
Transforming Growth Factor Beta and Alveolar Rhabdomyosarcoma: A Challenge of Tumor Differentiation and Chemotherapy Response
 , and
, and 
Abstract
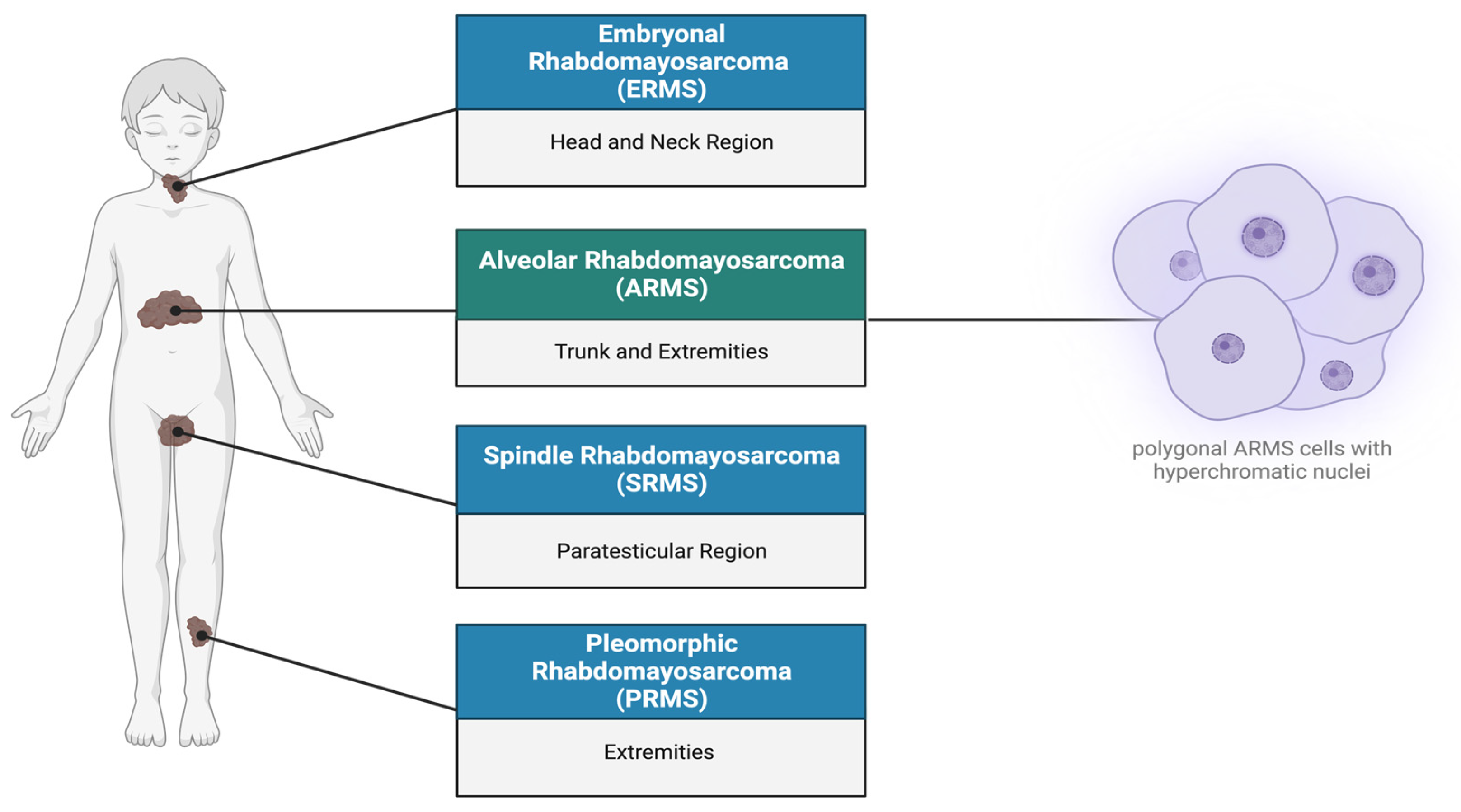
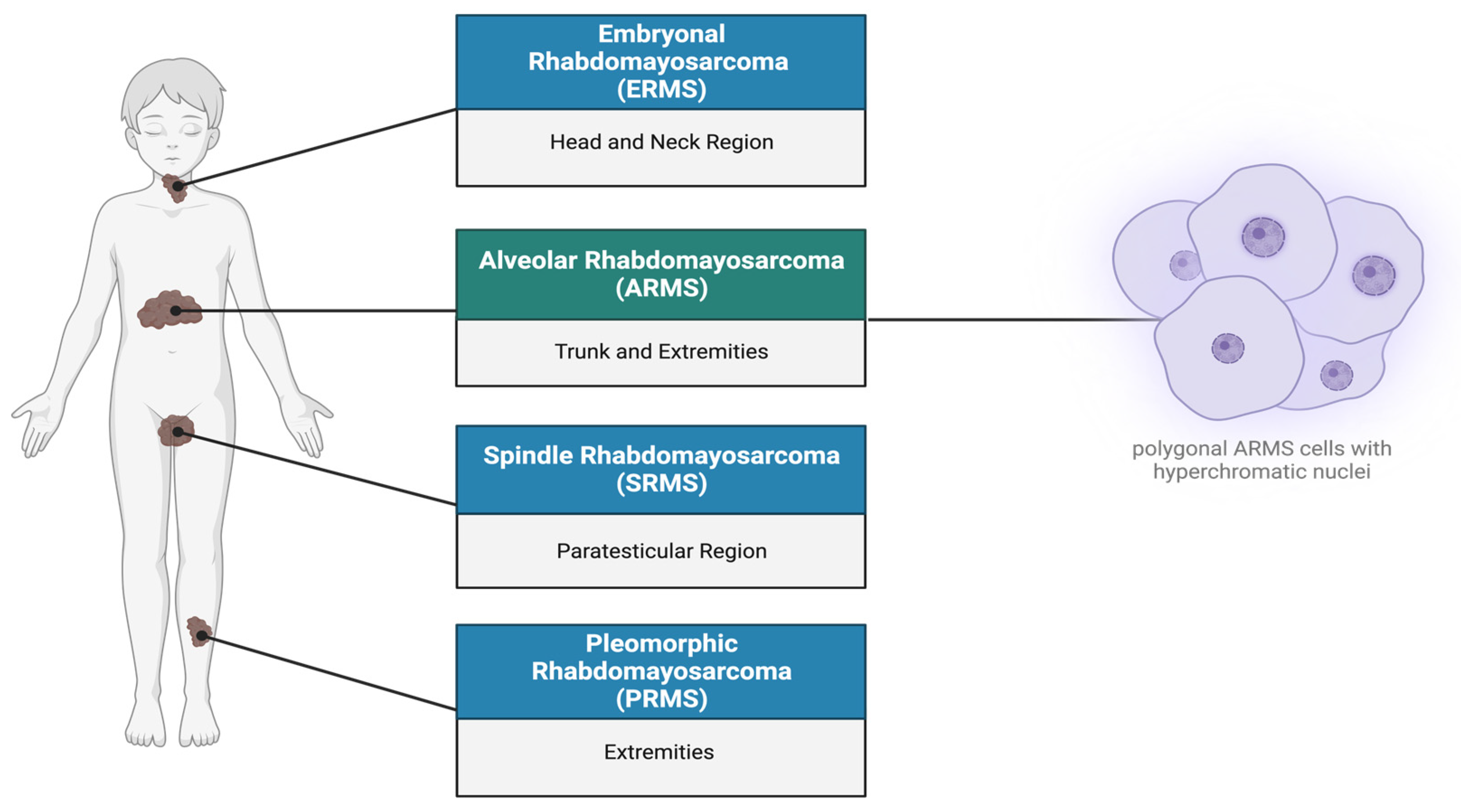
