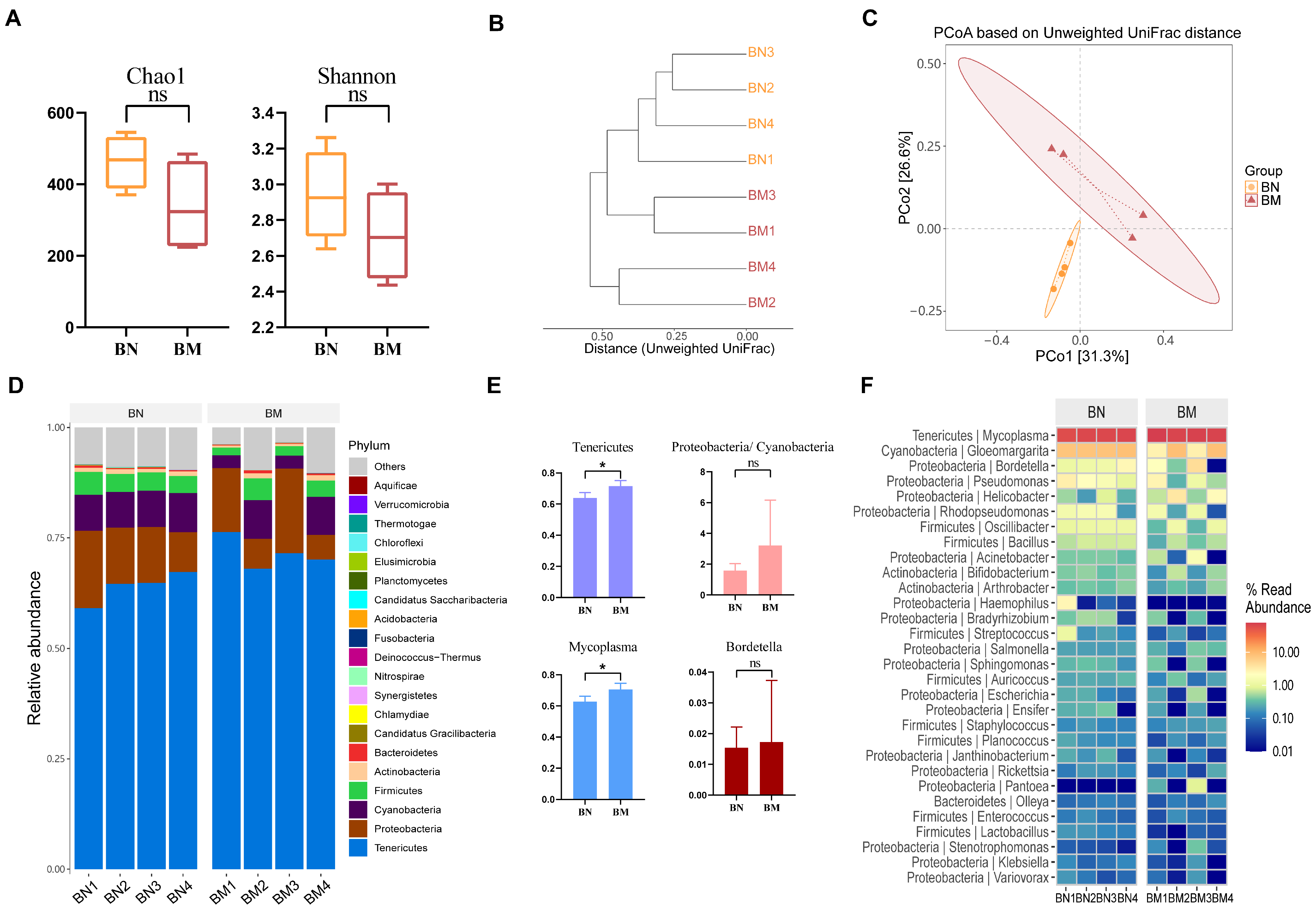
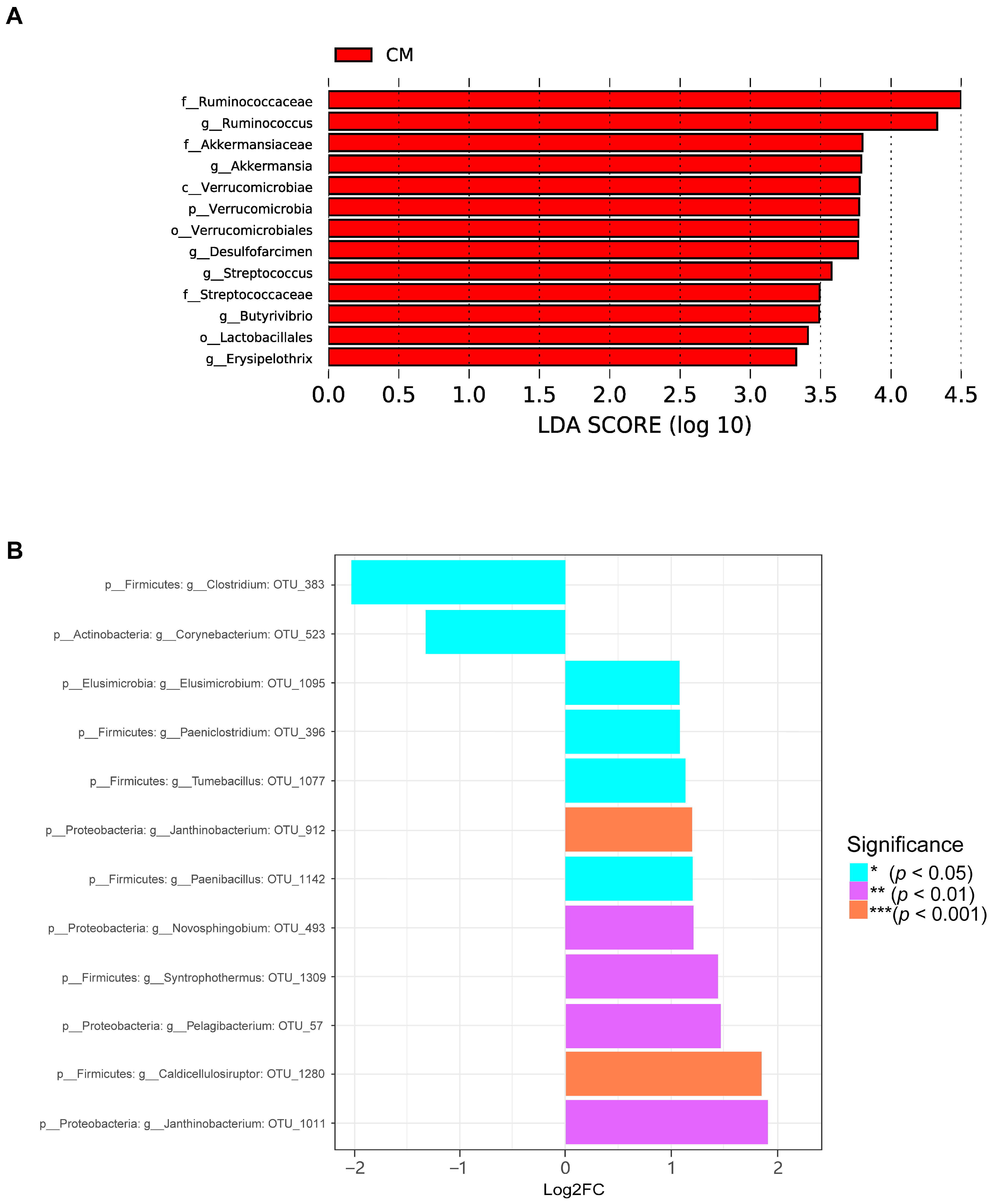
2.6. Microbial Composition of the Colon of Guinea Pigs Varied Minimally in the Three Different Ecological Niches
2.7. Evaluation of Microbial Composition in Three Different Ecological Niches of CVA Guinea Pigs
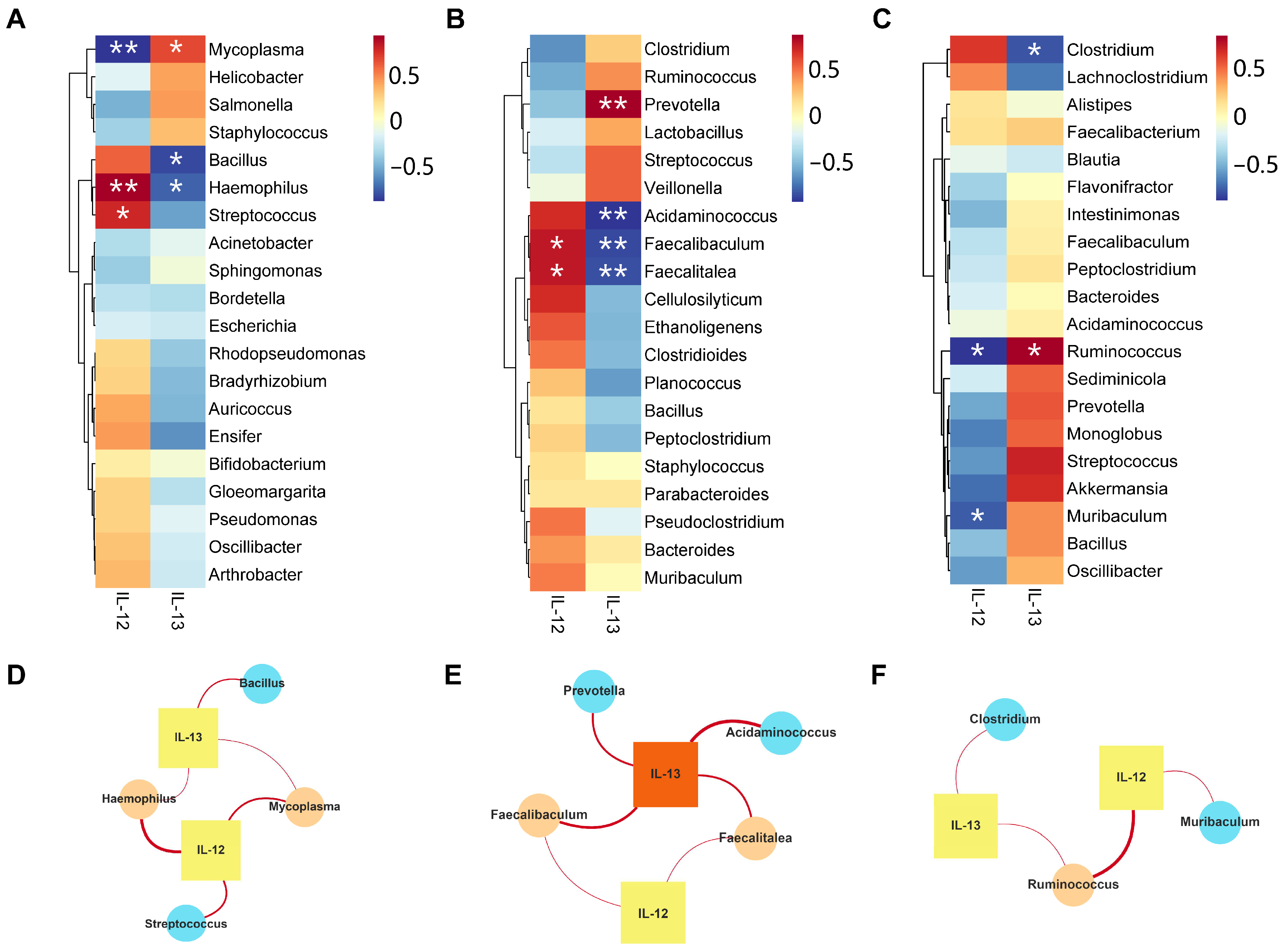
2.8. Correlation Analysis of Microbiota and Inflammatory Factors
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Animal
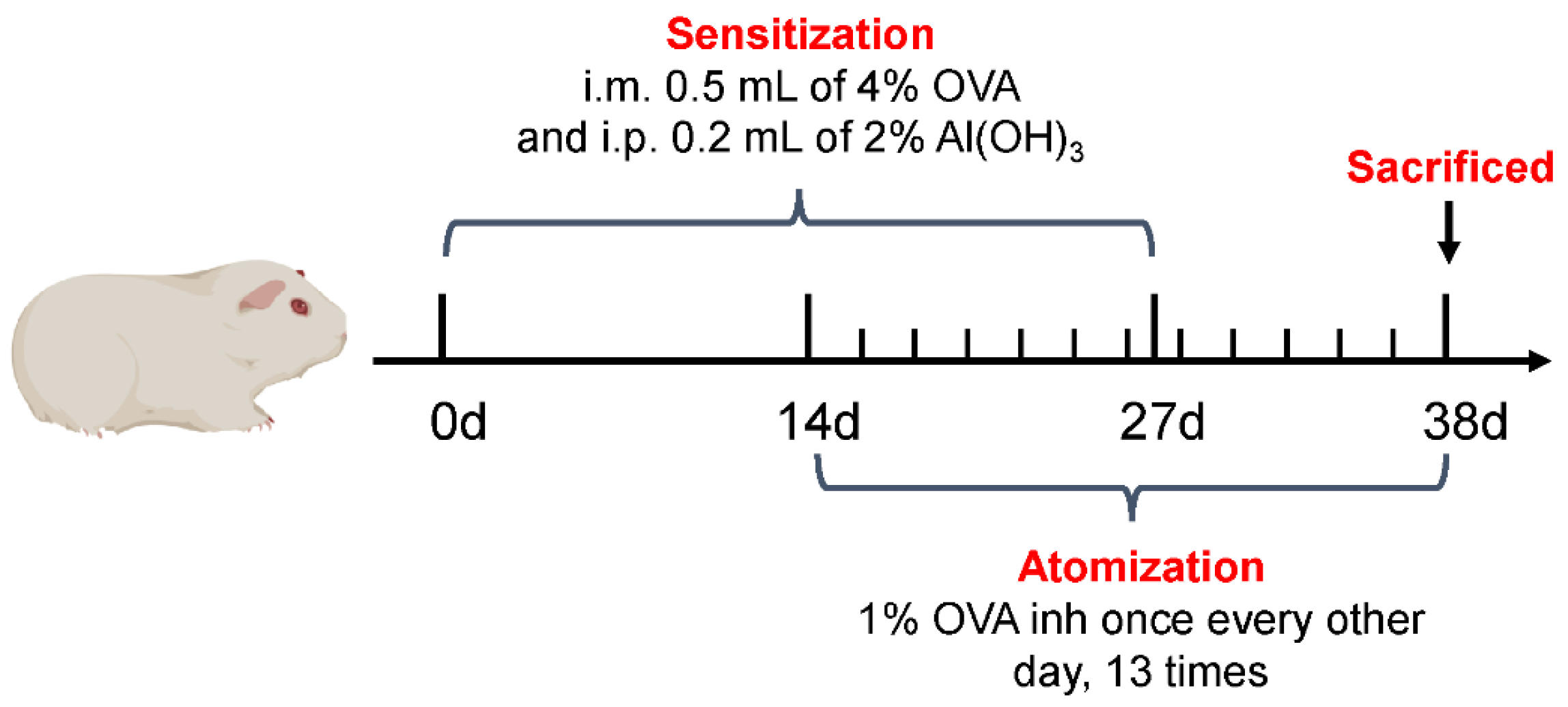
4.3. Construction of CVA Guinea Pig Model
4.4. ELISA of Inflammatory Cytokines in BALF
4.5. Analysis of 16S rDNA of Microbiota
4.6. Bioinformatics Analysis
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morice, A.H.; Millqvist, E.; Bieksiene, K.; Birring, S.S.; Dicpinigaitis, P.; Domingo Ribas, C.; Hilton Boon, M.; Kantar, A.; Lai, K.; McGarvey, L.; et al. ERS guidelines on the diagnosis and treatment of chronic cough in adults and children. Eur. Respir. J. 2020, 55, 1901136. [Google Scholar] [CrossRef]
- Kang, H.; Koh, Y.Y.; Yoo, Y.; Yu, J.; Kim, D.K.; Kim, C.K. Maximal airway response to methacholine in cough-variant asthma: Comparison with classic asthma and its relationship to peak expiratory flow variability. Chest 2005, 128, 3881–3887. [Google Scholar] [CrossRef]
- Gao, J.; Wu, F.; Wu, S.; Yang, X. Inflammatory Subtypes in Classic Asthma and Cough Variant Asthma. J. Inflamm. Res. 2020, 13, 1167–1173. [Google Scholar] [CrossRef]
- Niimi, A.; Matsumoto, H.; Minakuchi, M.; Kitaichi, M.; Amitani, R. Airway remodelling in cough-variant asthma. Lancet 2000, 356, 564–565. [Google Scholar] [CrossRef]
