
Transcriptome Analysis of Compensatory Growth and Meat Quality Alteration after Varied Restricted Feeding Conditions in Beef Cattle

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Animal Performance of Growth and Meat
2.2. Evaluation of RNA-Seq Data
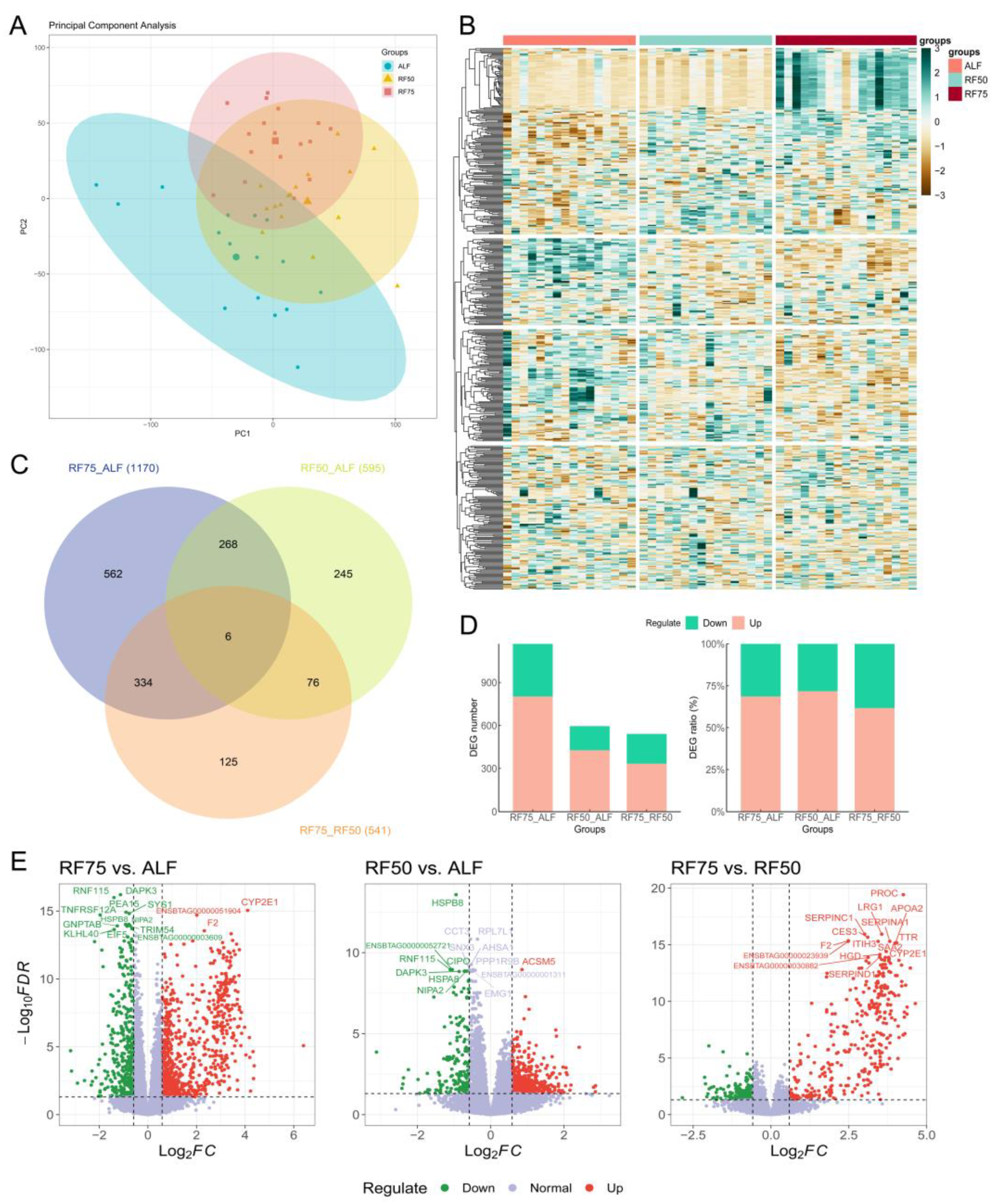
2.3. Identification of DEGs in Longissimus Dorsi Muscle
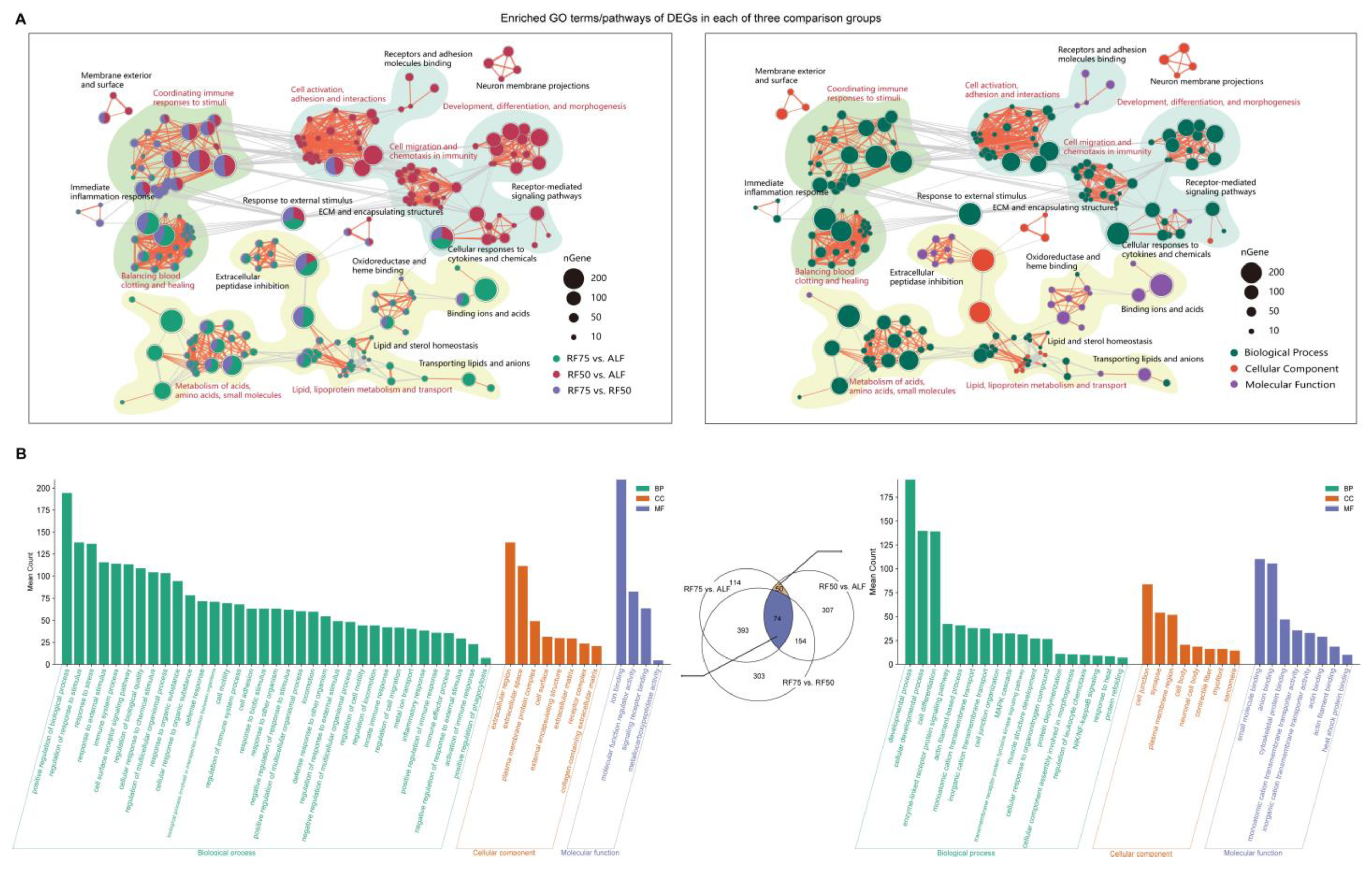
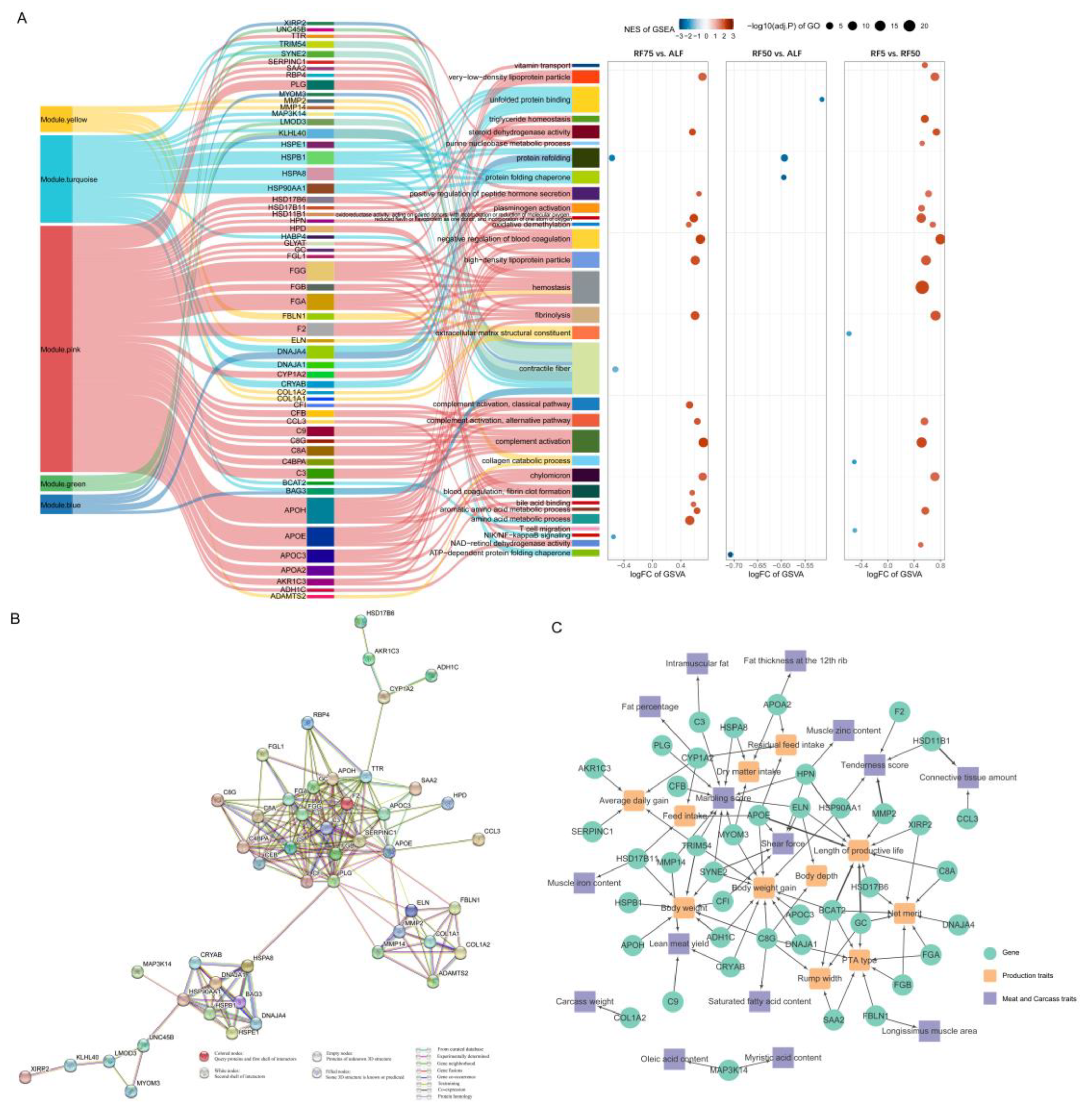
2.4. Functional Enrichment Analysis of DEGs
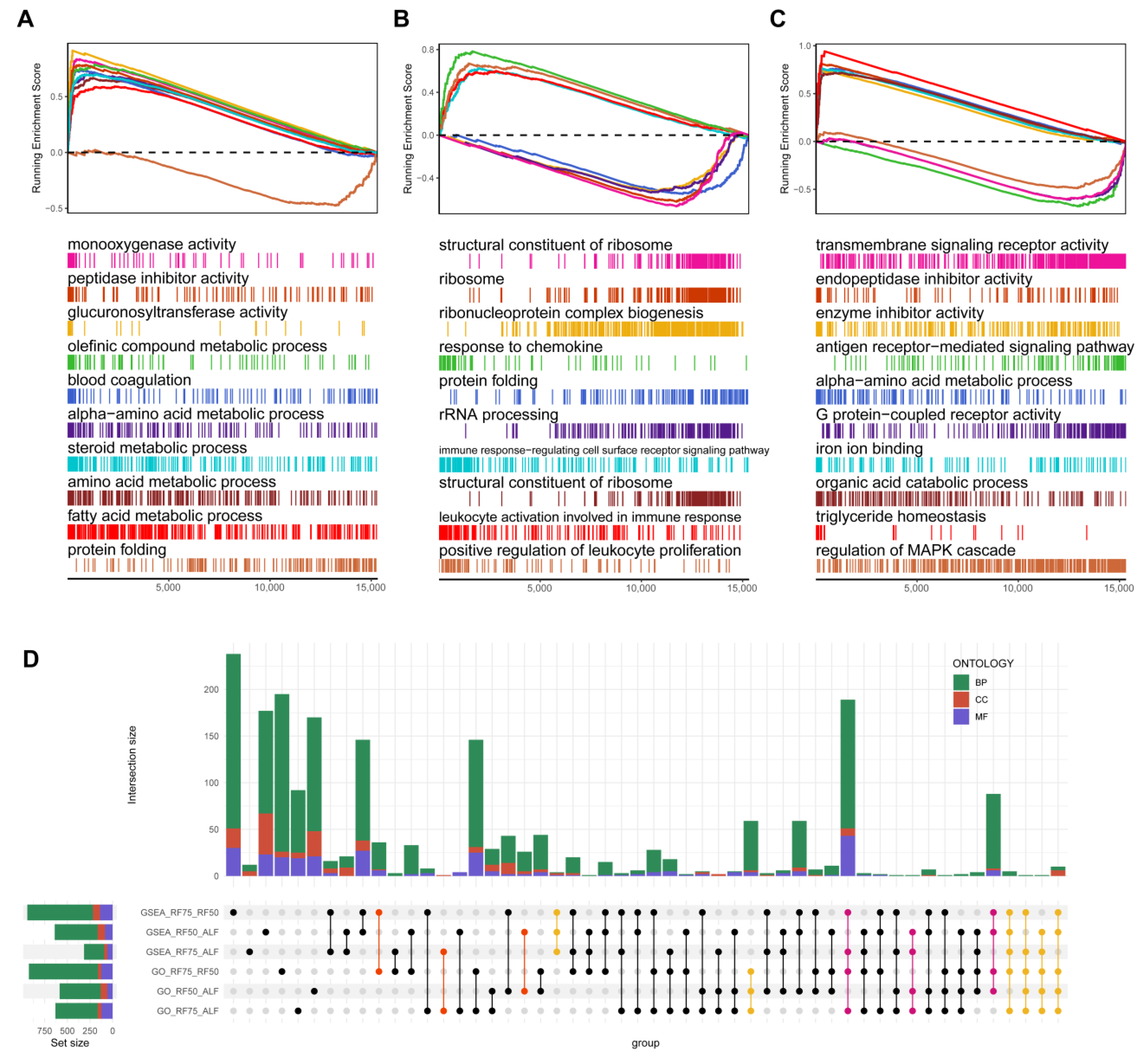
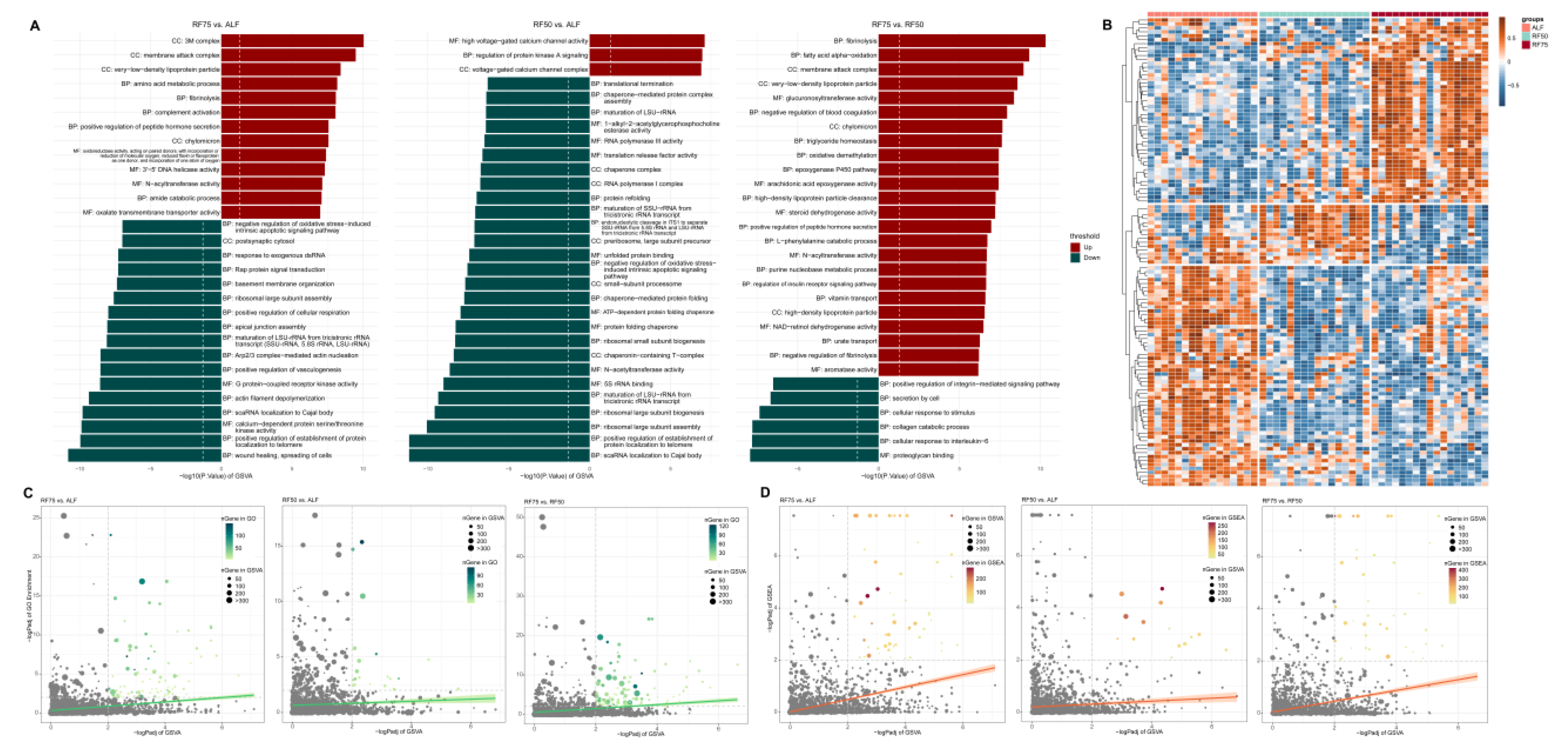
2.5. Gene Set Enrichment Analysis (GSEA) and Gene Set Variation Analysis (GSVA)
2.6. Weighted Gene Co-Expression Network Analysis
2.7. Integrated Analysis to Identify Key Genes
3. Discussion
4. Materials and Methods
4.1. Animal and Experiment Design
4.2. Meta Quality and Composition
4.2.1. pH
4.2.2. Shear Force
4.2.3. Water Holding Capacity
4.2.4. Cooking Loss
4.2.5. Protein Content
4.2.6. Fat
4.2.7. Moisture
4.3. RNA Extraction and Sequencing
4.4. Transcriptomic Data Analysis
4.5. Developing the GO Database
4.6. Gene Ontology Enrichment Analysis of DEGs
4.7. Gene Set Enrichment Analysis
4.8. Gene Set Variation Analysis
4.9. Co-Expression Analysis Based on WGCNA
4.10. Screening of Key Genes and PPI Network
4.11. Statistical Analyses and Graphs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hornick, J.L.; Van Eenaeme, C.; Gérard, O.; Dufrasne, I.; Istasse, L. Mechanisms of reduced and compensatory growth. Domest. Anim. Endocrinol. 2000, 19, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Drouillard, J.S.; Ferrell, C.L.; Klopfenstein, T.J.; Britton, R.A. Compensatory growth following metabolizable protein or energy restrictions in beef steers. J. Anim. Sci. 1991, 69, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Keogh, K.; Waters, S.M.; Kelly, A.K.; Kenny, D.A. Feed restriction and subsequent realimentation in Holstein Friesian bulls: I. Effect on animal performance; muscle, fat, and linear body measurements; and slaughter characteristics. J. Anim. Sci. 2015, 93, 3578–3589. [Google Scholar] [CrossRef] [PubMed]