1. Rhabdomyosarcoma Overview
2. Tumor Growth and Differentiation in ARMS
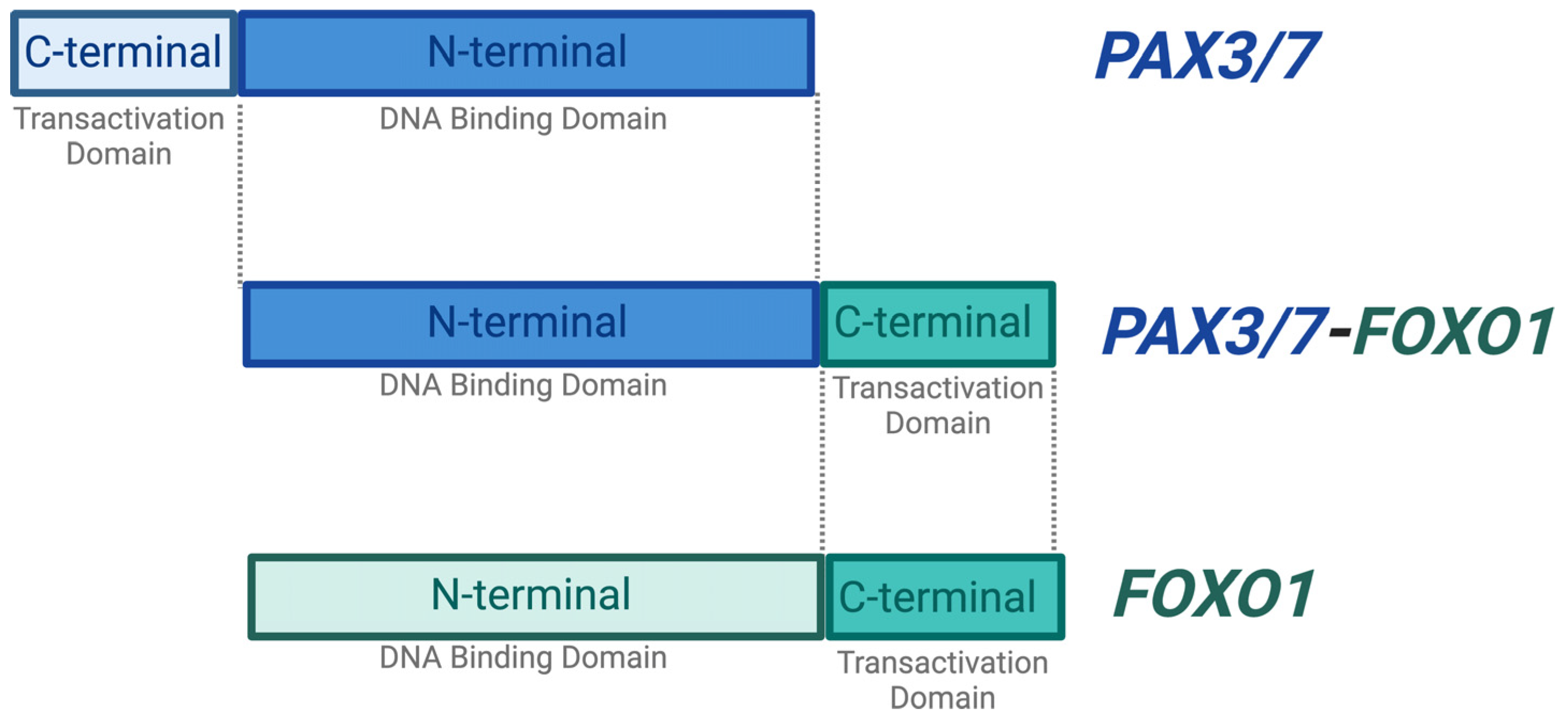
