
Hemodynamic and Clinical Profiles of Pulmonary Arterial Hypertension Patients with GDF2 and BMPR2 Variants
 ,
,
Abstract
:1. Introduction
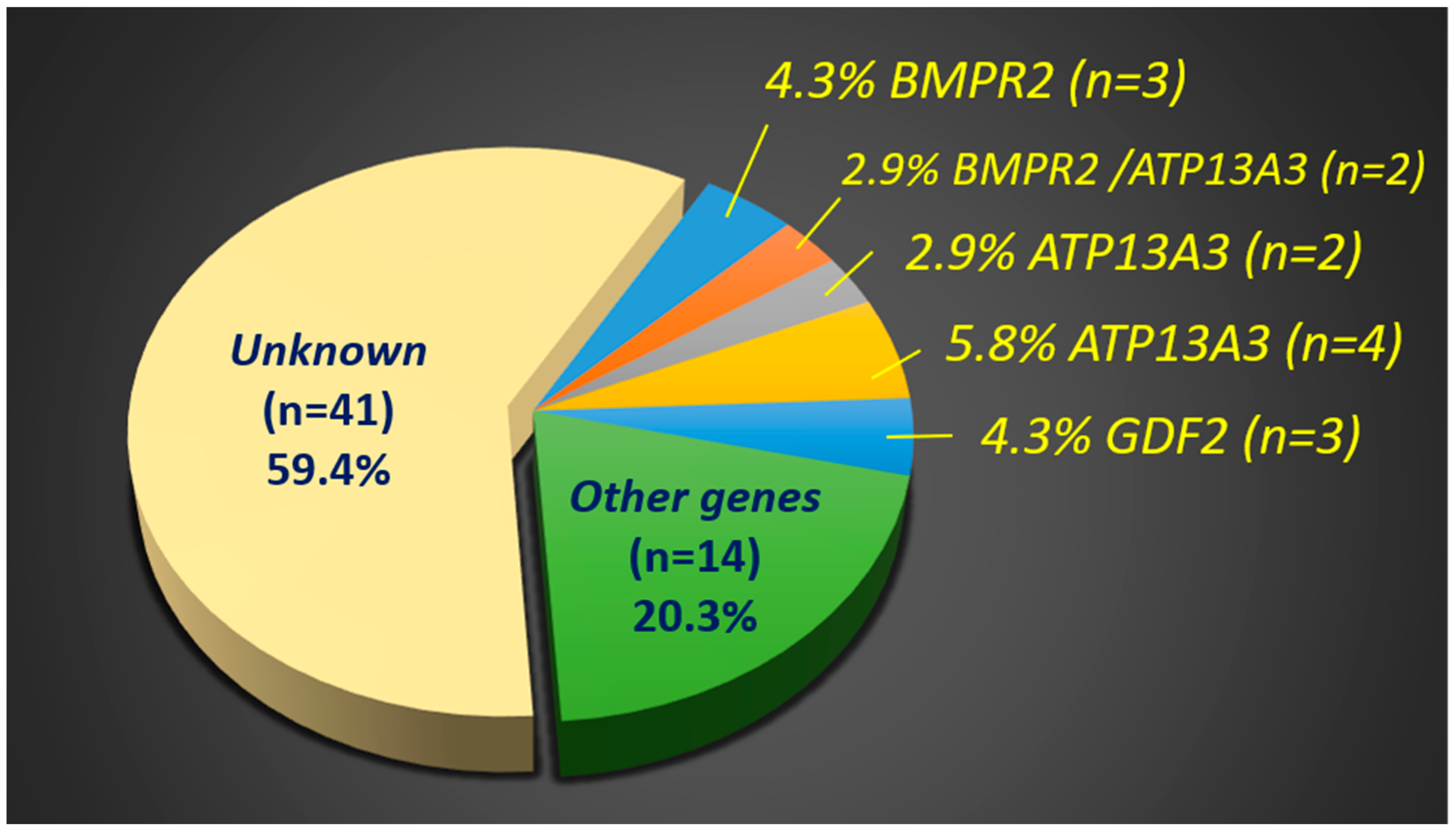
2. Results
3. Discussion
3.1. Clinical Manifestations of PAH Patients with BMPR2 Variants
3.2. Potential Mechanisms of the GDF2 Variant and PAH
3.3. Genetic Variants Other Than BMPR2 and GDF2 in PAH Patients
3.4. Limitations and Strengths
4. Materials and Methods
4.1. Serum GDF2 (BMP9) and BMP10 of PAH Patients
4.2. Hemodynamics and Cardiopulmonary Function Tests
4.3. DNA Extraction, Whole Exome Sequencing, and Data Analysis
4.4. Validation of Variants by Polymerase Chain Reaction and Sanger Sequencing
4.5. Generation of PAH-Specific iPS Cell Lines and Assessment of Function Analysis
4.6. Generation and Characterization of iPSC-Derived Patient-Specific Endothelial Progenitor Cells (EPCs) and Endothelial Cells (ECs)
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, C.Y.; Hung, C.C.; Chiang, C.H.; Tsa, Y.C.; Fu, Y.J.; Wang, C.L.; Tsai, F.T.; Tai, H.Y.; Lin, K.C.; Hung, W.T.; et al. Pulmonary arterial hypertension in the elderly population. J. Chin. Med. Assoc. 2022, 85, 18–23. [Google Scholar] [CrossRef]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.; Brida, M.; Carlsen, J.; Coats, A.J.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: Developed by the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Endorsed by the International Society for Heart and Lung Transplantation (ISHLT) and the European Reference Network on rare respiratory diseases (ERN-LUNG). Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar]
- Lau, E.M.; Tamura, Y.; McGoon, M.D.; Sitbon, O. The 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: A practical chronicle of progress. Eur. Respir. J. 2015, 46, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.T.; Charng, M.J.; Chi, P.L.; Cheng, C.C.; Hung, C.C.; Huang, W.C. Gene Mutation Annotation and Pedigree for Pulmonary Arterial Hypertension Patients in Han Chinese Patients. Glob. Heart 2021, 16, 70. [Google Scholar] [CrossRef]
- Jing, Z.-C.; Xu, X.-Q.; Han, Z.-Y.; Wu, Y.; Deng, K.-W.; Wang, H.; Wang, Z.-W.; Cheng, X.-S.; Xu, B.; Hu, S.-S.; et al. Registry and survival study in chinese patients with idiopathic and familial pulmonary arterial hypertension. Chest 2007, 132, 373–379. [Google Scholar] [CrossRef]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801899. [Google Scholar] [CrossRef]
- Evans, J.D.W.; Girerd, B.; Montani, D.; Wang, X.-J.; Galiè, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grünig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Guignabert, C.; Humbert, M. Targeting transforming growth factor-beta receptors in pulmonary hypertension. Eur. Respir. J. 2021, 57, 2002341. [Google Scholar] [CrossRef]
- Zhang, H.-S.; Liu, Q.; Piao, C.-M.; Zhu, Y.; Li, Q.-Q.; Du, J.; Gu, H. Genotypes and Phenotypes of Chinese Pediatric Patients With Idiopathic and Heritable Pulmonary Arterial Hypertension—A Single-Center Study. Can. J. Cardiol. 2019, 35, 1851–1856. [Google Scholar] [CrossRef]
- Wang, X.-J.; Lian, T.-Y.; Jiang, X.; Liu, S.-F.; Li, S.-Q.; Jiang, R.; Wu, W.-H.; Ye, J.; Cheng, C.-Y.; Du, Y.; et al. Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801609. [Google Scholar] [CrossRef]
- Jang, A.Y.; Kim, B.-G.; Kwon, S.; Seo, J.; Kim, H.K.; Chang, H.-J.; Chang, S.-A.; Cho, G.-Y.; Rhee, S.J.; Jung, H.O.; et al. Prevalence and clinical features of bone morphogenetic protein receptor type 2 mutation in Korean idiopathic pulmonary arterial hypertension patients: The PILGRIM explorative cohort. PLoS ONE 2020, 15, e0238698. [Google Scholar] [CrossRef]
- Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Lane, K.B.; Morgan, N.V.; Wheeler, L.; Phillips, J.A., 3rd; Newman, J.; Williams, D.; Galiè, N.; et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am. J. Hum. Genet. 2001, 68, 92–102. [Google Scholar] [CrossRef]
- Portillo, K.; Santos, S.; Madrigal, I.; Blanco, I.; Paré, C.; Borderías, L.; Peinado, V.I.; Roca, J.; Milà, M.; Barberà, J.A. Estudio del gen BMPR2 en pacientes con hipertensión arterial pulmonar [Study of the BMPR2 gene in patients with pulmonary arterial hypertension]. Arch. Bronconeumol. 2010, 46, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Gómez, J.; Reguero, J.R.; Alvarez, C.; Junquera, M.R.; Arango, A.; Morís, C.; Coto, E. A Semiconductor Chip-Based Next Generation Sequencing Procedure for the Main Pulmonary Hypertension Genes. Lung 2015, 193, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; Southgate, L.; Eichstaedt, C.A.; Aldred, M.A.; Austin, E.D.; Best, D.H.; Chung, W.K.; Benjamin, N.; Elliott, C.G.; Eyries, M.; et al. Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum. Mutat. 2015, 36, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Gamou, S.; Kataoka, M.; Aimi, Y.; Chiba, T.; Momose, Y.; Isobe, S.; Hirayama, T.; Yoshino, H.; Fukuda, K.; Satoh, T. Genetics in pulmonary arterial hypertension in a large homogeneous Japanese population. Clin. Genet. 2018, 94, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Yagi, H.; Matsuoka, R.; Akimoto, K.; Furutani, M.; Imamura, S.-I.; Uehara, R.; Nakayama, T.; Takao, A.; Nakazawa, M.; et al. Implications of mutations of activin receptor-like kinase 1 gene (ALK1) in addition to bone morphogenetic protein receptor II gene (BMPR2) in children with pulmonary arterial hypertension. Circ. J. 2008, 72, 127–133. [Google Scholar] [CrossRef]
- Chida, A.; Shintani, M.; Yagi, H.; Fujiwara, M.; Kojima, Y.; Sato, H.; Imamura, S.; Yokozawa, M.; Onodera, N.; Horigome, H.; et al. Outcomes of childhood pulmonary arterial hypertension in BMPR2 and ALK1 mutation carriers. Am. J. Cardiol. 2012, 110, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Gräf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef]
- Simonneau, G.; Galiè, N.; Rubin, L.J.; Langleben, D.; Seeger, W.; Domenighetti, G.; Gibbs, S.; Lebrec, D.; Speich, R.; Beghetti, M.; et al. Clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2004, 43, 5s–12s. [Google Scholar] [CrossRef]
- Pfarr, N.; Szamalek-Hoegel, J.; Fischer, C.; Hinderhofer, K.; Nagel, C.; Ehlken, N.; Tiede, H.; Olschewski, H.; Reichenberger, F.; Ghofrani, A.H.; et al. Hemodynamic and clinical onset in patients with hereditary pulmonary arterial hypertension and BMPR2 mutations. Respir. Res. 2011, 12, 99. [Google Scholar] [CrossRef]
- Sztrymf, B.; Coulet, F.; Girerd, B.; Yaici, A.; Jais, X.; Sitbon, O.; Montani, D.; Souza, R.; Simonneau, G.; Soubrier, F.; et al. Clinical outcomes of pulmonary arterial hypertension in carriers of BMPR2 mutation. Am. J. Respir. Crit. Care Med. 2008, 177, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Zhu, T.; Zhang, X.; Liu, Y.; Wang, Y.; Zhang, W. Gender differences in pulmonary arterial hypertension patients with BMPR2 mutation: A meta-analysis. Respir. Res. 2020, 21, 44. [Google Scholar] [CrossRef] [PubMed]