- Keady, S.M.; Waters, S.M.; Hamill, R.M.; Dunne, P.G.; Keane, M.G.; Richardson, R.I.; Kenny, D.A.; Moloney, A.P. Compensatory growth in crossbred Aberdeen Angus and Belgian Blue steers: Effects on the colour, shear force and sensory characteristics of longissimus muscle. Meat Sci. 2017, 125, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.; Therkildsen, M.; Byrne, D.V. Effects of a compensatory growth strategy on sensory and physical properties of meat from young bulls. Meat Sci. 2006, 74, 628–643. [Google Scholar] [CrossRef]
- Moloney, A.P.; Keane, M.G.; Mooney, M.T.; Rezek, K.; Smulders, F.J.; Troy, D.J. Energy supply patterns for finishing steers: Feed conversion efficiency, components of bodyweight gain and meat quality. Meat Sci. 2008, 79, 86–97. [Google Scholar] [CrossRef]
- Andersen, H.J.; Oksbjerg, N.; Young, J.F.; Therkildsen, M. Feeding and meat quality—A future approach. Meat Sci. 2005, 70, 543–554. [Google Scholar] [CrossRef]
- Coleman, S.W.; Evans, B.C. Effect of nutrition, age and size on compensatory growth in two breeds of steers. J. Anim. Sci. 1986, 63, 1968–1982. [Google Scholar] [CrossRef] [PubMed]
- Miszura, A.A.; Ferraz, M.V.C.; Cardoso, R.C.; Polizel, D.M.; Oliveira, G.B.; Barroso, J.P.R.; Gobato, L.G.M.; Nogueira, G.P.; Biava, J.S.; Ferreira, E.M.; et al. Implications of growth rates and compensatory growth on puberty attainment in Nellore heifers. Domest. Anim. Endocrinol. 2021, 74, 106526. [Google Scholar] [CrossRef]
- Menegat, M.B.; Dritz, S.S.; Tokach, M.D.; Woodworth, J.C.; DeRouchey, J.M.; Goodband, R.D. Phase-feeding strategies based on lysine specifications for grow-finish pigs1. J. Anim. Sci. 2020, 98, skz366. [Google Scholar] [CrossRef]
- Keogh, K.; Kenny, D.A.; Waters, S.M. Gene co-expression networks contributing to the expression of compensatory growth in metabolically active tissues in cattle. Sci. Rep. 2019, 9, 6093. [Google Scholar] [CrossRef]
- Keogh, K.; Kenny, D.A.; Cormican, P.; Kelly, A.K.; Waters, S.M. Effect of dietary restriction and subsequent re-alimentation on the transcriptional profile of hepatic tissue in cattle. BMC Genom. 2016, 17, 244. [Google Scholar] [CrossRef]