- Niimi, A.; Torrego, A.; Nicholson, A.G.; Cosio, B.G.; Oates, T.B.; Chung, K.F. Nature of airway inflammation and remodeling in chronic cough. J. Allergy Clin. Immunol. 2005, 116, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, H.; Niimi, A.; Matsumoto, H.; Takemura, M.; Ueda, T.; Yamaguchi, M.; Jinnai, M.; Inoue, H.; Ito, I.; Chin, K.; et al. Inflammatory subtypes in cough-variant asthma: Association with maintenance doses of inhaled corticosteroids. Chest 2010, 138, 1418–1425. [Google Scholar] [CrossRef]
- De Diego, A.; Martínez, E.; Perpiñá, M.; Nieto, L.; Compte, L.; Macián, V.; Senent, L. Airway inflammation and cough sensitivity in cough-variant asthma. Allergy 2005, 60, 1407–1411. [Google Scholar] [CrossRef]
- Hufnagl, K.; Pali-Schöll, I.; Roth-Walter, F.; Jensen-Jarolim, E. Dysbiosis of the gut and lung microbiome has a role in asthma. Semin. Immunopathol. 2020, 42, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How colonization by microbiota in early life shapes the immune system. Science 2016, 352, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Tuganbaev, T.; Skelly, A.N.; Honda, K. T Cell Responses to the Microbiota. Annu. Rev. Immunol. 2022, 40, 559–587. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Laxman, B.; Naureckas, E.T.; Hogarth, D.K.; Sperling, A.I.; Solway, J.; Ober, C.; Gilbert, J.A.; White, S.R. Associations between fungal and bacterial microbiota of airways and asthma endotypes. J. Allergy Clin. Immunol. 2019, 144, 1214–1227. [Google Scholar] [CrossRef] [PubMed]
- Logan, A.C.; Jacka, F.N.; Prescott, S.L. Immune-Microbiota Interactions: Dysbiosis as a Global Health Issue. Curr. Allergy Asthma Rep. 2016, 16, 13. [Google Scholar] [CrossRef]
