
The Role of Opioid Receptor Antagonists in Regulation of Blood Pressure and T-Cell Activation in Mice Selected for High Analgesia Induced by Swim Stress
 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
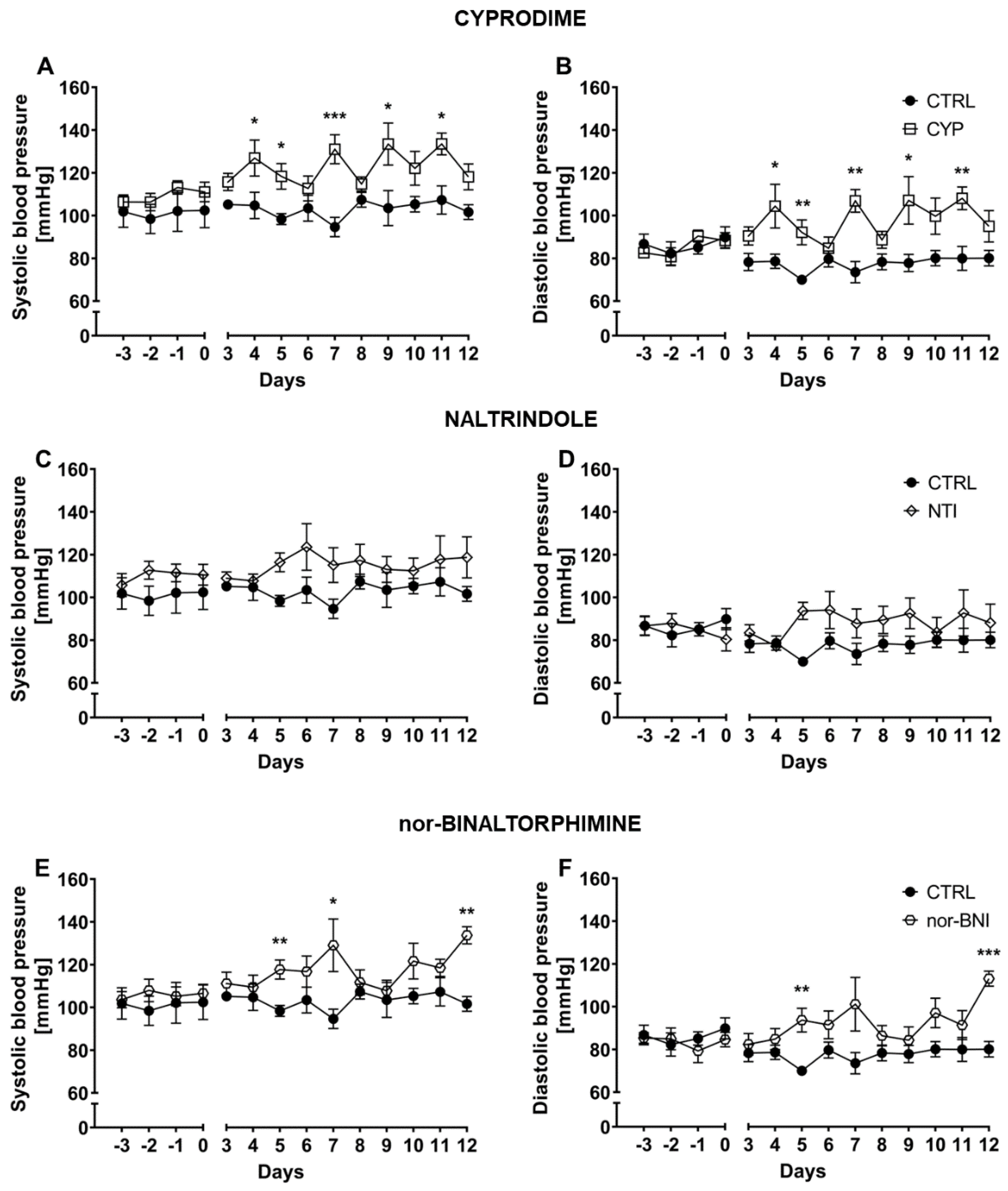
2.1. Effect of the Non-Selective Blockade of the Opioid System on Systolic and Diastolic Blood Pressure
2.2. Effect of Selective Opioid Receptor Antagonists on Systolic and Diastolic Blood Pressure
2.3. Effect of Non-Selective Blockade of the Opioid System on the T-Cells Subpopulations
2.4. Effect of the Selective Blockade of the Opioid System on the T-Cell Subpopulation
3. Discussion
4. Methods
4.1. Animals
4.2. Antagonist Administration
4.3. Measurement of Blood Pressure
4.4. Analysis of T-Cell Subpopulation and Activation
4.5. Data and Statistical Analysis
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Headrick, J.P.; Pepe, S.; Peart, J.N. Non-analgesic effects of opioids: Cardiovascular effects of opioids and their receptor systems. Curr. Pharm. Des. 2012, 18, 6090–6100. [Google Scholar] [CrossRef]
- Tanaka, G.Y. Letter: Hypertensive reaction to naloxone. JAMA 1974, 228, 25–26. [Google Scholar] [CrossRef] [PubMed]
- Britch, S.C.; Walsh, S.L. Treatment of opioid overdose: Current approaches and recent advances. Psychopharmacology 2022, 239, 2063–2081. [Google Scholar] [CrossRef] [PubMed]
- Bruehl, S.; Chung, O.Y.; Jirjis, J.N.; Biridepalli, S. Prevalence of clinical hypertension in patients with chronic pain compared to nonpain general medical patients. Clin. J. Pain. 2005, 21, 147–153. [Google Scholar] [CrossRef]
- King, J.W.; Bair, E.; Duggan, D.; Maixner, W.; Khan, A.A. The relationship between resting arterial blood pressure and acute postoperative pain in endodontic patients. J. Orofac. Pain. 2012, 26, 321–327. [Google Scholar] [PubMed]
- Deschaumes, C.; Devoize, L.; Sudrat, Y.; Baudet-Pommel, M.; Dualé, C.; Dallel, R. The relationship between resting arterial blood pressure and oral postsurgical pain. Clin. Oral. Investig. 2015, 19, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.V.; Chapleau, M.W.; Harwani, S.C.; Abboud, F.M. The immune system and hypertension. Immunol. Res. 2014, 59, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Cant, R.; Dalgleish, A.G.; Allen, R.L. Naltrexone Inhibits IL-6 and TNFα Production in Human Immune Cell Subsets following Stimulation with Ligands for Intracellular Toll-Like Receptors. Front. Immunol. 2017, 8, 809. [Google Scholar] [CrossRef] [PubMed]
- Panocka, I.; Marek, P.; Sadowski, B. Differentiation of neurochemical basis of stress-induced analgesia in mice by selective breeding. Brain Res. 1986, 397, 156–160. [Google Scholar] [CrossRef]
- Poznanski, P.; Lesniak, A.; Korostynski, M.; Szklarczyk, K.; Lazarczyk, M.; Religa, P.; Bujalska-Zadrozny, M.; Sadowski, B.; Sacharczuk, M. Delta-opioid receptor antagonism leads to excessive ethanol consumption in mice with enhanced activity of the endogenous opioid system. Neuropharmacology 2017, 118, 90–101. [Google Scholar] [CrossRef]