- Keogh, K.; Kenny, D.A.; Cormican, P.; McCabe, M.S.; Kelly, A.K.; Waters, S.M. Effect of Dietary Restriction and Subsequent Re-Alimentation on the Transcriptional Profile of Bovine Skeletal Muscle. PLoS ONE 2016, 11, e0149373. [Google Scholar] [CrossRef]
- Ali, M.; Nicieza, A.; Wootton, R.J. Compensatory growth in fishes: A response to growth depression. Fish Fish. 2003, 4, 147–190. [Google Scholar] [CrossRef]
- Whitaker, K.W.; Totoki, K.; Reyes, T.M. Metabolic adaptations to early life protein restriction differ by offspring sex and post-weaning diet in the mouse. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 1067–1074. [Google Scholar] [CrossRef]
- Stolzenbach, S.; Therkildsen, M.; Oksbjerg, N.; Lazarotti, R.; Ertbjerg, P.; Lametsch, R.; Byrne, D.V. Compensatory growth response as a strategy to enhance tenderness in entire male and female pork M. longissimus thoracis. Meat Sci. 2009, 81, 163–170. [Google Scholar] [CrossRef]
- Winchester, C.F.; Ellis, N.R. Delayed Growth of Beef Cattle; US Department of Agriculture: Washington, DC, USA, 1957. [Google Scholar]
- Levy, D.; Folman, Y.; Holzer, Z.; Drori, D. Compensatory Growth in Intensively Raised Bull Calves. J. Anim. Sci. 1971, 33, 1078–1085. [Google Scholar] [CrossRef]
- Therkildsen, M. Muscle protein degradation in bull calves with compensatory growth. Livest. Prod. Sci. 2005, 98, 205–218. [Google Scholar] [CrossRef]
- Jones, S.J.; Starkey, D.L.; Calkins, C.R.; Crouse, J.D. Myofibrillar protein turnover in feed-restricted and realimented beef cattle. J. Anim. Sci. 1990, 68, 2707–2715. [Google Scholar] [CrossRef] [PubMed]
- Hornick, J.L.; Van Eenaeme, C.; Clinquart, A.; Diez, M.; Istasse, L. Different periods of feed restriction before compensatory growth in Belgian Blue bulls: I. animal performance, nitrogen balance, meat characteristics, and fat composition. J. Anim. Sci. 1998, 76, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Purslow, P.P. New developments on the role of intramuscular connective tissue in meat toughness. Annu. Rev. Food Sci. Technol. 2014, 5, 133–153. [Google Scholar] [CrossRef]
