
Functional and Compositional Changes in Sirex noctilio Gut Microbiome in Different Habitats: Unraveling the Complexity of Invasive Adaptation
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
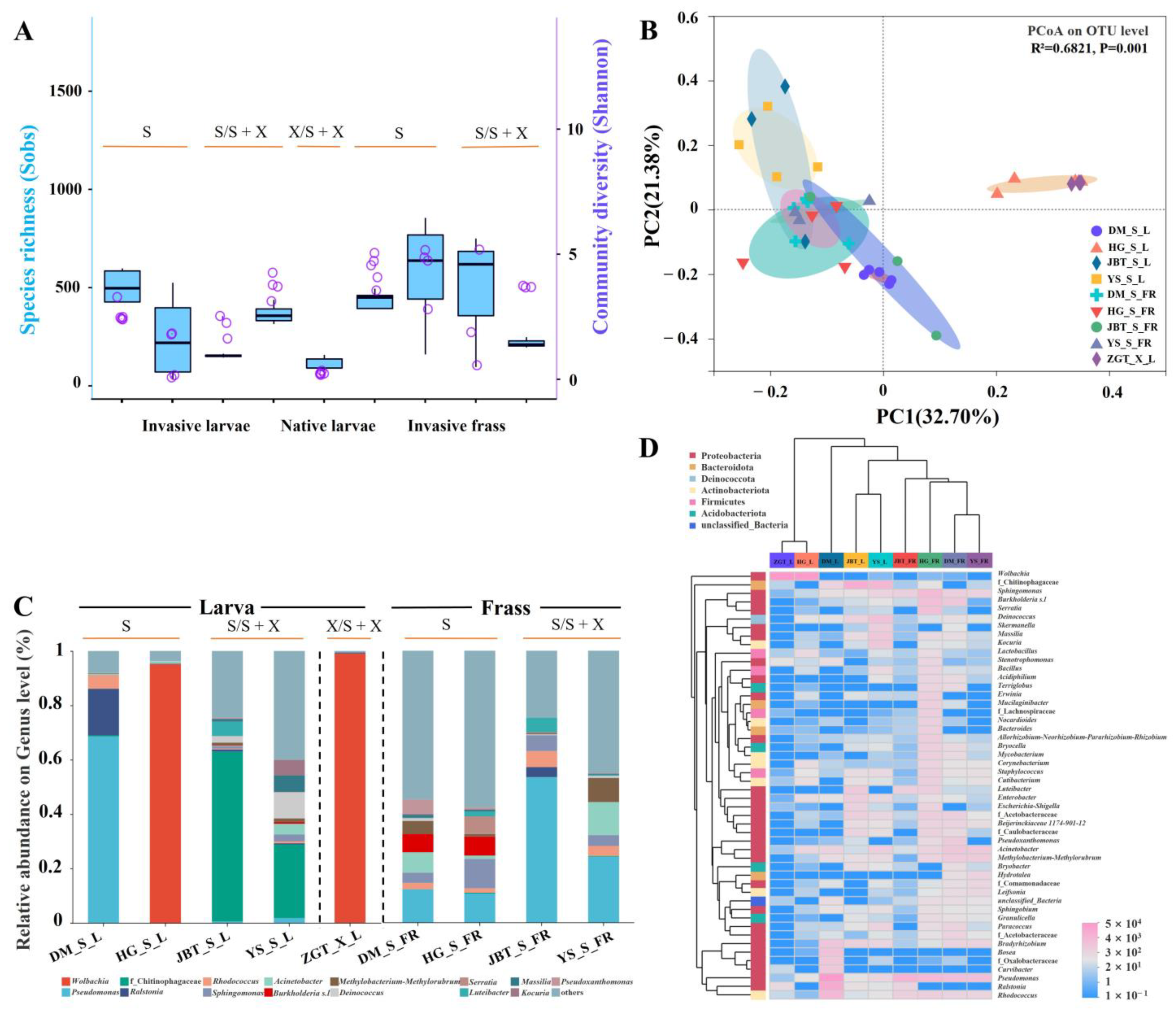
2.1. Microbial Communities Associated with Invasive and Native Woodwasp Species
2.1.1. Different Composition of the Gut Microbiome in the Two Sirex Species
2.1.2. Composition of the S. noctilio Gut Microbiome in Different Geographic Locations
2.1.3. Sirex noctilio Gut Microbiome Are Different from Frass
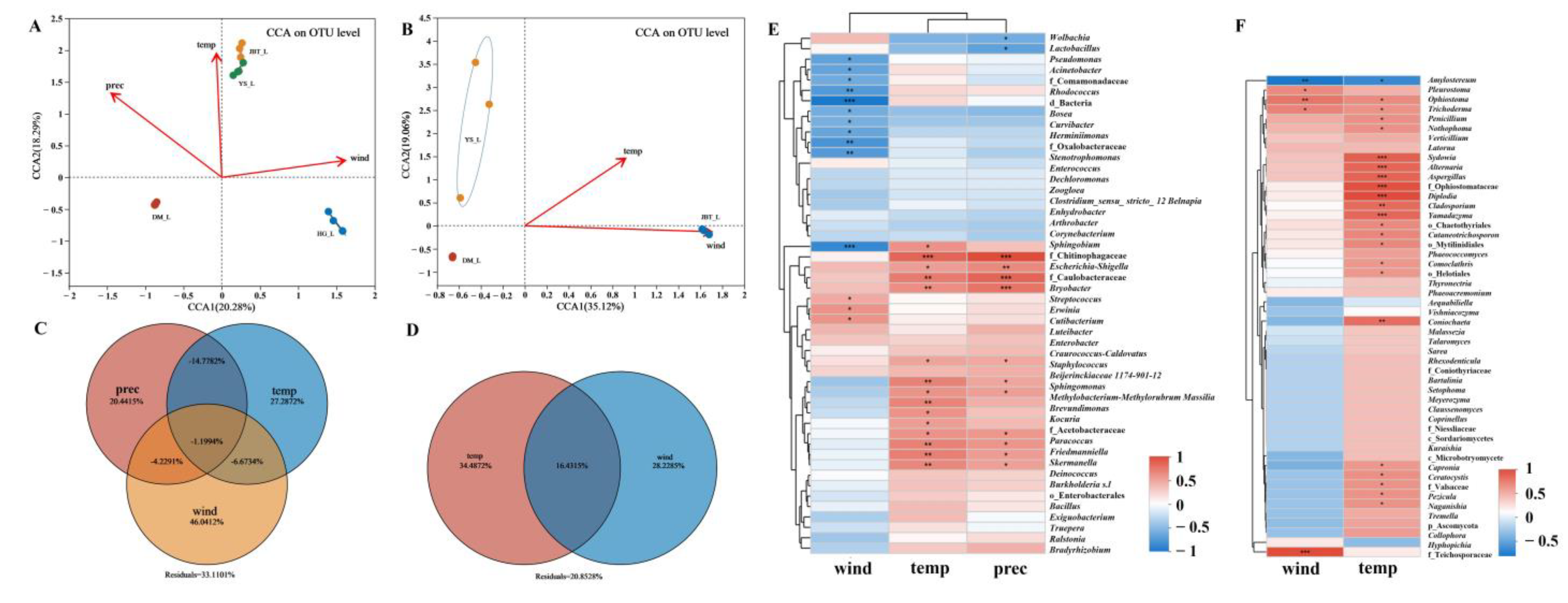
2.2. Potential Factors Affecting Microbial Communities
2.2.1. Effects of Meteorological Factors on S. noctilio Gut Microbiome
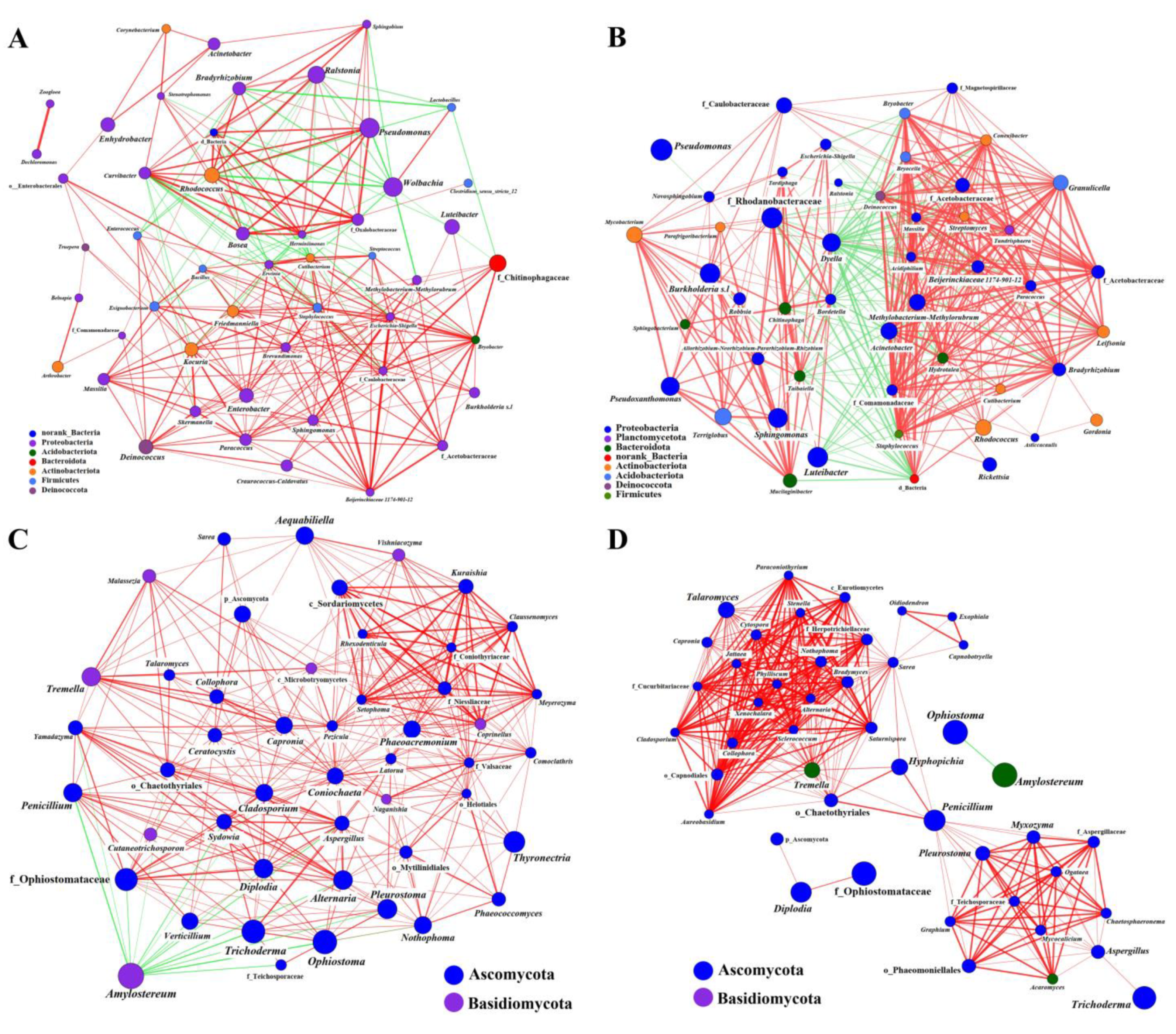
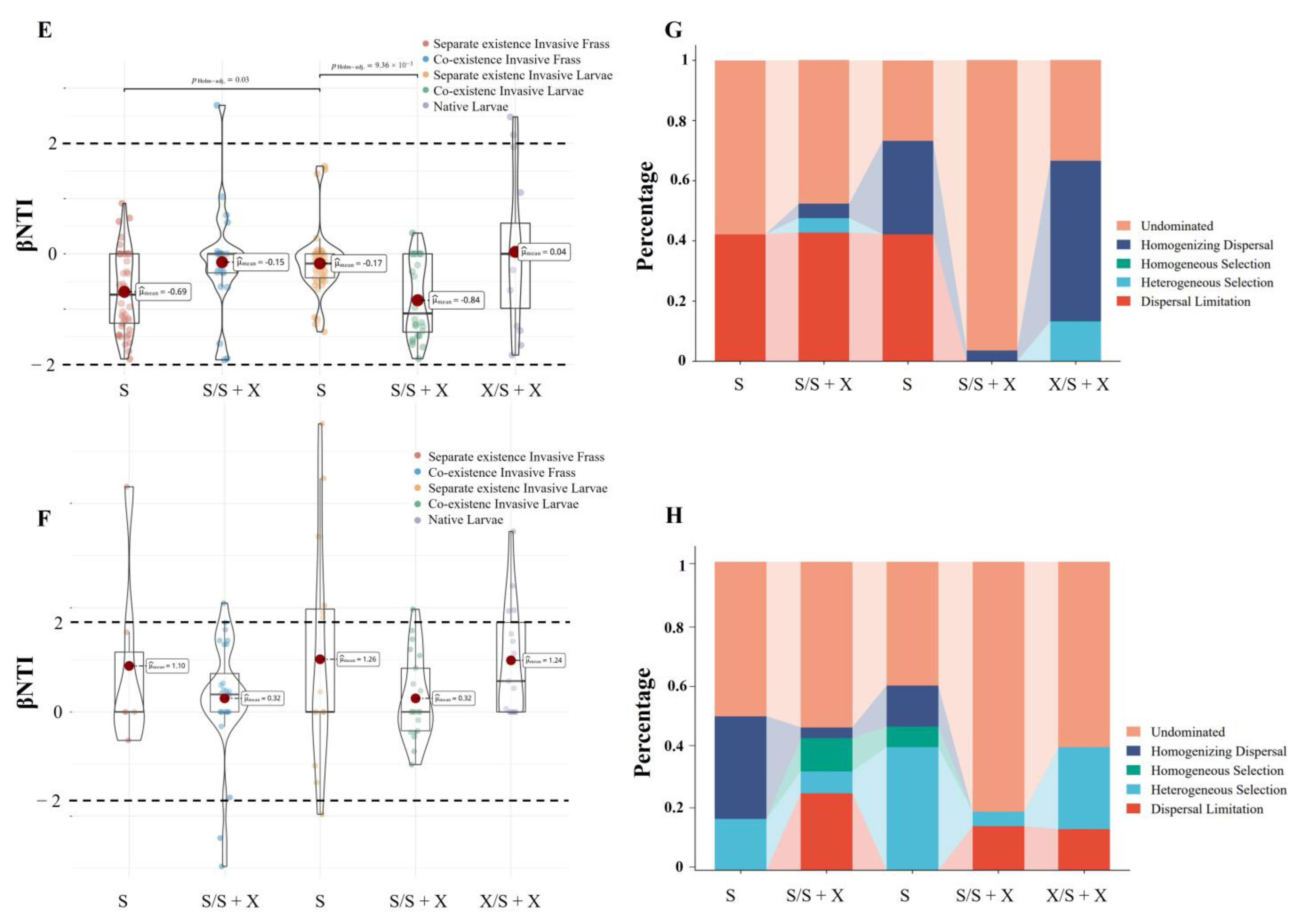
2.2.2. Coexistence of the Two Sirex Niches Enriched S. noctilio Gut Microbiome
2.3. Functional Differences between Woodwasp Larval Gut and Frass
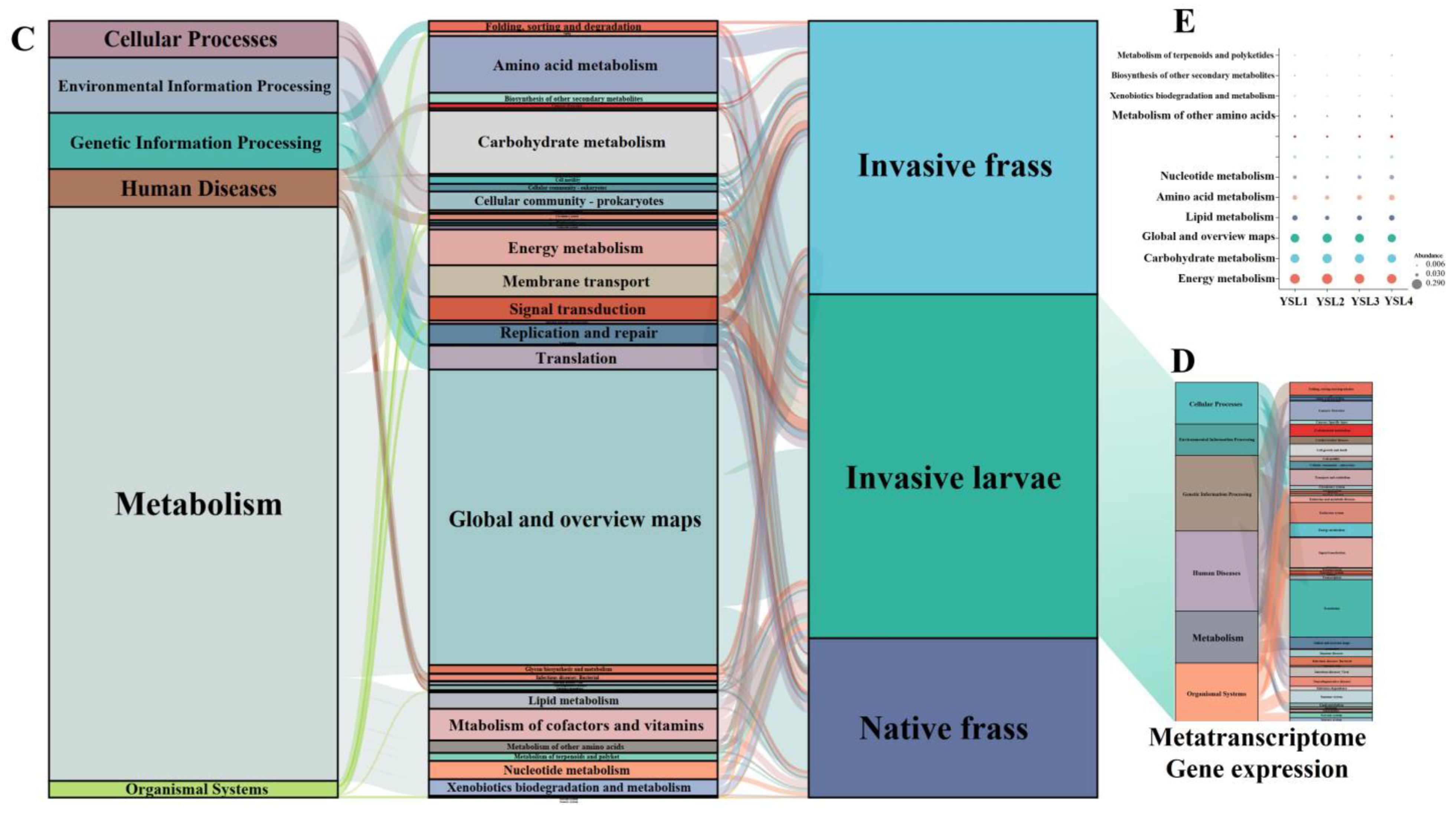
2.3.1. Functional Prediction Based on Amplifier Sequencing
2.3.2. Microbial Community Composition and CAZymes Identified in the Metatranscriptomes of the S. noctilio Larval Gut Microbiome
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Larval Identification
4.2. Total DNA and RNA Extraction, Library Preparation, and Sequencing
4.3. Amplicon Sequence Data Processing and Statistical Analysis
4.4. Metatranscriptome Sequence Data Processing and Statistical Analysis
4.5. Environmental Data and Statistical Analysis
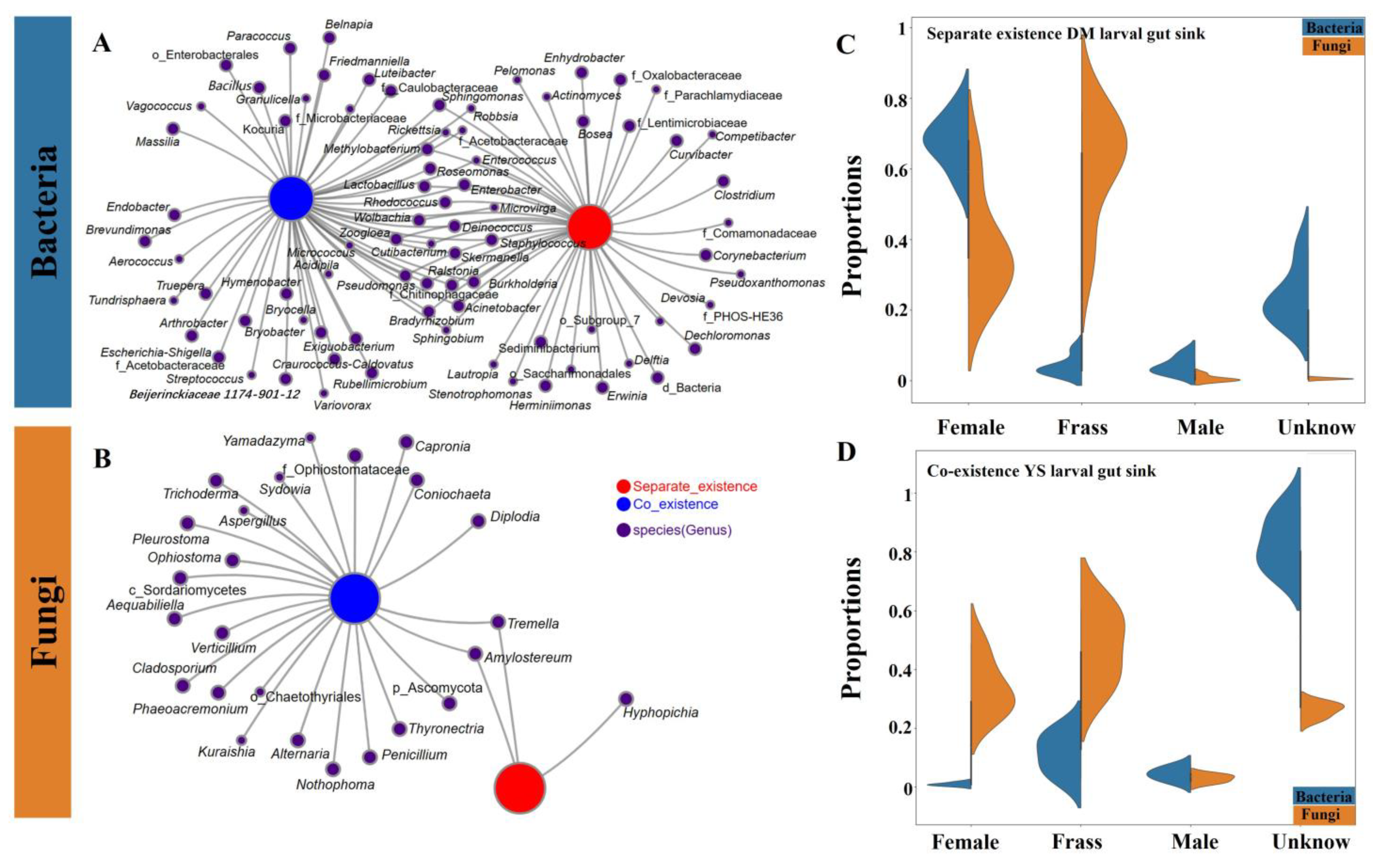
4.6. Estimation of Ecological Processes and Source Tracking of the Larval Gut Microbiome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haack, R.A. Intercepted Scolytidae (Coleoptera) at US ports of entry: 1985–2000. Integr. Pest Manag. Rev. 2001, 6, 253–282. [Google Scholar] [CrossRef]