- Cukrowska, B.; Bierła, J.B.; Zakrzewska, M.; Klukowski, M.; Maciorkowska, E. The Relationship between the Infant Gut Microbiota and Allergy. The Role of Bifidobacterium breve and Prebiotic Oligosaccharides in the Activation of Anti-Allergic Mechanisms in Early Life. Nutrients 2020, 12, 946. [Google Scholar] [CrossRef]
- Cahenzli, J.; Köller, Y.; Wyss, M.; Geuking, M.B.; McCoy, K.D. Intestinal microbial diversity during early-life colonization shapes long-term IgE levels. Cell Host Microbe 2013, 14, 559–570. [Google Scholar] [CrossRef]
- Su, Z.; Jia, X.; Fan, Y.; Zhao, F.; Qiao, Y. Progress of Research on the Relationship between Lung Microbiome and Lung Cancer. Zhongguo Fei Ai Za Zhi Chin. J. Lung Cancer 2022, 25, 40–45. [Google Scholar] [CrossRef]
- Gollwitzer, E.S.; Saglani, S.; Trompette, A.; Yadava, K.; Sherburn, R.; McCoy, K.D.; Nicod, L.P.; Lloyd, C.M.; Marsland, B.J. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat. Med. 2014, 20, 642–647. [Google Scholar] [CrossRef]
- Russell, S.L.; Gold, M.J.; Willing, B.P.; Thorson, L.; McNagny, K.M.; Finlay, B.B. Perinatal antibiotic treatment affects murine microbiota, immune responses and allergic asthma. Gut Microbes 2013, 4, 158–164. [Google Scholar] [CrossRef]
- Thorsen, J.; Rasmussen, M.A.; Waage, J.; Mortensen, M.; Brejnrod, A.; Bønnelykke, K.; Chawes, B.L.; Brix, S.; Sørensen, S.J.; Stokholm, J.; et al. Infant airway microbiota and topical immune perturbations in the origins of childhood asthma. Nat. Commun. 2019, 10, 5001. [Google Scholar] [CrossRef]
- Rosenberg, H.F.; Druey, K.M. Modeling asthma: Pitfalls, promises, and the road ahead. J. Leukoc. Biol. 2018, 104, 41–48. [Google Scholar] [CrossRef]