- Larkin, E.K.; Newman, J.H.; Austin, E.D.; Hemnes, A.R.; Wheeler, L.; Robbins, I.M.; West, J.D.; Phillips, J.A.; Hamid, R.; Loyd, J.E. Longitudinal Analysis Casts Doubt on the Presence of Genetic Anticipation in Heritable Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Cogan, J.D.; West, J.D.; Hedges, L.K.; Hamid, R.; Dawson, E.P.; Wheeler, L.A.; Parl, F.F.; Loyd, J.E.; Phillips, J.A. Alterations in oestrogen metabolism: Implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur. Respir. J. 2009, 34, 1093. [Google Scholar] [CrossRef]
- Yang, H.; Zeng, Q.; Ma, Y.; Liu, B.; Chen, Q.; Li, W.; Xiong, C.; Zhou, Z. Genetic analyses in a cohort of 191 pulmonary arterial hypertension patients. Respir. Res. 2018, 19, 87. [Google Scholar] [CrossRef]
- Wang, G.; Fan, R.; Ji, R.; Zou, W.; Penny, D.J.; Varghese, N.P.; Fan, Y. Novel homozygous BMP9 nonsense mutation causes pulmonary arterial hypertension: A case report. BMC Pulm. Med. 2016, 16, 17. [Google Scholar] [CrossRef]
- Gallego, N.; Cruz-Utrilla, A.; Guillén, I.; Bonora, A.M.; Ochoa, N.; Arias, P.; Lapunzina, P.; Escribano-Subias, P.; Nevado, J.; Tenorio-Castaño, J. Expanding the Evidence of a Semi-Dominant Inheritance in GDF2 Associated with Pulmonary Arterial Hypertension. Cells 2021, 10, 3178. [Google Scholar] [CrossRef]
- Gelinas, S.M.; Benson, C.E.; Khan, M.A.; Berger, R.M.F.; Trembath, R.C.; Machado, R.D.; Southgate, L. Whole Exome Sequence Analysis Provides Novel Insights into the Genetic Framework of Childhood-Onset Pulmonary Arterial Hypertension. Genes 2020, 11, 1328. [Google Scholar] [CrossRef]
- Welch, C.L.; Aldred, M.A.; Balachandar, S.; Dooijes, D.; Eichstaedt, C.A.; Gräf, S.; Houweling, A.C.; Machado, R.D.; Pandya, D.; Prapa, M.; et al. Defining the clinical validity of genes reported to cause pulmonary arterial hypertension. Genet. Med. 2023, 25, 100925. [Google Scholar] [CrossRef]
- Harrison, R.; Flanagan, J.A.; Sankelo, M.; Abdalla, S.A.; Rowell, J.; Machado, R.D.; Elliott, C.G.; Robbins, I.M.; Olschewski, H.; McLaughlin, V.; et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J. Med. Genet. 2003, 40, 865–871. [Google Scholar] [CrossRef]
- Gallione, C.J.; Klaus, D.J.; Yeh, E.Y.; Stenzel, T.T.; Xue, Y.; Anthony, K.B.; McAllister, K.A.; Baldwin, M.A.; Berg, J.N.; Lux, A.; et al. Mutation and expression analysis of the endoglin gene in hereditary hemorrhagic telangiectasia reveals null alleles. Hum. Mutat. 1998, 11, 286–294. [Google Scholar] [CrossRef]
- Morty, R.E.; Nejman, B.; Kwapiszewska, G.; Hecker, M.; Zakrzewicz, A.; Kouri, F.M.; Peters, D.M.; Dumitrascu, R.; Seeger, W.; Knaus, P.; et al. Dysregulated Bone Morphogenetic Protein Signaling in Monocrotaline-Induced Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1072–1078. [Google Scholar] [CrossRef]
- Humbert, M.; Mclaughlin, V.; Gibbs, S.; Gomberg-Maitland, M.; Hoeper, M.; Preston, I.; Souza, R.; Waxman, A.; Ghofrani, H.; Subias, P.E.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef]
- David, L.; Mallet, C.; Mazerbourg, S.; Feige, J.-J.; Bailly, S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007, 109, 1953–1961. [Google Scholar] [CrossRef]
- Theilmann, A.L.; Hawke, L.G.; Hilton, L.R.; Whitford, M.K.; Cole, D.V.; Mackeil, J.L.; Dunham-Snary, K.J.; Mewburn, J.; James, P.D.; Maurice, D.H.; et al. Endothelial BMPR2 Loss Drives a Proliferative Response to BMP (Bone Morphogenetic Protein) 9 via Prolonged Canonical Signaling. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2605–2618. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Shao, N.Y.; Sa, S.; Li, D.; Termglinchan, V.; Ameen, M.; Karakikes, I.; Sosa, G.; Grubert, F.; Lee, J.; et al. Patient-Specific iPSC-Derived Endothelial Cells Uncover Pathways that Protect against Pulmonary Hypertension in BMPR2 Mutation Carriers. Cell Stem Cell 2017, 20, 490.e5–504.e5. [Google Scholar] [CrossRef] [PubMed]