- Lantschner, M.V.; Villacide, J.M.; Garnas, J.R.; Croft, P.; Carnegie, A.J.; Liebhold, A.M.; Corley, J.C. Temperature explains variable spread rates of the invasive woodwasp Sirex noctilio in the Southern Hemisphere. Biol. Invasions 2014, 16, 329–339. [Google Scholar] [CrossRef]
- Ramsfield, T.; Bentz, B.; Faccoli, M.; Jactel, H.; Brockerhoff, E. Forest health in a changing world: Effects of globalization and climate change on forest insect and pathogen impacts. Forestry 2016, 89, 245–252. [Google Scholar] [CrossRef]
- Hajek, A.E.; Tobin, P.C.; Kroll, S.A.; Long, S.J. Symbionts mediate oviposition behaviour in invasive and native woodwasps. Agric. For. Entomol. 2018, 20, 442–450. [Google Scholar] [CrossRef]
- Douglas, A.E. Omics and the metabolic function of insect–microbial symbioses. Curr. Opin. Insect Sci. 2018, 29, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Engel, P.; Moran, N.A. The gut microbiota of insects–diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Duan, J.; Gao, M.; Wang, Y.; Wang, X.; Zhao, K. Diversity, roles, and biotechnological applications of symbiotic microorganisms in the gut of termite. Curr. Microbiol. 2019, 76, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Berasategui, A.; Axelsson, K.; Nordlander, G.; Schmidt, A.; Borg-Karlson, A.K.; Gershenzon, J.; Terenius, O.; Kaltenpoth, M. The gut microbiota of the pine weevil is similar across Europe and resembles that of other conifer-feeding beetles. Mol. Ecol. 2016, 25, 4014–4031. [Google Scholar] [CrossRef]
- Janusz, G.; Pawlik, A.; Sulej, J.; Świderska-Burek, U.; Jarosz-Wilkołazka, A.; Paszczyński, A. Lignin degradation: Microorganisms, enzymes involved, genomes analysis and evolution. FEMS Microbiol. Rev. 2017, 41, 941–962. [Google Scholar] [CrossRef]
- Qu, Y.-F.; Wu, Y.-Q.; Zhao, Y.-T.; Lin, L.-H.; Du, Y.; Li, P.; Li, H.; Ji, X. The invasive red-eared slider turtle is more successful than the native Chinese three-keeled pond turtle: Evidence from the gut microbiota. PeerJ 2020, 8, e10271. [Google Scholar] [CrossRef]