- Jiao, H.Y.; Su, W.W.; Li, P.B.; Liao, Y.; Zhou, Q.; Zhu, N.; He, L.L. Therapeutic effects of naringin in a guinea pig model of ovalbumin-induced cough-variant asthma. Pulm. Pharmacol. Ther. 2015, 33, 59–65. [Google Scholar] [CrossRef]
- Wen, L.; Zhang, T.; Chen, F.; Hu, L.; Dou, C.; Ding, X.; Altamirano, A.; Wei, G.; Yan, Z. Modified Dingchuan Decoction treats cough-variant asthma by suppressing lung inflammation and regulating the lung microbiota. J. Ethnopharmacol. 2023, 306, 116171. [Google Scholar] [CrossRef]
- Mendez, R.; Banerjee, S.; Bhattacharya, S.K.; Banerjee, S. Lung inflammation and disease: A perspective on microbial homeostasis and metabolism. IUBMB Life 2019, 71, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.F. Airway microbial dysbiosis in asthmatic patients: A target for prevention and treatment? J. Allergy Clin. Immunol. 2017, 139, 1071–1081. [Google Scholar] [CrossRef]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Hu, X.; Wang, F.; Yang, S.; Yuan, X.; Yang, T.; Zhou, Y.; Li, Y. Rabbit microbiota across the whole body revealed by 16S rRNA gene amplicon sequencing. BMC Microbiol. 2021, 21, 312. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Luo, J.L.; Ali, M.K.; Spiekerkoetter, E.; Nicolls, M.R. The Human Respiratory Microbiome: Current Understandings and Future Directions. Am. J. Respir. Cell Mol. Biol. 2023, 68, 245–255. [Google Scholar] [CrossRef]
- Wypych, T.P.; Wickramasinghe, L.C.; Marsland, B.J. The influence of the microbiome on respiratory health. Nat. Immunol. 2019, 20, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Geng, J.; Tang, X.; Fan, H.; Xu, J.; Wen, X.; Ma, Z.S.; Shi, P. Spatial heterogeneity and co-occurrence patterns of human mucosal-associated intestinal microbiota. ISME J. 2014, 8, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef]