- Desroches-Castan, A.; Tillet, E.; Ricard, N.; Ouarné, M.; Mallet, C.; Feige, J.J.; Bailly, S. Differential Consequences of Bmp9 Deletion on Sinusoidal Endothelial Cell Differentiation and Liver Fibrosis in 129/Ola and C57BL/6 Mice. Cells 2019, 8, 1079. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.; Ruiz-Llorente, L.; McDonald, J.; Quarrell, O.; Ugonna, K.; Bentham, J.; Mason, R.; Martin, J.; Moore, D.; Bergstrom, K.; et al. Homozygous GDF2 nonsense mutations result in a loss of circulating BMP9 and BMP10 and are associated with either PAH or an “HHT-like” syndrome in children. Mol. Genet. Genomic. Med. 2021, 9, e1685. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, J.; Tang, X.; Li, H.; Shen, Y.; Gu, W.; Zhao, S. Homozygous GDF2-Related Hereditary Hemorrhagic Telangiectasia in a Chinese Family. Pediatrics 2020, 146, e20191970. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.E.-A.; Moyou-Somo, R.; Oyono, J.L.E.; Kamgang, R. Identifying Potential Mutations Responsible for Cases of Pulmonary Arterial Hypertension. Appl. Clin. Genet. 2021, 14, 113–124. [Google Scholar] [CrossRef]
- Hodgson, J.; Swietlik, E.M.; Salmon, R.M.; Hadinnapola, C.; Nikolic, I.; Wharton, J.; Guo, J.; Liley, J.; Haimel, M.; Bleda, M.; et al. Characterization of GDF2 Mutations and Levels of BMP9 and BMP10 in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2020, 201, 575–585. [Google Scholar] [CrossRef]
- Morris, H.E.; Neves, K.B.; Montezano, A.C.; MacLean, M.R.; Touyz, R.M. Notch3 signalling and vascular remodelling in pulmonary arterial hypertension. Clin. Sci. 2019, 133, 2481–2498. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Leathers, R.; Makino, A.; Huang, C.; Parsa, P.; Macias, J.; Yuan, J.X.; Jamieson, S.W.; Thistlethwaite, P.A. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat. Med. 2009, 15, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Padhye, A.A.; Sahay, S. An adult case of NOTCH3 mutation in pulmonary artery hypertension. Pulm. Circ. 2022, 12, e12050. [Google Scholar] [CrossRef]
- Chida, A.; Shintani, M.; Matsushita, Y.; Sato, H.; Eitoku, T.; Nakayama, T.; Furutani, Y.; Hayama, E.; Kawamura, Y.; Inai, K.; et al. Mutations of NOTCH3 in childhood pulmonary arterial hypertension. Mol. Genet. Genom. Med. 2014, 2, 229–239. [Google Scholar] [CrossRef]
- Southgate, L.; Machado, R.D.; Gräf, S.; Morrell, N.W. Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 2020, 17, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; Welch, C.L.; Haimel, M.; Bleda, M.; Colglazier, E.; Coulson, J.D.; Debeljak, M.; Ekstein, J.; Fineman, J.R.; Golden, W.C.; et al. Biallelic variants of ATP13A3 cause dose-dependent childhood-onset pulmonary arterial hypertension characterised by extreme morbidity and mortality. J. Med. Genet. 2022, 59, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Eichstaedt, C.A.; Song, J.; Benjamin, N.; Harutyunova, S.; Fischer, C.; Grünig, E.; Hinderhofer, K. EIF2AK4 mutation as “second hit” in hereditary pulmonary arterial hypertension. Respir. Res. 2016, 17, 141. [Google Scholar] [CrossRef]