- Foelker, C.J. Beneath the bark: Associations among Sirex noctilio development, bluestain fungi, and pine host species in North America. Ecol. Entomol. 2016, 41, 676–684. [Google Scholar] [CrossRef]
- Spradbery, J. A comparative study of the phytotoxic effects of siricid woodwasps on conifers. Ann. Appl. Biol. 1973, 75, 309–320. [Google Scholar] [CrossRef]
- Li, J.; Li, C.; Wang, M.; Wang, L.; Liu, X.; Gao, C.; Ren, L.; Luo, Y. Gut Structure and microbial communities in Sirex noctilio (Hymenoptera: Siricidae) and their predicted contribution to larval nutrition. Front. Microbiol. 2021, 12, 641141. [Google Scholar] [CrossRef]
- Schloss, P.D.; Delalibera, I., Jr.; Handelsman, J.; Raffa, K.F. Bacteria associated with the guts of two wood-boring beetles: Anoplophora glabripennis and Saperda vestita (Cerambycidae). Environ. Entomol. 2006, 35, 625–629. [Google Scholar] [CrossRef]
- Ryan, K.; Hurley, B.P. Life history and biology of Sirex noctilio. In The Sirex Woodwasp and Its Fungal Symbiont: Research and Management of a Worldwide Invasive Pest; Springer: Berlin/Heidelberg, Germany, 2011; pp. 15–30. [Google Scholar]
- Li, D.; Shi, J.; Lu, M.; Ren, L.; Zhen, C.; Luo, Y. Detection and identification of the invasive Sirex noctilio (Hymenoptera: Siricidae) fungal symbiont, Amylostereum areolatum (Russulales: Amylostereacea), in China and the stimulating effect of insect venom on laccase production by A. areolatum YQL03. J. Econ. Entomol. 2015, 108, 1136–1147. [Google Scholar] [CrossRef]
- Sun, X.; Tao, J.; Ren, L.; Shi, J.; Luo, Y. Identification of Sirex noctilio (Hymenoptera: Siricidae) using a species-specific cytochrome Coxidase subunit I PCR assay. J. Econ. Entomol. 2016, 109, 1424–1430. [Google Scholar] [CrossRef]
- Sun, X.; Xu, Q.; Luo, Y. A maximum entropy model predicts the potential geographic distribution of Sirex noctilio. Forests 2020, 11, 175. [Google Scholar] [CrossRef]