- Lesniak, A.; Chmielewska, D.; Poznanski, P.; Bujalska-Zadrozny, M.; Strzemecka, J.; Sacharczuk, M. Divergent Response to Cannabinoid Receptor Stimulation in High and Low Stress-Induced Analgesia Mouse Lines Is Associated with Differential G-Protein Activation. Neuroscience 2019, 404, 246–258. [Google Scholar] [CrossRef]
- Poznański, P.; Lesniak, A.; Bujalska-Zadrozny, M.; Strzemecka, J.; Sacharczuk, M. Bidirectional selection for high and low stress-induced analgesia affects G-protein activity. Neuropharmacology 2019, 144, 37–42. [Google Scholar] [CrossRef]
- Skiba, D.S.; Szczepaniak, P.; Siedliński, M.; Poznański, P.; Łazarczyk, M.; Jaskuła, K.; Religa, P.; Sacharczuk, M.; Gaciong, Z. Hypertensive Effect of Downregulation of the Opioid System in Mouse Model of Different Activity of the Endogenous Opioid System. Int. J. Mol. Sci. 2021, 22, 4179. [Google Scholar] [CrossRef]
- Perekopskiy, D.; Afzal, A.; Jackson, S.N.; Muller, L.; Woods, A.S.; Kiyatkin, E.A. The Role of Peripheral Opioid Receptors in Triggering Heroin-induced Brain Hypoxia. Sci. Rep. 2020, 10, 833. [Google Scholar] [CrossRef]
- Hernandez, J.; Pérez-Ojeda, E.; Serrano, J.S.; Castillo, J.R.; Serrano, M.I. Possible involvement of epinephrine in the cardiovascular effect of naloxone in humans. Clin. Ther. 1985, 7, 418–423. [Google Scholar]
- Cohen, M.R.; Cohen, R.M.; Pickar, D.; Murphy, D.L.; Bunney, W.E., Jr. Physiological effects of high dose naloxone administration to normal adults. Life Sci. 1982, 30, 2025–2031. [Google Scholar] [CrossRef] [PubMed]
- al’Absi, M.; Wittmers, L.E.; Ellestad, D.; Nordehn, G.; Kim, S.W.; Kirschbaum, C.; Grant, J.E. Sex differences in pain and hypothalamic-pituitary-adrenocortical responses to opioid blockade. Psychosom. Med. 2004, 66, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, M.; Maggi, M.; De Feo, M.L.; Cuomo, S.; Delitala, G.; Giusti, G.; Serio, M. Effects of naloxone on catecholamine plasma levels in adult men. A dose-response study. Acta Endocrinol. 1984, 106, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Grossman, A.; Bouloux, P.; Price, P.; Drury, P.L.; Lam, K.S.; Turner, T.; Thomas, J.; Besser, G.M.; Sutton, J. The role of opioid peptides in the hormonal responses to acute exercise in man. Clin. Sci. 1984, 67, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.A.; Gustafson, A.B.; Garthwaite, T.L.; Kalkhoff, R.K.; Cowley, A.W., Jr.; Morgan, W.P. Influence of endogenous opioids on the response of selected hormones to exercise in humans. J. Appl. Physiol. 1986, 61, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Ray, L.A.; Bujarski, S.; Courtney, K.E.; Moallem, N.R.; Lunny, K.; Roche, D.; Leventhal, A.M.; Shoptaw, S.; Heinzerling, K.; London, E.D.; et al. The Effects of Naltrexone on Subjective Response to Methamphetamine in a Clinical Sample: A Double-Blind, Placebo-Controlled Laboratory Study. Neuropsychopharmacology 2015, 40, 2347–2356. [Google Scholar] [CrossRef]