- Wang, X.-J.; Xu, X.-Q.; Sun, K.; Liu, K.-Q.; Li, S.-Q.; Jiang, X.; Zhao, Q.-H.; Wang, L.; Peng, F.-H.; Ye, J.; et al. Association of Rare PTGIS Variants with Susceptibility and Pulmonary Vascular Response in Patients with Idiopathic Pulmonary Arterial Hypertension. JAMA Cardiol. 2020, 5, 677–684. [Google Scholar] [CrossRef]
- Liang, K.-W.; Chang, S.-K.; Chen, Y.-W.; Lin, W.-W.; Tsai, W.-J.; Wang, K.-Y. Whole Exome Sequencing of Patients With Heritable and Idiopathic Pulmonary Arterial Hypertension in Central Taiwan. Front. Cardiovasc. Med. 2022, 9, 911649. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.-C.A.; Chen, C.-Y.; Chen, T.-T.; Kuo, P.-H.; Hsu, Y.-H.; Yang, H.-I.; Chen, W.J.; Su, M.-W.; Chu, H.-W.; Shen, C.-Y.; et al. Taiwan Biobank: A rich biomedical research database of the Taiwanese population. Cell Genom. 2022, 2, 100197. [Google Scholar] [CrossRef] [PubMed]
- Lewicka, E.; Dabrowska-Kugacka, A.; Chmara, M.; Wasąg, B. Genetics and genetic testing in pulmonary arterial hypertension (RCD code: II-1A.2). J. Rare Cardiovasc. Dis. 2018, 3, 226. [Google Scholar] [CrossRef]
- Kunig, A.M.; Parker, T.A.; Nogee, L.M.; Abman, S.H.; Kinsella, J.P. ABCA3 deficiency presenting as persistent pulmonary hypertension of the newborn. J. Pediatr. 2007, 151, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhu, G.; Jiang, D.; Rhen, J.; Li, X.; Liu, H.; Lyu, Y.; Tsai, P.; Rose, Y.; Nguyen, T.; et al. Reduced Notch1 Cleavage Promotes the Development of Pulmonary Hypertension. Hypertension 2022, 79, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gonzalez, I.; Tenorio-Castano, J.; Ochoa-Parra, N.; Gallego, N.; Pérez-Olivares, C.; Lago-Docampo, M.; Doza, J.P.; Valverde, D.; Lapunzina, P.; Escribano-Subias, P. Novel Genetic and Molecular Pathways in Pulmonary Arterial Hypertension Associated with Connective Tissue Disease. Cells. 2021, 10, 1488. [Google Scholar] [CrossRef]
- Hurst, L.A.; Dunmore, B.J.; Long, L.; Crosby, A.; Al-Lamki, R.; Deighton, J.; Southwood, M.; Yang, X.; Nikolic, M.Z.; Herrera, B.; et al. TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun. 2017, 8, 14079. [Google Scholar] [CrossRef]
- Sahoo, S.; Li, Y.; de Jesus, D.; Sembrat, J.; Rojas, M.M.; Goncharova, E.; Cifuentes-Pagano, E.; Straub, A.C.; Pagano, P.J. Notch2 suppression mimicking changes in human pulmonary hypertension modulates Notch1 and promotes endothelial cell proliferation. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H542–H557. [Google Scholar] [CrossRef]
- Gómez, J.; Reguero, J.R.; Junquera, M.R.; Alvarez, C.; Morís, C.; Alonso, B.; Iglesias, S.; Coto, E. Next generation sequencing of the NOTCH3 gene in a cohort of pulmonary hypertension patients. Int. J. Cardiol. 2016, 209, 149–150. [Google Scholar] [CrossRef]
- Lago-Docampo, M.; Tenorio, J.; Hernández-González, I.; Pérez-Olivares, C.; Escribano-Subías, P.; Pousada, G.; Baloira, A.; Arenas, M.; Lapunzina, P.; Valverde, D. Characterization of rare ABCC8 variants identified in Spanish pulmonary arterial hypertension patients. Sci. Rep. 2020, 10, 15135. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, M.S.; Ma, L.; Zhu, N.; Qi, H.; McClenaghan, C.; Gonzaga-Jauregui, C.; Dewey, F.E.; Overton, J.D.; Reid, J.G.; Shuldiner, A.R.; et al. Loss-of-Function ABCC8 Mutations in Pulmonary Arterial Hypertension. Circ. Genom. Precis. Med. 2018, 11, e002087. [Google Scholar] [CrossRef] [PubMed]