- Gao, T.; Xu, Q.; Liu, Y.; Zhao, J.; Shi, J. Predicting the potential geographic distribution of Sirex nitobei in China under climate change using maximum entropy model. Forests 2021, 12, 151. [Google Scholar] [CrossRef]
- He, S.; Ivanova, N.; Kirton, E.; Allgaier, M.; Bergin, C.; Scheffrahn, R.H.; Kyrpides, N.C.; Warnecke, F.; Tringe, S.G.; Hugenholtz, P. Comparative metagenomic and metatranscriptomic analysis of hindgut paunch microbiota in wood-and dung-feeding higher termites. PLoS ONE 2013, 8, e61126. [Google Scholar] [CrossRef] [PubMed]
- Martinson, V.G.; Danforth, B.N.; Minckley, R.L.; Rueppell, O.; Tingek, S.; Moran, N.A. A simple and distinctive microbiota associated with honey bees and bumble bees. Mol. Ecol. 2011, 20, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-F.; Bakker, M.G.; Judd, T.M.; Reardon, K.F.; Vivanco, J.M. Variations in diversity and richness of gut bacterial communities of termites (Reticulitermes flavipes) fed with grassy and woody plant substrates. Microb. Ecol. 2013, 65, 531–536. [Google Scholar] [CrossRef]
- Chen, B.; Du, K.; Sun, C.; Vimalanathan, A.; Liang, X.; Li, Y.; Wang, B.; Lu, X.; Li, L.; Shao, Y. Gut bacterial and fungal communities of the domesticated silkworm (Bombyx mori) and wild mulberry-feeding relatives. ISME J. 2018, 12, 2252–2262. [Google Scholar] [CrossRef]
- Kudo, R.; Masuya, H.; Endoh, R.; Kikuchi, T.; Ikeda, H. Gut bacterial and fungal communities in ground-dwelling beetles are associated with host food habit and habitat. ISME J. 2019, 13, 676–685. [Google Scholar] [CrossRef]
- Jones, A.G.; Mason, C.J.; Felton, G.W.; Hoover, K. Host plant and population source drive diversity of microbial gut communities in two polyphagous insects. Sci. Rep. 2019, 9, 2792. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, Y.; Li, H.; Yu, X.; Shi, W.; Zhai, J. Comparison of microbial communities in Colorado potato beetles (Leptinotarsa decemlineata say) collected from different sources in China. Front. Microbiol. 2021, 12, 639913. [Google Scholar] [CrossRef]