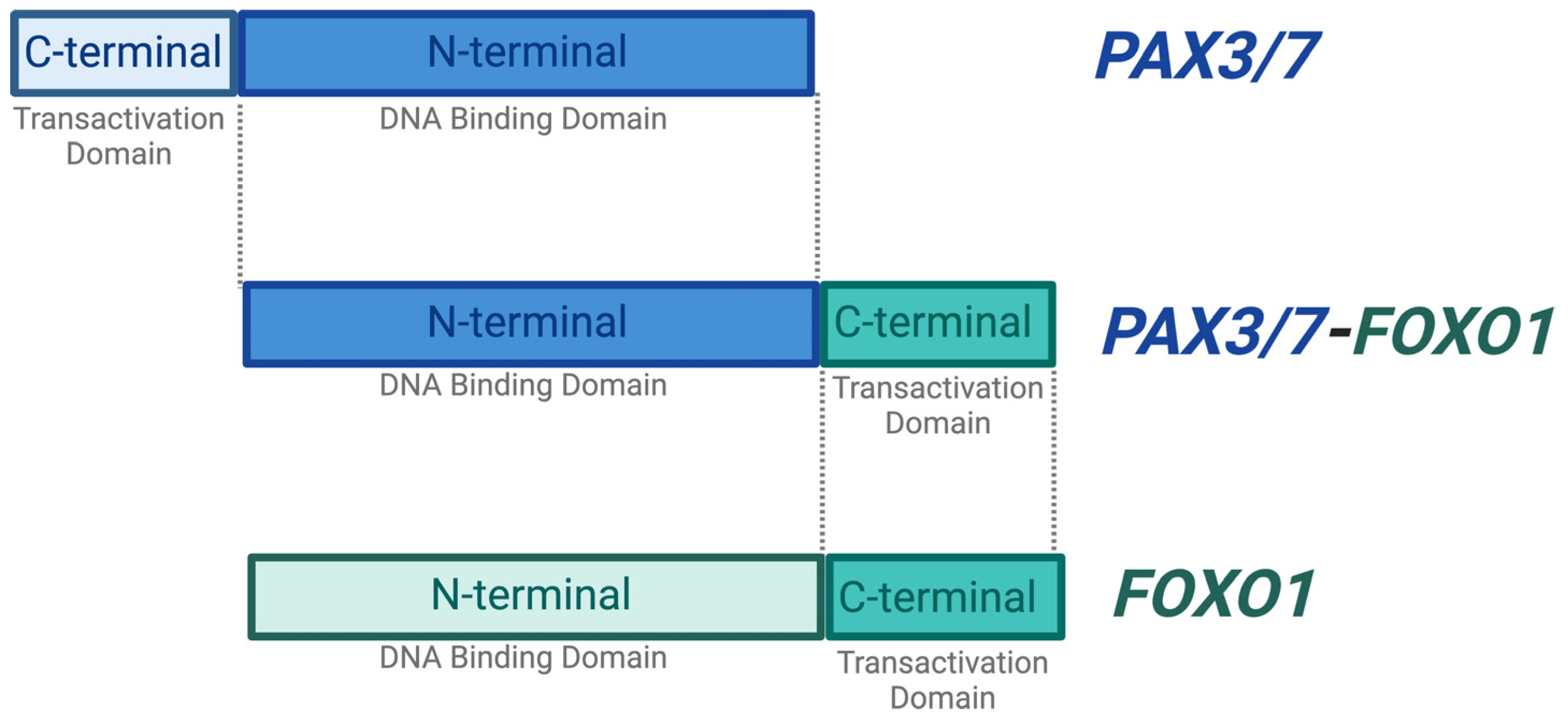
2.1. PAX3/7 and FOXO1
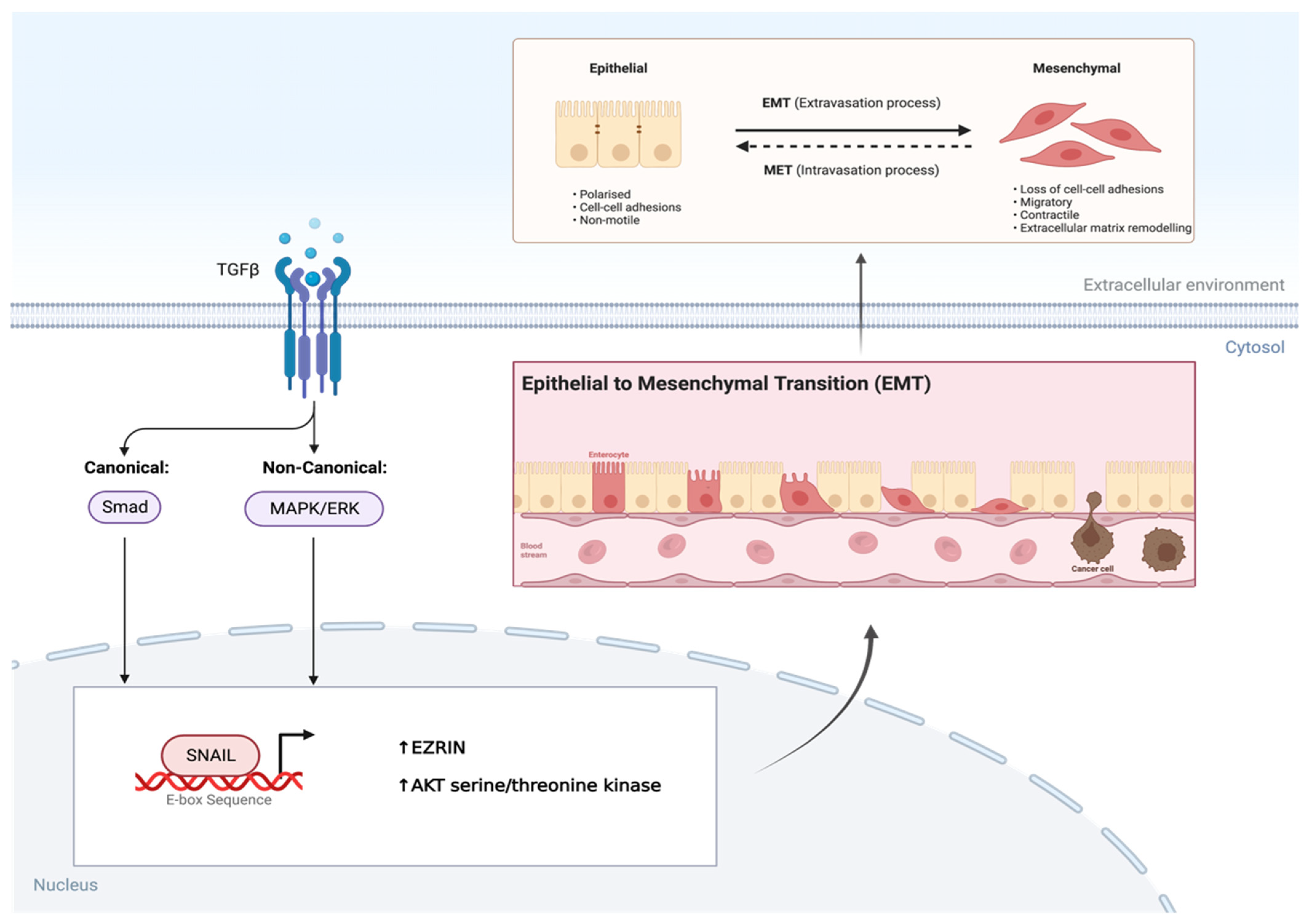