- Eyries, M.; Montani, D.; Nadaud, S.; Girerd, B.; Levy, M.; Bourdin, A.; Trésorier, R.; Chaouat, A.; Cottin, V.; Sanfiorenzo, C.; et al. Widening the landscape of heritable pulmonary hypertension mutations in paediatric and adult cases. Eur. Respir. J. 2019, 53, 1801371. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Swietlik, E.M.; Welch, C.L.; Pauciulo, M.W.; Hagen, J.J.; Zhou, X.; Guo, Y.; Karten, J.; Pandya, D.; Tilly, T.; et al. Rare variant analysis of 4241 pulmonary arterial hypertension cases from an international consortium implicates FBLN2, PDGFD, and rare de novo variants in PAH. Genome Med. 2021, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Gong, D.; Wang, W. Soluble JAGGED1 inhibits pulmonary hypertension by attenuating notch signaling. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2733–2739. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Pauciulo, M.W.; Welch, C.L.; Lutz, K.A.; Coleman, A.W.; Gonzaga-Jauregui, C.; Wang, J.; Grimes, J.M.; Martin, L.J.; He, H. Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension. Genome Med. 2019, 11, 69. [Google Scholar] [CrossRef]
- Nasim, T.; Ogo, T.; Ahmed, M.; Randall, R.; Chowdhury, H.M.; Snape, K.M.; Bradshaw, T.Y.; Southgate, L.; Lee, G.J.; Jackson, I.; et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum. Mutat. 2011, 32, 1385–1389. [Google Scholar] [CrossRef]
- Maron, B.A. Clarifying the Pulmonary Arterial Hypertension Molecular Landscape Using Functional Genetics. Am. J. Respir. Crit. Care Med. 2020, 202, 488–490. [Google Scholar] [CrossRef]
- Belostotsky, R.; Ben-Shalom, E.; Rinat, C.; Becker-Cohen, R.; Feinstein, S.; Zeligson, S.; Segel, R.; Elpeleg, O.; Nassar, S.; Frishberg, Y. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am. J. Hum. Genet. 2011, 88, 193–200. [Google Scholar] [CrossRef]
- Paulin, R.; Dromparis, P.; Sutendra, G.; Gurtu, V.; Zervopoulos, S.; Bowers, L.; Haromy, A.; Webster, L.; Provencher, S.; Bonnet, S.; et al. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab. 2014, 20, 827–839. [Google Scholar] [CrossRef]
- Garcia-Rivas, G.; Jerjes-Sánchez, C.; Rodriguez, D.; Garcia-Pelaez, J.; Trevino, V. A systematic review of genetic mutations in pulmonary arterial hypertension. BMC Med. Genet. 2017, 18, 82. [Google Scholar] [CrossRef] [PubMed]
- Pienkos, S.; Gallego, N.; Condon, D.F.; Cruz-Utrilla, A.; Ochoa, N.; Nevado, J.; Arias, P.; Agarwal, S.; Patel, H.; Chakraborty, A.; et al. Novel TNIP2 and TRAF2 Variants Are Implicated in the Pathogenesis of Pulmonary Arterial Hypertension. Front. Med. 2021, 8, 625763. [Google Scholar] [CrossRef] [PubMed]
- de Jesus Perez, V.A.; Yuan, K.; Lyuksyutova, M.A.; Dewey, F.; Orcholski, M.E.; Shuffle, E.M.; Mathur, M.; Yancy, L.J.; Rojas, V.; Li, C.G.; et al. Whole-exome sequencing reveals TopBP1 as a novel gene in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 1260–1272. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.Y.; Stebbings, E.; Grigoriadou, S.; Wood, N.W.; Hambleton, S.; Burns, S.O.; Thrasher, A.J.; et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | ||
|---|---|---|
| Female (n, %) | 51 (73.9) | |
| Age at diagnosis, years | 50 ± 20 | |
| Six-minute walking distance (m) | 332 ± 127 | |
| Mean pulmonary arterial pressure (mmHg) | 41 ± 16 | |
| Pulmonary arterial wedge pressure (mmHg) | 14 ± 4 | |
| Pulmonary vascular resistance (Wood units) | 8 ± 7 | |
| Peak tricuspid regurgitation peak gradient (mmHg) | 52 ± 30 | |
| Peak oxygen consumption (mL/min/kg) | 12 ± 4 | |
| Ventilatory equivalents for carbon dioxide (VE/VCO2) | 39 ± 14 | |
| Right atrial pressure (mmHg) | 13 ± 10 | |
| Cardiac index (L/min/m2) | 3 ± 1.1 | |
| Pulmonary artery saturation (%) | 66 ± 12 | |