- McCubbin, J.A.; Cheung, R.; Montgomery, T.B.; Bulbulian, R.; Wilson, J.F. Aerobic Fitness and Opioidergic Inhibition of Cardiovascular Stress Reactivity. Psychophysiology 1992, 29, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Herman, B.H.; Hammock, M.K.; Arthur-Smith, A.; Kuehl, K.; Appelgate, K. Effects of acute administration of naltrexone on cardiovascular function, body temperature, body weight and serum concentrations of liver enzymes in autistic children. Dev. Pharmacol. Ther. 1989, 12, 118–127. [Google Scholar] [CrossRef] [PubMed]
- McCaul, M.E.; Wand, G.S.; Stauffer, R.; Lee, S.M.; Rohde, C.A. Naltrexone dampens ethanol-induced cardiovascular and hypothalamic- pituitary-adrenal axis activation. Neuropsychopharmacology 2001, 25, 537–547. [Google Scholar] [CrossRef]
- Maguire, D.R.; Gerak, L.R.; Cami-Kobeci, G.; Husbands, S.M.; France, C.P.; Belli, B.; Flynn, P. OREX-1019: A Novel Treatment of Opioid Use Disorder and Relapse Prevention. J. Pharmacol. Exp. Ther. 2020, 372, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Byrd, L.D. Cardiovascular effects of naloxone, naltrexone and morphine in the squirrel monkey. Life Sci. 1983, 32, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Anand-Srivastava, M.B. Role of Gi proteins in the regulation of blood pressure and vascular remodeling. Biochem. Pharmacol. 2023, 208, 115384. [Google Scholar] [CrossRef] [PubMed]
- Samuels, E.R.; Szabadi, E. Functional neuroanatomy of the noradrenergic locus coeruleus: Its roles in the regulation of arousal and autonomic function part I: Principles of functional organisation. Curr. Neuropharmacol. 2008, 6, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Cuthbert, B.N.; Holaday, J.W.; Meyerhoff, J.; Li, C.H. Intravenous beta-endorphin: Behavioral and physiological effects in conscious monkeys. Peptides 1989, 10, 729–734. [Google Scholar] [CrossRef]
- Sitsen, J.M.; Van Ree, J.M.; De Jong, W. Cardiovascular and respiratory effects of beta-endorphin in anesthetized and conscious rats. J. Cardiovasc. Pharmacol. 1982, 4, 883–888. [Google Scholar] [CrossRef]
- Fontana, F.; Bernardi, P.; Spampinato, S.; Boschi, S.; De Iasio, R.; Grossi, G. Pressor effects of endogenous opioid system during acute episodes of blood pressure increases in hypertensive patients. Hypertension 1997, 29, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Barron, B.A. Opioid peptides and the heart. Cardiovasc. Res. 1999, 43, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Zimlichman, R.; Gefel, D.; Eliahou, H.; Matas, Z.; Rosen, B.; Gass, S.; Ela, C.; Eilam, Y.; Vogel, Z.; Barg, J. Expression of opioid receptors during heart ontogeny in normotensive and hypertensive rats. Circulation 