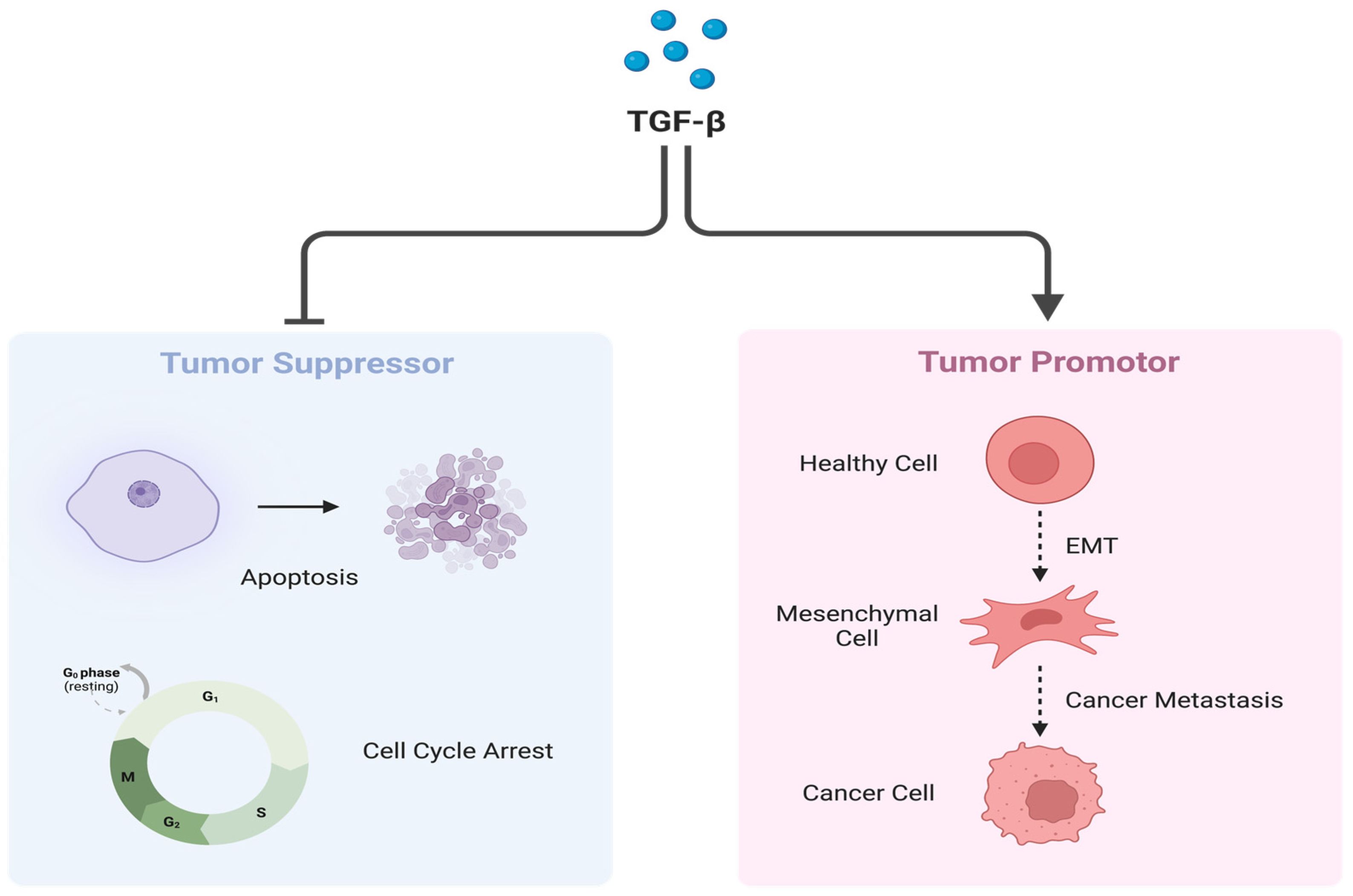