- Li, D.; Chen, H.; Mao, B.; Yang, Q.; Zhao, J.; Gu, Z.; Zhang, H.; Chen, Y.Q.; Chen, W. Microbial Biogeography and Core Microbiota of the Rat Digestive Tract. Sci. Rep. 2017, 8, 45840. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, Y.; Wang, X.; Kong, L.; Johnston, L.J.; Lu, L.; Ma, X. Dietary nutrients shape gut microbes and intestinal mucosa via epigenetic modifications. Crit. Rev. Food Sci. Nutr. 2022, 62, 783–797. [Google Scholar] [CrossRef]
- Arrieta, M.C.; Stiemsma, L.T.; Dimitriu, P.A.; Thorson, L.; Russell, S.; Yurist-Doutsch, S.; Kuzeljevic, B.; Gold, M.J.; Britton, H.M.; Lefebvre, D.L.; et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci. Transl. Med. 2015, 7, 307ra152. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, T.; Wang, X.; Yu, C.; Qin, T.; Li, J.; Zhang, M.; Li, Z.; Cui, X.; Xu, X.; et al. Cangma Huadu granules attenuate H1N1 virus-induced severe lung injury correlated with repressed apoptosis and altered gut microbiome. Front. Microbiol. 2022, 13, 947112. [Google Scholar] [CrossRef]
- Ma, Q.; Li, Y.; Wang, J.; Li, P.; Duan, Y.; Dai, H.; An, Y.; Cheng, L.; Wang, T.; Wang, C.; et al. Investigation of gut microbiome changes in type 1 diabetic mellitus rats based on high-throughput sequencing. Biomed. Pharmacother. 2020, 124, 109873. [Google Scholar] [CrossRef]