1996, 93, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.C.; Shieh, J.P.; Chung, H.H.; Hung, C.H.; Lin, H.J.; Cheng, J.T. Activation of peripheral opioid µ-receptors in blood vessel may lower blood pressure in spontaneously hypertensive rats. Pharmacology 2011, 87, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.M.; Shan, J. Modulation of sympathetic actions on the heart by opioid receptor stimulation. J. Biomed. Sci. 2001, 8, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Headrick, J.P.; See Hoe, L.E.; Du Toit, E.F.; Peart, J.N. Opioid receptors and cardioprotection-’opioidergic conditioning’ of the heart. Br. J. Pharmacol. 2015, 172, 2026–2050. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, J.E.; Ferrario, C.M. Cardiovascular effects of chronic naloxone infusion in normal dogs. Neuroendocrinology 1986, 43, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Hahn, E.F.; Martucci, C.P.; Gilligan, J.P.; Spector, S. Early exposure to naloxone increases blood pressure in normotensive and hypertensive rats. Biochem. Biophys. Res. Commun. 1982, 107, 707–713. [Google Scholar] [CrossRef]
- Romualdi, P.; Lesa, G.; Donatini, A.; Ferri, S. Long-term exposure to opioid antagonists up-regulates prodynorphin gene expression in rat brain. Brain Res. 1995, 672, 42–47. [Google Scholar] [CrossRef]
- Tempel, A.; Gardner, E.L.; Zukin, R.S. Neurochemical and functional correlates of naltrexone-induced opiate receptor up-regulation. J Pharmacol Exp Ther 1985, 232, 439–444. [Google Scholar]
- Grasing, K.; Szeto, H. Naloxone causes a dose-dependent increase in total power and delta wave activity in the EEG of opioid-naive rats. J. Pharmacol. Exp. Ther. 1991, 259, 464–469. [Google Scholar] [PubMed]
- Farsang, C.; Kunos, G. Naloxone reverses the antihypertensive effect of clonidine. Br. J. Pharmacol. 1979, 67, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Moura, E.; Afonso, J.; Serrão, M.P.; Vieira-Coelho, M.A. Effect of clonidine on tyrosine hydroxylase activity in the adrenal medulla and brain of spontaneously hypertensive rats. Basic. Clin. Pharmacol. Toxicol. 2009, 104, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Bramnert, M. The effect of naloxone on blood pressure, heart rate, plasma catecholamines, renin activity and aldosterone following exercise in healthy males. Regul. Pept. 1988, 22, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Nakao, K.; Itoh, H.; Shirakami, G.; Sugawara, A.; Saito, Y.; Mukoyama, M.; Arai, H.; Hosoda, K.; Shiono, S.; et al. Effects of naloxone on vasopressin secretion in conscious rats: Evidence for inhibitory role of endogenous opioid peptides in vasopressin secretion. Endocrinology 1989, 125, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Strahlendorf, J.C.; Strahlendorf, H.K.; McKown-Pulliam, R.; Hughes, M.J.; Lang, I.M. Chronic administration of naltrexone alters central catecholamine levels but not the development of hypertension in spontaneously hypertensive rats. Neuropharmacology 1982, 21, 1195–1198. [Google Scholar] [CrossRef]