| N-terminal prohormone of brain natriuretic peptide (ng/L) | 1869 ± 2988 | |
| World Health Organization functional class (N, %) | I | 4 (5.8) |
| II | 19 (27.5) | |
| III | 44 (63.8) | |
| IV | 2 (2.9) | |
| Progression of symptoms | No | 26 (37.7) |
| Slow | 34 (49.3) | |
| Rapid | 9 (13.0) | |
| No. | ID | Sex | Gene | cDNA | Amino Acid | Variant | Genotype | PolyPhen2/SIFT | ACMG 2015 * | MAF in EAS |
|---|---|---|---|---|---|---|---|---|---|---|
| Category A. PAH patients with BMPR2 variants (N = 5) | ||||||||||
| 1 | A100553 | F | BMPR2 | c.877_888del | p. Leu293_Ser296del | Inframe_del | Het | N/A ($) | LP | 0.00000 |
| 2 | A100593 | F | BMPR2 | c.1750C>T | p. Arg584* | Nonsense | Het | N/A ($) [12] | P | 0.00000 |
| ATP13A3 | c.970+1G>A | Splicing | Het | N/A ($) | LP | 0.00009 | ||||
| ABCA3 | c.635G>A | p. Arg212Gln | Missense | Het | PD/T | VUS | 0.00022 | |||
| 3 | A100655 | F | BMPR2 | c.994C>T | p. Arg332* | Nonsense | Het | N/A ($) [13] | P | 0.00000 |
| 4 | A100719 | F | BMPR2 | c.1750C>T | p. Arg584* | Nonsense | Het | N/A ($) [12] | P | 0.00000 |
| ATP13A3 | c.970+1G>A | Splicing | Het | N/A ($) | LP | 0.00000 | ||||
| 5 | A110118 | F | BMPR2 | c.1478C>T | p. Thr493Ile | Missense | Het | PD/D [14] | LP | 0.00000 |
| ENG | c.278G>A | p. Arg93Gln | Missense | Het | B/T | VUS | 0.00019 | |||
| Category B. PAH patients with GDF2 variants (N = 3) | ||||||||||
| 6 | A100537 | M | GDF2 | c.1207G>A | p. Val403Ile | Missense | Het | -/T ($) | VUS | 0.00000 |
| 7 | A100692 | M | GDF2 | c.38T>C | p. Leu13Pro | Missense | Het | -/D ($) | VUS | 0.00000 |
| BMP10 | c.475G>A | p. Asp159Asn | Missense | Het | B/T | VUS | 0.00019 | |||
| 8 | A110160 | M | GDF2 | c.259C>T | p. Gln87* | Nonsense | Het | N/A ($) | LP | 0.00000 |
| Category C. PAH patients with variants of other PAH-related genes (N = 20) | ||||||||||
| 9 | A100538 | F | PTGIS | c.592G>A | p. Val198Ile | Missense | Het | B/T | VUS | 0.00038 |
| 10 | A100540 | M | ATP13A3 | c.260G>A | p. Arg87His | Missense | Het | B/T | VUS | 0.00000 |
| 11 | A100554 | F | ATP13A3 | c.3040A>T | p. Ile1014Phe | Missense | Het | B/D | VUS | 0.00000 |
| 12 | A100559 | F | KLK1 | c.119C>T | p. Ala40Val | Missense | Het | B/T | VUS | 0.00019 |
| 13 | A100588 | F | SIRT3 | c.415A>G | p. Arg139Gly | Missense | Het | PD/D | VUS | 0.00019 |
| 14 | A100591 | F | NOTCH2 | c.7177G>C | p. Ala2393Pro | Missense | Het | PD/T | VUS | 0.00000 |
| 15 | A100607 | F | THBS1 | c.3272G>A | p. Arg1091His | Missense | Het | PD/D | VUS | 0.00019 |
| KCNA3 | c.283G>A | p. Ala95Thr | Missense | Het | B/T | VUS | 0.00034 | |||
| 16 | A100641 | F | PTGIS | c.635G>A | p. Arg212Gln | Missense | Het | B/T | VUS | 0.00019 |
| 17 | A100643 | M | TNIP2 | c.248C>G | p. Ser83Trp | Missense | Het | -/D ($) | VUS | 0.00019 |
| 18 | A100653 | F | BMPR1B | c.664A>G | p. Lys222Glu | Missense | Het | PD/T | VUS | 0.00000 |
| NOTCH1 | c.3549G>T | p. Glu1183Asp | Missense | Het | B/T | VUS | 0.00019 | |||
| PTGIS | c.592G>A | p. Val198Ile | Missense | Het | B/T | VUS | 0.00038 | |||
| 19 | A100654 | F | JAG2 | c.320C>G | p. Pro107Arg | Missense | Het | PD/T | VUS | 0.00059 |
| 20 | A100683 | F | JAG2 | c.359G>C | p. Arg120Pro | Missense | Het | PD/D | VUS | 0.00063 |
| NOTCH1 | c.3973G>A | p. Ala1325Thr | Missense | Het | PD/T | VUS | 0.00019 | |||
| 21 | A100697 | F | SMAD4 | c.554C>T | p. Pro185Leu | Missense | Het | PD/D | VUS | 0.00019 |
| NOTCH3 | c.67C>T | p. Pro23Ser | Missense | Het | B/T | VUS | 0.00000 | |||