- Ma, J.; Piao, X.; Mahfuz, S.; Long, S.; Wang, J. The interaction among gut microbes, the intestinal barrier and short chain fatty acids. Anim. Nutr. Zhongguo Xu Mu Shou Yi Xue Hui 2022, 9, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, Y.; Li, Q.; Ye, X.; Guo, X.; Sun, L.; Zou, J.; Shen, Y.; Mao, Y.; Li, C.; et al. Tetrahydrocurcumin alleviates allergic airway inflammation in asthmatic mice by modulating the gut microbiota. Food Funct. 2021, 12, 6830–6840. [Google Scholar] [CrossRef] [PubMed]
- Nowak, J.K.; Dworacka, M.; Gubaj, N.; Dossimov, A.; Dossimov, Z.; Walkowiak, J. Expression profiling of ileal mucosa in asthma reveals upregulation of innate immunity and genes characteristic of Paneth and goblet cells. Allergy Asthma Clin. Immunol. 2021, 17, 82. [Google Scholar] [CrossRef]
- Hamilton, M.K.; Boudry, G.; Lemay, D.G.; Raybould, H.E. Changes in intestinal barrier function and gut microbiota in high-fat diet-fed rats are dynamic and region dependent. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G840–G851. [Google Scholar] [CrossRef]
- Avalos-Fernandez, M.; Alin, T.; Métayer, C.; Thiébaut, R.; Enaud, R.; Delhaes, L. The respiratory microbiota alpha-diversity in chronic lung diseases: First systematic review and meta-analysis. Respir. Res. 2022, 23, 214. [Google Scholar] [CrossRef]
- Zhang, Q.; Cox, M.; Liang, Z.; Brinkmann, F.; Cardenas, P.A.; Duff, R.; Bhavsar, P.; Cookson, W.; Moffatt, M.; Chung, K.F. Airway Microbiota in Severe Asthma and Relationship to Asthma Severity and Phenotypes. PLoS ONE 2016, 11, e0152724. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, J.; Xi, Z.; Shi, Y.; Wang, L.; Lu, A. Characteristics of Lung Microbiota in Children’s Refractory Mycoplasma pneumoniae Pneumonia Coinfected with Human Adenovirus B. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 7065890. [Google Scholar] [CrossRef] [PubMed]