- Makulska-Nowak, H.E.; Gumulka, S.W.; Kolaric, S. Effects of Naloxone Hydrochloride and Naloxone Methiodide on the Development and Severity of Hypertension in SHR During Experimental Inflammation. Analgesia 1998, 3, 291–295. [Google Scholar] [CrossRef]
- Yin, X.; Zhu, Y.H.; Xu, S.F. Distributions of mu and delta opioid receptors in central nervous system of SHR rats and normotensive WKY rats. Zhongguo Yao Li Xue Bao 1996, 17, 28–31. [Google Scholar]
- Munro, T.A.; Huang, X.P.; Inglese, C.; Perrone, M.G.; Van’t Veer, A.; Carroll, F.I.; Béguin, C.; Carlezon, W.A., Jr.; Colabufo, N.A.; Cohen, B.M.; et al. Selective κ opioid antagonists nor-BNI, GNTI and JDTic have low affinities for non-opioid receptors and transporters. PLoS ONE 2013, 8, e70701. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Ingenito, A.J. Chronic blockade of hippocampal kappa receptors increases arterial pressure in conscious spontaneously hypertensive rats but not in normotensive Wistar Kyoto rats. Clin. Exp. Hypertens. 2000, 22, 507–519. [Google Scholar] [CrossRef]
- Wright, R.C.; McConnaughey, M.M.; Phan, T.A.; Ingenito, A.J. Kappa-opioid receptor antisense oligonucleotide injected into rat hippocampus causes hypertension. Eur. J. Pharmacol. 1999, 377, 57–61. [Google Scholar] [CrossRef]
- Wright, R.C.; Ingenito, A.J. Blockade of dorsal hippocampal kappa-opioid receptors increases blood pressure in normotensive and isolation-induced hypertensive rats. Neuropeptides 2003, 37, 127–132. [Google Scholar] [CrossRef]
- Moss, I.R.; Faltus, R.E.; Inman, J.D.; Laferrière, A. Cardiorespiratory and sleep-wake behavior in developing swine: Kappa-opioid influence. Respir. Physiol. 1995, 101, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.I.; Ferreira, H.S.; Saraiva, R.M.; Almeida, T.S.; Fregoneze, J.B. Central kappa opioid receptors modulate salt appetite in rats. Physiol. Behav. 2012, 106, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Kraft, K.; Diehl, J.; Stumpe, K.O. Influence of chronic opioid delta receptor antagonism on blood pressure development and tissue contents of catecholamines and endogenous opioids in spontaneously hypertensive rats. Clin. Exp. Hypertens. A 1991, 13, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.B.; Gordon, F.J.; Holtzman, S.G. Naltrindole, a selective delta-opioid receptor antagonist, potentiates the lethal effects of cocaine by a central mechanism of action. Eur. J. Pharmacol. 1997, 333, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Sezen, S.F.; Kenigs, V.A.; Kapusta, D.R. Renal excretory responses produced by the delta opioid agonist, BW373U86, in conscious rats. J. Pharmacol. Exp. Ther. 1998, 287, 238–245. [Google Scholar] [PubMed]