- Velázquez, D.E.; Latorre, M.E. Physicochemical, thermal and mechanical characterization study of perimysial collagen of two bovine muscles. Int. J. Biol. Macromol. 2019, 136, 404–409. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Rahmatallah, Y.; Emmert-Streib, F.; Glazko, G. Gene set analysis approaches for RNA-seq data: Performance evaluation and application guideline. Brief. Bioinform. 2016, 17, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Goeman, J.J.; Bühlmann, P. Analyzing gene expression data in terms of gene sets: Methodological issues. Bioinformatics 2007, 23, 980–987. [Google Scholar] [CrossRef]
- Chen, J.W.; Shrestha, L.; Green, G.; Leier, A.; Marquez-Lago, T.T. The hitchhikers’ guide to RNA sequencing and functional analysis. Brief. Bioinform. 2023, 24, bbac529. [Google Scholar] [CrossRef] [PubMed]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Hu, C.; Yang, J.; Qi, Z.; Wu, H.; Wang, B.; Zou, F.; Mei, H.; Liu, J.; Wang, W.; Liu, Q. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. MedComm 2022, 3, e161. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Carstens, G.E.; Johnson, D.E.; Ellenberger, M.A.; Tatum, J.D. Physical and chemical components of the empty body during compensatory growth in beef steers. J. Anim. Sci. 1991, 69, 3251–3264. [Google Scholar] [CrossRef]
- Connor, E.E.; Kahl, S.; Elsasser, T.H.; Parker, J.S.; Li, R.W.; Van Tassell, C.P.; Baldwin, R.L.t.; Barao, S.M. Enhanced mitochondrial complex gene function and reduced liver size may mediate improved feed efficiency of beef cattle during compensatory growth. Funct. Integr. Genom. 2010, 10, 39–51. [Google Scholar] [CrossRef]
- Wang, Y.; Tian, X.; Liu, X.; Xing, J.; Guo, C.; Du, Y.; Zhang, H.; Wang, W. Focusing on intramuscular connective tissue: Effect of cooking time and temperature on physical, textual, and structural properties of yak meat. Meat Sci. 2022, 184, 108690. [Google Scholar] [CrossRef]
- Roy, B.C.; Sedgewick, G.; Aalhus, J.L.; Basarab, J.A.; Bruce, H.L. Modification of mature non-reducible collagen cross-link concentrations in bovine m. gluteus medius and semitendinosus with steer age at slaughter, breed cross and growth promotants. Meat Sci. 2015, 110, 109–117. [Google Scholar] [CrossRef]