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18933–18938. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Wang, J.; Huang, J.; Zhang, S.; Vogler, A.P.; Liu, Q.; Li, Y.; Yang, M.; Li, Y.; Zhou, X. Host phylogeny and diet shape gut microbial communities within bamboo-feeding insects. Front. Microbiol. 2021, 12, 633075. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Zeng, Q.; Li, J.; Li, J.; Tan, X.; Gao, X.; Huang, P.; Wu, S. Vertical variation in prokaryotic community composition and co-occurrence patterns in sediments of the Three Gorges Reservoir, China. Environ. Res. 2023, 237, 116927. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, O.A.; Villacide, J.M.; Bernstein, C.; Corley, J.C. Flight variability in the woodwasp Sirex noctilio (Hymenoptera: Siricidae): An analysis of flight data using wavelets. J. Exp. Biol. 2009, 212, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Ireland, K.B.; Bulman, L.; Hoskins, A.J.; Pinkard, E.A.; Mohammed, C.; Kriticos, D.J. Estimating the potential geographical range of Sirex noctilio: Comparison with an existing model and relationship with field severity. Biol. Invasions 2018, 20, 2599–2622. [Google Scholar] [CrossRef]
- Ge, Y.; Jing, Z.; Diao, Q.; He, J.-Z.; Liu, Y.-J. Host species and geography differentiate honeybee gut bacterial communities by changing the relative contribution of community assembly processes. mBio 2021, 12, e0075121. [Google Scholar] [CrossRef] [PubMed]
- Werren, J.H.; Baldo, L.; Clark, M.E. Wolbachia: Master manipulators of invertebrate biology. Nat. Rev. Microbiol. 2008, 6, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Michalik, A.; Franco, D.C.; Deng, J.; Szklarzewicz, T.; Stroiński, A.; Kobiałka, M.; Łukasik, P. Variable organization of symbiont-containing tissue across planthoppers hosting different heritable endosymbionts. Front. Physiol. 2023, 14, 471. [Google Scholar] [CrossRef] [PubMed]