- Morilak, D.; Drolet, G.; Chalmers, J. A lack of potency for the delta-opioid antagonist naltrindole after microinjection into the rostral ventrolateral medulla of rabbits. Clin. Exp. Pharmacol. Physiol. 1990, 17, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef]
- Plein, L.M.; Rittner, H.L. Opioids and the immune system-friend or foe. Br. J. Pharmacol. 2018, 175, 2717–2725. [Google Scholar] [CrossRef]
- Manfredi, B.; Sacerdote, P.; Bianchi, M.; Locatelli, L.; Veljic-Radulovic, J.; Panerai, A.E. Evidence for an opioid inhibitory effect on T cell proliferation. J. Neuroimmunol. 1993, 44, 43–48. [Google Scholar] [CrossRef]
- Ferry, A.; Weill, B.; Amiridis, I.; Laziry, F.; Rieu, M. Splenic immunomodulation with swimming-induced stress in rats. Immunol. Lett. 1991, 29, 261–264. [Google Scholar] [CrossRef]
- Singh, V.K.; Bajpai, K.; Narayan, P.; Yadav, V.S.; Dhawan, V.C.; Haq, W.; Mathur, K.B.; Agarwal, S.S. Delta-opioid receptor antagonist inhibits immunomodulation by Met-enkephalin analogs. Neuroimmunomodulation 1999, 6, 355–360. [Google Scholar] [CrossRef]
- Jiao, X.; Wang, X.; Wang, R.; Geng, J.; Liu, N.; Chen, H.; Griffin, N.; Shan, F. Rules to activate CD8(+)T cells through regulating subunits of opioid receptors by methionine enkephalin (MENK). Int. Immunopharmacol. 2018, 65, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Meng, Y.; Plotnikoff, N.P.; Youkilis, G.; Griffin, N.; Shan, F. Low dose naltrexone (LDN) enhances maturation of bone marrow dendritic cells (BMDCs). Int. Immunopharmacol. 2013, 17, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, T.K. The Role of Opioid Receptors in Immune System Function. Front. Immunol. 2019, 10, 2904. [Google Scholar] [CrossRef] [PubMed]
- Rahim, R.T.; Meissler, J.J., Jr.; Cowan, A.; Rogers, T.J.; Geller, E.B.; Gaughan, J.; Adler, M.W.; Eisenstein, T.K. Administration of mu-, kappa- or delta2-receptor agonists via osmotic minipumps suppresses murine splenic antibody responses. Int. Immunopharmacol. 2001, 1, 2001–2009. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Townsend, R.; Eisenstein, T.K.; Adler, M.W.; Rogers, T.J. Both T cells and macrophages are targets of kappa-opioid-induced immunosuppression. Brain Behav. Immun. 1994, 8, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Sharp, B.M.; McKean, D.J.; McAllen, K.; Shahabi, N.A. Signaling through delta opioid receptors on murine splenic T cells and stably transfected Jurkat cells. Ann. N. Y. Acad. Sci. 1998, 840, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Sharp, B.M.; Shahabi, N.A.; Heagy, W.; McAllen, K.; Bell, M.; Huntoon, C.; McKean, D.J. Dual signal transduction through delta opioid receptors in a transfected human T-cell line. Proc. Natl. Acad. Sci. USA 1996, 93, 8294–8299. [Google Scholar] [CrossRef]