- Lindholm-Perry, A.K.; Kern, R.J.; Keel, B.N.; Snelling, W.M.; Kuehn, L.A.; Freetly, H.C. Profile of the Spleen Transcriptome in Beef Steers with Variation in Gain and Feed Intake. Front. Genet. 2016, 7, 127. [Google Scholar] [CrossRef]
- Cunningham-Rundles, S.; McNeeley, D.F.; Moon, A. Mechanisms of nutrient modulation of the immune response. J. Allergy Clin. Immunol. 2005, 115, 1119–1128; quiz 1129. [Google Scholar] [CrossRef]
- Coss, S.L.; Zhou, D.; Chua, G.T.; Aziz, R.A.; Hoffman, R.P.; Wu, Y.L.; Ardoin, S.P.; Atkinson, J.P.; Yu, C.Y. The complement system and human autoimmune diseases. J. Autoimmun. 2023, 137, 102979. [Google Scholar] [CrossRef] [PubMed]
- Phieler, J.; Garcia-Martin, R.; Lambris, J.D.; Chavakis, T. The role of the complement system in metabolic organs and metabolic diseases. Semin. Immunol. 2013, 25, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Iyer, A.; Suen, J.Y.; Seow, V.; Reid, R.C.; Brown, L.; Fairlie, D.P. C5aR and C3aR antagonists each inhibit diet-induced obesity, metabolic dysfunction, and adipocyte and macrophage signaling. FASEB J. 2013, 27, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Scantlebury, T.; Maslowska, M.; Cianflone, K. Chylomicron-specific enhancement of acylation stimulating protein and precursor protein C3 production in differentiated human adipocytes. J. Biol. Chem. 1998, 273, 20903–20909. [Google Scholar] [CrossRef]
- Paglialunga, S.; Fisette, A.; Yan, Y.; Deshaies, Y.; Brouillette, J.F.; Pekna, M.; Cianflone, K. Acylation-stimulating protein deficiency and altered adipose tissue in alternative complement pathway knockout mice. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E521–E529. [Google Scholar] [CrossRef]
- Sekine, H.; Machida, T.; Fujita, T. Factor D. Immunol. Rev. 2023, 313, 15–24. [Google Scholar] [CrossRef]
- Moreno-Navarrete, J.M.; Fernández-Real, J.M. The complement system is dysfunctional in metabolic disease: Evidences in plasma and adipose tissue from obese and insulin resistant subjects. Semin. Cell Dev. Biol. 2019, 85, 164–172. [Google Scholar] [CrossRef]
- Ohinata, K.; Yoshikawa, M. Food intake regulation by central complement system. Adv. Exp. Med. Biol. 2008, 632, 35–46. [Google Scholar]