- Sohn, K.H.; Baek, M.G.; Choi, S.M.; Bae, B.; Kim, R.Y.; Kim, Y.C.; Kim, H.Y.; Yi, H.; Kang, H.R. Alteration of Lung and Gut Microbiota in IL-13-Transgenic Mice Simulating Chronic Asthma. J. Microbiol. Biotechnol. 2020, 30, 1819–1826. [Google Scholar] [CrossRef]
- Kumar, S.; Roy, R.D.; Sethi, G.R.; Saigal, S.R. Mycoplasma pneumoniae infection and asthma in children. Trop. Dr. 2019, 49, 117–119. [Google Scholar] [CrossRef]
- Zhao, J.L.; Wang, X.; Wang, Y.S. Relationships between Th1/Th2 cytokine profiles and chest radiographic manifestations in childhood Mycoplasma pneumoniae pneumonia. Ther. Clin. Risk Manag. 2016, 12, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.X.; Yuan, H.Y.; Li, Y.N.; Wei, Z.; Liu, Y.; Liang, J. Ephedra sinica polysaccharide alleviates airway inflammations of mouse asthma-like induced by PM2.5 and ovalbumin via the regulation of gut microbiota and short chain fatty acid. J. Pharm. Pharmacol. 2022, 74, 1784–1796. [Google Scholar] [CrossRef]
- Park, Y.M.; Lee, S.Y.; Kang, M.J.; Kim, B.S.; Lee, M.J.; Jung, S.S.; Yoon, J.S.; Cho, H.J.; Lee, E.; Yang, S.I.; et al. Imbalance of Gut Streptococcus, Clostridium, and Akkermansia Determines the Natural Course of Atopic Dermatitis in Infant. Allergy Asthma Immunol. Res. 2020, 12, 322–337. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lin, W.; Gu, H.; Ying, K.; Li, T.; Shao, H. Study on the Relationship Between Bronchoalveolar Lavage Fluid Cell Count, Th1/Th2 Cytokines and Pulmonary Function in Patients with Cough Variant Asthma. J. Asthma Allergy 2022, 15, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Wills-Karp, M. IL-12/IL-13 axis in allergic asthma. J. Allergy Clin. Immunol. 