- Schiff, N.M.; Goulet, H.; Smith, D.R.; Boudreault, C.; Wilson, A.D.; Scheffler, B.E. Siricidae (Hymenoptera: Symphyta: Siricoidea) of the western hemisphere. Can. J. Arthropod Identif. 2012, 21, 1–305. [Google Scholar]
- Marčiulynas, A.; Lynikienė, J.; Gedminas, A.; Povilaitienė, A.; Menkis, A. Fungal Communities Associated with Siricid Wood Wasps: Focus on Sirex juvencus, Urocerus gigas, and Tremex fuscicornis. Insects 2024, 15, 49. [Google Scholar] [CrossRef]
- Barcoto, M.O.; Carlos-Shanley, C.; Fan, H.; Ferro, M.; Nagamoto, N.S.; Bacci, M.; Currie, C.R.; Rodrigues, A. Fungus-growing insects host a distinctive microbiota apparently adapted to the fungiculture environment. Sci. Rep. 2020, 10, 12384. [Google Scholar] [CrossRef]
- Kwong, W.K.; Moran, N.A. Gut microbial communities of social bees. Nat. Rev. Microbiol. 2016, 14, 374–384. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Yang, R.; Wang, X.Y.; Wen, T.; Gong, M.H.; Shen, Y.; Xu, J.Y.; Zhao, D.S.; Du, Y.Z. Gut microbiota composition in the sympatric and diet-sharing Drosophila simulans and Dicranocephalus wallichii bowringi shaped largely by community assembly processes rather than regional species pool. iMeta 2022, 1, e57. [Google Scholar] [CrossRef]
- Zhang, S.; Li, Z.; Shu, J.; Xue, H.; Guo, K.; Zhou, X. Soil-derived bacteria endow Camellia weevil with more ability to resist plant chemical defense. Microbiome 2022, 10, 97. [Google Scholar] [CrossRef]
- Antwis, R.E.; Griffiths, S.M.; Harrison, X.A.; Aranega-Bou, P.; Arce, A.; Bettridge, A.S.; Brailsford, F.L.; de Menezes, A.; Devaynes, A.; Forbes, K.M. Fifty important research questions in microbial ecology. FEMS Microbiol. Ecol. 2017, 93, fix044. [Google Scholar] [CrossRef]
- Chen, L.; Dini-Andreote, F.; Liu, H.; Wang, H.; Dumbrell, A.; Wang, Z.; Chen, X.; Chen, F.; Chen, X.; Wu, L. Integrating variation in bacterial-fungal co-occurrence network with soil carbon dynamics. J. Appl. Ecol. 2023, 61, 36–50. [Google Scholar] [CrossRef]