- Keragala, C.B.; Draxler, D.F.; McQuilten, Z.K.; Medcalf, R.L. Haemostasis and innate immunity—A complementary relationship: A review of the intricate relationship between coagulation and complement pathways. Br. J. Haematol. 2018, 180, 782–798. [Google Scholar] [CrossRef]
- Chana-Muñoz, A.; Jendroszek, A.; Sønnichsen, M.; Wang, T.; Ploug, M.; Jensen, J.K.; Andreasen, P.A.; Bendixen, C.; Panitz, F. Origin and diversification of the plasminogen activation system among chordates. BMC Evol. Biol. 2019, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Mutch, N.J.; Medcalf, R.L. The fibrinolysis renaissance. J. Thromb. Haemost. 2023, 21, 3304–3316. [Google Scholar] [CrossRef]
- Medcalf, R.L.; Keragala, C.B. Fibrinolysis: A Primordial System Linked to the Immune Response. Int. J. Mol. Sci. 2021, 22, 3406. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.H. Plasmin(ogen) at the Nexus of Fibrinolysis, Inflammation, and Complement. Semin. Thromb. Hemost. 2017, 43, 135–142. [Google Scholar] [CrossRef]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Rebeck, G.W. The role of APOE on lipid homeostasis and inflammation in normal brains. J. Lipid Res. 2017, 58, 1493–1499. [Google Scholar] [CrossRef]
- Wang, M.; Yang, B.; Shao, P.; Jie, F.; Yang, X.; Lu, B. Sterols and Sterol Oxidation Products: Effect of Dietary Intake on Tissue Distribution in ApoE-Deficient Mice. J. Agric. Food Chem. 2021, 69, 11867–11877. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Tso, P.; Woods, S.C.; Clegg, D.J.; Barber, K.L.; Carey, K.; Liu, M. Brain apolipoprotein E: An important regulator of food intake in rats. Diabetes 2008, 57, 2092–2098. [Google Scholar] [CrossRef] [PubMed]
- Raz, A.; Goodman, D.S. The interaction of thyroxine with human plasma prealbumin and with the prealbumin-retinol-binding protein complex. J. Biol. Chem. 1969, 244, 3230–3237. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.; Saraiva, M.J. Transthyretin: A multifaceted protein. Biomol. Concepts 2014, 5, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Fleming, C.E.; Saraiva, M.J.; Sousa, M.M. Transthyretin enhances nerve regeneration. J. Neurochem. 