- House, R.V.; Thomas, P.T.; Kozak, J.T.; Bhargava, H.N. Suppression of immune function by non-peptidic delta opioid receptor antagonists. Neurosci. Lett. 1995, 198, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, K.; Akami, T.; Okamoto, M.; Akioka, K.; Nakai, I.; Oka, T.; Nagase, H. Immunosuppression by delta opioid receptor antagonist. Transplant. Proc. 1993, 25, 738–740. [Google Scholar]
- Miller, B. delta opioid receptor expression is induced by concanavalin A in CD4+ T cells. J. Immunol. 1996, 157, 5324–5328. [Google Scholar] [CrossRef] [PubMed]
- Börner, C.; Stumm, R.; Höllt, V.; Kraus, J. Comparative analysis of mu-opioid receptor expression in immune and neuronal cells. J. Neuroimmunol. 2007, 188, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Balasubramanian, S.; Sumandeep, S.; Charboneau, R.; Wang, J.; Melnyk, D.; Beilman, G.J.; Vatassery, R.; Barke, R.A. Morphine directs T cells toward T(H2) differentiation. Surgery 2001, 130, 304–309. [Google Scholar] [CrossRef]
- Rogers, T.J.; Steele, A.D.; Howard, O.M.; Oppenheim, J.J. Bidirectional heterologous desensitization of opioid and chemokine receptors. Ann. N. Y. Acad. Sci. 2000, 917, 19–28. [Google Scholar] [CrossRef]
- Panocka, I.; Marek, P.; Sadowski, B. Inheritance of stress-induced analgesia in mice. Selective breeding study. Brain Res. 1986, 397, 152–155. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skiba, D.; Jaskuła, K.; Nawrocka, A.; Poznański, P.; Łazarczyk, M.; Szymański, Ł.; Żera, T.; Sacharczuk, M.; Cudnoch-Jędrzejewska, A.; Gaciong, Z. The Role of Opioid Receptor Antagonists in Regulation of Blood Pressure and T-Cell Activation in Mice Selected for High Analgesia Induced by Swim Stress. Int. J. Mol. Sci. 2024, 25, 2618. https://doi.org/10.3390/ijms25052618
Skiba D, Jaskuła K, Nawrocka A, Poznański P, Łazarczyk M, Szymański Ł, Żera T, Sacharczuk M, Cudnoch-Jędrzejewska A, Gaciong Z. The Role of Opioid Receptor Antagonists in Regulation of Blood Pressure and T-Cell Activation in Mice Selected for High Analgesia Induced by Swim Stress. International Journal of Molecular Sciences. 2024; 25(5):2618. https://doi.org/10.3390/ijms25052618
Chicago/Turabian StyleSkiba, Dominik, Kinga Jaskuła, Agata Nawrocka, Piotr Poznański, Marzena Łazarczyk, Łukasz Szymański, Tymoteusz Żera, Mariusz Sacharczuk, Agnieszka Cudnoch-Jędrzejewska, and Zbigniew Gaciong. 2024. "The Role of Opioid Receptor Antagonists in Regulation of Blood Pressure and T-Cell Activation in Mice Selected for High Analgesia Induced by Swim Stress" International Journal of Molecular Sciences 25, no. 5: 2618. https://doi.org/10.3390/ijms25052618
APA StyleSkiba, D., Jaskuła, K., Nawrocka, A., Poznański, P., Łazarczyk, M., Szymański, Ł., Żera, T., Sacharczuk, M., Cudnoch-Jędrzejewska, A., & Gaciong, Z. (2024). The Role of Opioid Receptor Antagonists in Regulation of Blood Pressure and T-Cell Activation in Mice Selected for High Analgesia Induced by Swim Stress. International Journal of Molecular Sciences, 25(5), 2618. https://doi.org/10.3390/ijms25052618