2001, 107, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Ingram, J.L.; Kraft, M. IL-13 in asthma and allergic disease: Asthma phenotypes and targeted therapies. J. Allergy Clin. Immunol. 2012, 130, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, T.; Zhao, X.; Wang, J.; Wang, Q. Plasma Metabolites and Gut Microbiota Are Associated with T cell Imbalance in BALB/c Model of Eosinophilic Asthma. Front. Pharmacol. 2022, 13, 819747. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Baldauf, S.L. Phylogeny for the faint of heart: A tutorial. Trends Genet. 2003, 19, 345–351. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dou, C.; Hu, L.; Ding, X.; Chen, F.; Li, X.; Wei, G.; Yan, Z. Microbiota Alterations in Lung, Ileum, and Colon of Guinea Pigs with Cough Variant Asthma. Int. J. Mol. Sci. 2024, 25, 2449. https://doi.org/10.3390/ijms25042449
Dou C, Hu L, Ding X, Chen F, Li X, Wei G, Yan Z. Microbiota Alterations in Lung, Ileum, and Colon of Guinea Pigs with Cough Variant Asthma. International Journal of Molecular Sciences. 2024; 25(4):2449. https://doi.org/10.3390/ijms25042449
Chicago/Turabian StyleDou, Chongyang, Lin Hu, Xian Ding, Fangfang Chen, Xi Li, Guihua Wei, and Zhiyong Yan. 2024. "Microbiota Alterations in Lung, Ileum, and Colon of Guinea Pigs with Cough Variant Asthma" International Journal of Molecular Sciences 25, no. 4: 2449. https://doi.org/10.3390/ijms25042449
APA StyleDou, C., Hu, L., Ding, X., Chen, F., Li, X., Wei, G., & Yan, Z. (2024). Microbiota Alterations in Lung, Ileum, and Colon of Guinea Pigs with Cough Variant Asthma. International Journal of Molecular Sciences, 25(4), 2449. https://doi.org/10.3390/ijms25042449