2007, 103, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Li, M.; Sun, X.; Plath, M.; Li, C.; Lan, X.; Lei, C.; Huang, Y.; Bai, Y.; Qi, X.; et al. Global Transcriptome Analysis during Adipogenic Differentiation and Involvement of Transthyretin Gene in Adipogenesis in Cattle. Front. Genet. 2018, 9, 463. [Google Scholar] [CrossRef] [PubMed]
- Marchais-Oberwinkler, S.; Henn, C.; Möller, G.; Klein, T.; Negri, M.; Oster, A.; Spadaro, A.; Werth, R.; Wetzel, M.; Xu, K.; et al. 17β-Hydroxysteroid dehydrogenases (17β-HSDs) as therapeutic targets: Protein structures, functions, and recent progress in inhibitor development. J. Steroid Biochem. Mol. Biol. 2011, 125, 66–82. [Google Scholar] [CrossRef]
- Saloniemi, T.; Jokela, H.; Strauss, L.; Pakarinen, P.; Poutanen, M. The diversity of sex steroid action: Novel functions of hydroxysteroid (17β) dehydrogenases as revealed by genetically modified mouse models. J. Endocrinol. 2012, 212, 27–40. [Google Scholar] [CrossRef]
- Stomby, A.; Otten, J.; Ryberg, M.; Andrew, R.; Walker, B.R.; Olsson, T. Diet-induced weight loss alters hepatic glucocorticoid metabolism in type 2 diabetes mellitus. Eur. J. Endocrinol. 2020, 182, 447–457. [Google Scholar] [CrossRef]
- Wang, M.X.; Peng, Z.G. 17β-hydroxysteroid dehydrogenases in the progression of nonalcoholic fatty liver disease. Pharmacol. Ther. 2023, 246, 108428. [Google Scholar] [CrossRef]
- Ikeda, S.; Saito, H.; Fukatsu, K.; Inoue, T.; Han, I.; Furukawa, S.; Matsuda, T.; Hidemura, A. Dietary restriction impairs neutrophil exudation by reducing CD11b/CD18 expression and chemokine production. Arch. Surg. 2001, 136, 297–304. [Google Scholar] [CrossRef]
- Alwarawrah, Y.; Kiernan, K.; MacIver, N.J. Changes in Nutritional Status Impact Immune Cell Metabolism and Function. Front. Immunol. 2018, 9, 1055. [Google Scholar] [CrossRef]
- Stegen, M.; Frey, U.H. The Role of G Protein-Coupled Receptor Kinase 6 Regulation in Inflammation and Pain. Int. J. Mol. Sci. 2022, 23, 15880. [Google Scholar] [CrossRef]
- NY/T 815-2004; Feeding Standard of Beef Cattle. The Standardization Administration of the People’s Republic of China: Beijing, China, 2004.