- Madden, J. Oviposition behaviour of the woodwasp, Sirex noctilio F. Aust. J. Zool. 1974, 22, 341–351. [Google Scholar] [CrossRef]
- Wang, M.; Fu, N.; Gao, C.; Wang, L.; Ren, L.; Luo, Y. Multilocus Genotyping and Intergenic Spacer Single Nucleotide Polymorphisms of Amylostereum areolatum (Russulales: Amylostereacea) Symbionts of Native and Non-Native Sirex Species. J. Fungi 2021, 7, 1065. [Google Scholar] [CrossRef] [PubMed]
- Voolstra, C.R.; Ziegler, M. Adapting with microbial help: Microbiome flexibility facilitates rapid responses to environmental change. BioEssays 2020, 42, 2000004. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37 (Suppl. 1), D233–D238. [Google Scholar] [CrossRef]
- He, J.; Yang, K.; Tang, W.; Lu, H.; Qin, J.; Chen, Y.; Li, X. The first high-resolution meteorological forcing dataset for land process studies over China. Sci. Data 2020, 7, 25. [Google Scholar] [CrossRef]
- Li, H.; Xia, W.; Liu, X.; Wang, X.; Liu, G.; Chen, H.; Zhu, L.; Li, D. Food provisioning results in functional, but not compositional, convergence of the gut microbiomes of two wild Rhinopithecus species: Evidence of functional redundancy in the gut microbiome. Sci. Total Environ. 2023, 858, 159957. [Google Scholar] [CrossRef]
- Knights, D.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Mozer, M.C.; Collman, R.G.; Bushman, F.D.; Knight, R.; Kelley, S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011, 8, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Konopka, A.E. Estimating and mapping ecological processes influencing microbial community assembly. Front. Microbiol. 2015, 6, 370. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, X.; Guo, L. Research progress on community assembly mechanisms of fungi. Mycosystema 2023, 42, 13–25. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAZy | Count of Family | Count of Gene | Major Family |
|---|---|---|---|
| GHs | 28 | 99 | GH16, GH18, GH20, GH47, GH13, GH38, GH31, GH109, GH15, GH22 |