| 22 | A100710 | F | TOPBP1 | c.1366A>G | p. Lys456Glu | Missense | Het | N/A ($) | VUS | 0.00000 |
| 23 | A100720 | M | JAG1 | c.3286C>T | p. Arg1096Trp | Missense | Het | PD/D | VUS | 0.00000 |
| 24 | A100740 | F | TOPBP1 | c.75A>T | p. Lys25Asn | Missense | Het | B/D | VUS | 0.00000 |
| 25 | A110041 | F | ABCA3 | c.143C>T | p. Ser48Leu | Missense | Het | PD/T | VUS | 0.00058 |
| 26 | A110136 | F | TNIP2 | c.281T>C | p. Ile94Thr | Missense | Het | PD/T | VUS | 0.00019 |
| 27 | A110138 | F | PTGIS | c.860A>G | p. Asn287Ser | Missense | Het | PD/T | VUS | 0.00039 |
| 28 | A110161 | M | KLK1 | c.469G>A | p. Gly157Ser | Missense | Het | PD/D | VUS | 0.00000 |
| Characteristics | BMPR2 (n = 5) | GDF2 (n = 3) | N-Gene (n = 61) | p-Value | ||
|---|---|---|---|---|---|---|
| BMPR2 vs. N-Genet | GDF2 vs. N-Gene | |||||
| Female (N, %) | 5 (100) | 0 (0) | 46 (75.4) | |||
| Age of onset, years | 43 ± 11 | 25 ± 13 | 51 ± 20 | 0.626 | 0.048 | |
| Six-minute walking distance (m) | 351 ± 165 | 490 ± 101 | 322 ± 122 | 0.851 | 0.051 | |
| Mean pulmonary arterial pressure (mmHg) | 66 ± 15 | 66 ± 13 | 38 ± 13 | <0.001 | 0.001 | |
| Pulmonary arterial wedge pressure (mmHg) | 11 ± 3 | 11 ± 3 | 14 ± 5 | 0.299 | 0.499 | |
| Pulmonary vascular resistance (woods) | 22 ± 9 | 15 ± 12 | 7 ± 5 | <0.001 | 0.048 | |
| Peak tricuspid regurgitation peak gradient (mmHg) | 64 ± 35 | 58 ± 23 | 50 ± 31 | 0.573 | 0.903 | |
| Peak oxygen consumption (mL/min/kg) | 12 ± 4 | 15 ± 6 | 12 ± 4 | 0.977 | 0.501 | |
| Ventilatory equivalents for carbon dioxide (VE/VCO2) | 50 ± 18 | 45 ± 4 | 38 ± 14 | 0.148 | 0.754 | |
| Right atrial pressure (mmHg) | 14 ± 9 | 11 ± 5 | 13 ± 10 | 0.995 | 0.902 | |
| Cardiac index (L/min/m2) | 1.6 ± 0.5 | 2.6 ± 1.1 | 2.8 ± 1.1 | 0.030 | 0.966 | |
| Pulmonary artery saturation (%) | 54 ± 13 | 68 ± 10 | 67 ± 11 | 0.045 | 0.956 | |
| N-terminal prohormone of brain natriuretic peptide (ng/L) | 4117 ± 5336 | 228 ± 46 | 1763 ± 2763 | 0.172 | 0.616 | |
| World Health Organization functional class (N, %) | I | 1 (20) | 0 (0) | 3 (4.9) | 0.469 | 0.389 |
| II | 1 (20) | 2 (66.7) | 16 (26.2) | |||
| III | 3 (60) | 1 (33.3) | 40 (65.6) | |||
| IV | 0 (0) | 0 (0) | 2 (3.3) | |||
| Progression of symptoms | No | 1 (20) | 1 (33.3) | 24 (39.3) | 0.417 | 0.523 |
| Slow | 4 (80) | 1 (33.3) | 29 (47.5) | |||
| Rapid | 0 (0) | 1 (33.3) | 8 (13.1) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.-T.; Weng, K.-P.; Chang, S.-K.; Huang, W.-C.; Chen, L.-W. Hemodynamic and Clinical Profiles of Pulmonary Arterial Hypertension Patients with GDF2 and BMPR2 Variants. Int. J. Mol. Sci. 2024, 25, 2734. https://doi.org/10.3390/ijms25052734
Wang M-T, Weng K-P, Chang S-K, Huang W-C, Chen L-W. Hemodynamic and Clinical Profiles of Pulmonary Arterial Hypertension Patients with GDF2 and BMPR2 Variants. International Journal of Molecular Sciences. 2024; 25(5):2734. https://doi.org/10.3390/ijms25052734
Chicago/Turabian StyleWang, Mei-Tzu, Ken-Pen Weng, Sheng-Kai Chang, Wei-Chun Huang, and Lee-Wei Chen. 2024. "Hemodynamic and Clinical Profiles of Pulmonary Arterial Hypertension Patients with GDF2 and BMPR2 Variants" International Journal of Molecular Sciences 25, no. 5: 2734. https://doi.org/10.3390/ijms25052734
APA StyleWang, M.-T., Weng, K.-P., Chang, S.-K., Huang, W.-C., & Chen, L.-W. (2024). Hemodynamic and Clinical Profiles of Pulmonary Arterial Hypertension Patients with GDF2 and BMPR2 Variants. International Journal of Molecular Sciences, 25(5), 2734. https://doi.org/10.3390/ijms25052734