- B/T 27643-2011; Beef Carcass and Cuts. The Standardization Administration of the People’s Republic of China: Beijing, China, 2011.
- GB 5009.5-2016; Determination of Protein in Food. The Standardization Administration of the People’s Republic of China: Beijing, China, 2016.
- GB 5009.6-2016; Determination of Fat in Foods. The Standardization Administration of the People’s Republic of China: Beijing, China, 2016.
- GB 5009.3-2016; Determination of Water Content in Foods Standards. The Standardization Administration of the People’s Republic of China: Beijing, China, 2016.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Kucera, M.; Isserlin, R.; Arkhangorodsky, A.; Bader, G.D. AutoAnnotate: A Cytoscape app for summarizing networks with semantic annotations. F1000Res 2016, 5, 1717. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Hu, Z.L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait/Variable | ALF | RF75 | RF50 | SED | PANOVA |
|---|---|---|---|---|---|
| Body weight (BW), kg | |||||
| Start, d0 | 375.76 | 379.41 | 375.74 | 42.95 | 0.991 |
| Mid of RF_d28 | 415.24 Aa1 | 391.43 a | 358.52 b | 55.16 | 0.001 |
| End of RF, d49 | 447.46 A | 401.43 B | 363.52 C | 50.46 | <0.001 |
| Realimentation_d135 | 538.33 Aa | 529.37 a | 495.07 Bb | 50.35 | 0.007 |
| Realimentation_d165 | 577.89 Aa | 572.32 a | 537.96 Bb | 50.06 | 0.01 |
| Slaughter, d380 | 767.18 | 774.05 | 759.9 | 59.00 | 0.751 |
| Average daily weight gain (ADG), kg/d | |||||
| RF_d0–28 | 1.52 A | 0.52 B | −0.61 C | 1.01 | <0.001 |
| RF_d28–49 | 1.4 A | 0.43 B | 0.22 B | 0.79 | <0.001 |
| RF, d0–49 | 1.46 A | 0.45 B | −0.25 B | 0.78 | <0.001 |
| Realimentation_d49_135 | 1.04 B | 1.47 A | 1.51 A | 0.31 | <0.001 |
| Realimentation_d135_165 | 1.32 | 1.46 | 1.43 | 0.45 | 0.552 |
| Realimentation_d165_380 | 0.82 | 0.93 | 0.96 | 0.28 | 0.196 |
| Realimentation, d49–380 | 0.93 B | 1.12 A | 1.15 A | 0.21 | <0.001 |
| Entire Period | 1.00 | 1.04 | 0.97 | 0.17 | 0.45 |
| Trait/Variable | ALF | RF75 | RF50 | SED | PANOVA |
|---|---|---|---|---|---|
| Carcass trait | |||||
| Carcass Weight, kg | 414.2 | 419.36 | 404.00 | 31.98 | 0.30 |
| Striploin 1, kg | 6.40 | 6.41 | 6.23 | 0.76 | 0.70 |
| High-rib, kg | 11.60 | 12.00 | 11.69 | 1.47 | 0.66 |
| Tenderloin, kg | 4.12 | 4.10 | 3.99 | 0.51 | 0.68 |
| EMA 3 | 104.6 | 108.71 | 106.25 | 11.16 | 0.5 |
| Meat Quality | |||||
| pH24 | 5.45 Aa2 | 5.80 Bb | 5.65 b | 0.29 | <0.001 |
| Shear Force | 9.21 | 9.01 | 9.19 | 2.70 | 0.966 |
| WHC | 0.38 | 0.36 | 0.37 | 0.04 | 0.208 |
| Cooking Loss | 0.3 b | 0.29 ab | 0.24 a | 0.06 | 0.029 |
| Meat Composition | |||||
| Protein, % | 23.55 a | 22.77 b | 23.26 ab | 0.78 | 0.014 |
| Fat, % | 2.98 a | 3.8 b | 3.34 ab | 0.93 | 0.042 |
| Moisture, % | 72.89 | 72.06 | 72.33 | 1.22 | 0.146 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, T.; Liang, M.; Du, L.; Li, K.; Li, J.; Qian, L.; Xue, Q.; Qiu, S.; Xu, L.; Zhang, L.; et al. Transcriptome Analysis of Compensatory Growth and Meat Quality Alteration after Varied Restricted Feeding Conditions in Beef Cattle. Int. J. Mol. Sci. 2024, 25, 2704. https://doi.org/10.3390/ijms25052704
Deng T, Liang M, Du L, Li K, Li J, Qian L, Xue Q, Qiu S, Xu L, Zhang L, et al. Transcriptome Analysis of Compensatory Growth and Meat Quality Alteration after Varied Restricted Feeding Conditions in Beef Cattle. International Journal of Molecular Sciences. 2024; 25(5):2704. https://doi.org/10.3390/ijms25052704
Chicago/Turabian StyleDeng, Tianyu, Mang Liang, Lili Du, Keanning Li, Jinnan Li, Li Qian, Qingqing Xue, Shiyuan Qiu, Lingyang Xu, Lupei Zhang, and et al. 2024. "Transcriptome Analysis of Compensatory Growth and Meat Quality Alteration after Varied Restricted Feeding Conditions in Beef Cattle" International Journal of Molecular Sciences 25, no. 5: 2704. https://doi.org/10.3390/ijms25052704