| GTs | 46 | 173 | GT31, GT27, GT2, GT92, GT49, GT8, GT10, GT7, GT1,GT22, GT4 |
| AAs | 4 | 13 | AA3, AA4, AA1, AA7 |
| CBMs | 19 | 127 | CBM14, CBM32, CBM13, CBM21, CBM48, CBM50, CBM37, CBM63 |
| CEs | 7 | 46 | CE10, CE1, CE4, CE14, CE11, CE5, CE9 |
| PLs | 2 | 2 | PL1, PL22 |
| 106 | 460 |
| Location | Geographic Coordinates | Developmental Stage | Collection Year/Month | |
|---|---|---|---|---|
| Larva | Frass | |||
| Duerbert Mongolian Autonomous County, Daqing City, Heilongjiang Province (DM) | 46.88° N 124.46° E | 50 | 50 | May 2018 |
| Hegang City, Heilongjiang Province (HG) | 47.12° N 130.17° E | 40 | 40 | May 2016 |
| Jinbaotun Town, Tongliao City, Inner Mongolia Autonomous Region (JBT) | 43.07° N 123.28° E | 35 | 35 | May 2018 |
| Yushu City, Jilin Province (YS) | 44.86° N 126.87° E | 60 | 35 | May 2018 |
| Zhanggutai Town, Zhangwu County, Fuxin City, Liaoning Province (ZGT) | 42.64° N 122.50° E | 30 | / | May 2018 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Fu, N.; Wang, M.; Gao, C.; Gao, B.; Ren, L.; Tao, J.; Luo, Y. Functional and Compositional Changes in Sirex noctilio Gut Microbiome in Different Habitats: Unraveling the Complexity of Invasive Adaptation. Int. J. Mol. Sci. 2024, 25, 2526. https://doi.org/10.3390/ijms25052526
Li J, Fu N, Wang M, Gao C, Gao B, Ren L, Tao J, Luo Y. Functional and Compositional Changes in Sirex noctilio Gut Microbiome in Different Habitats: Unraveling the Complexity of Invasive Adaptation. International Journal of Molecular Sciences. 2024; 25(5):2526. https://doi.org/10.3390/ijms25052526
Chicago/Turabian StyleLi, Jiale, Ningning Fu, Ming Wang, Chenglong Gao, Bingtao Gao, Lili Ren, Jing Tao, and Youqing Luo. 2024. "Functional and Compositional Changes in Sirex noctilio Gut Microbiome in Different Habitats: Unraveling the Complexity of Invasive Adaptation" International Journal of Molecular Sciences 25, no. 5: 2526. https://doi.org/10.3390/ijms25052526