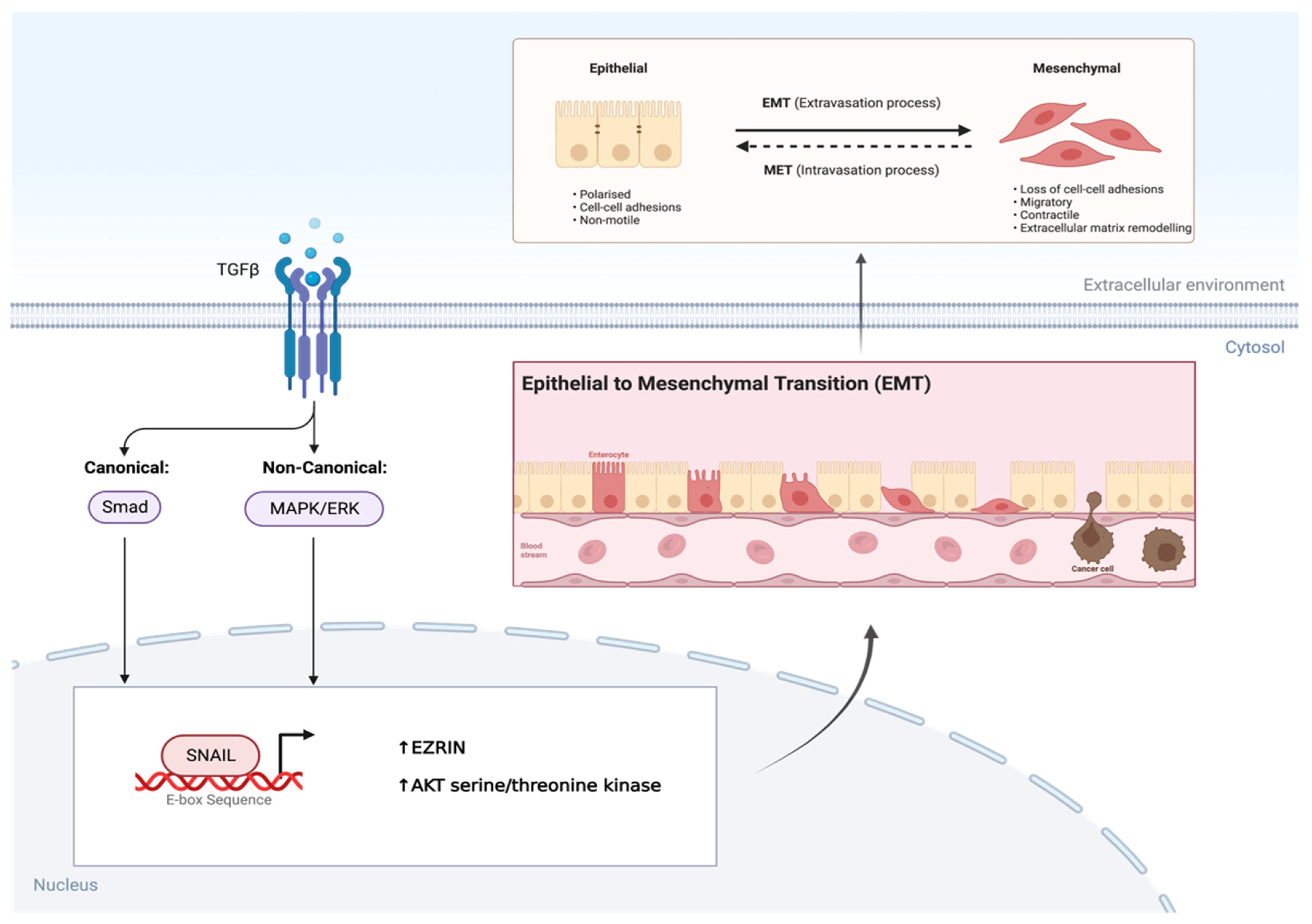
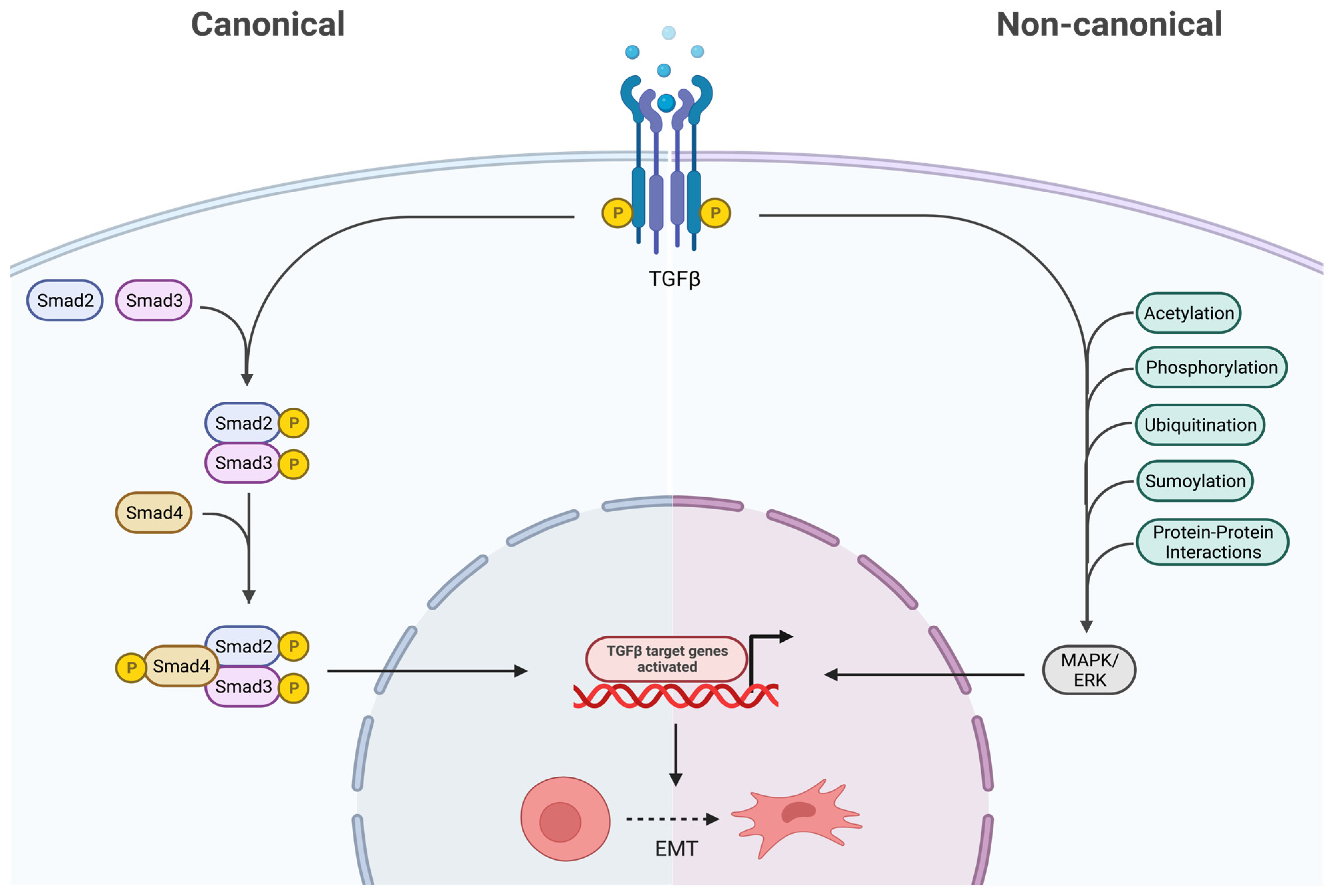
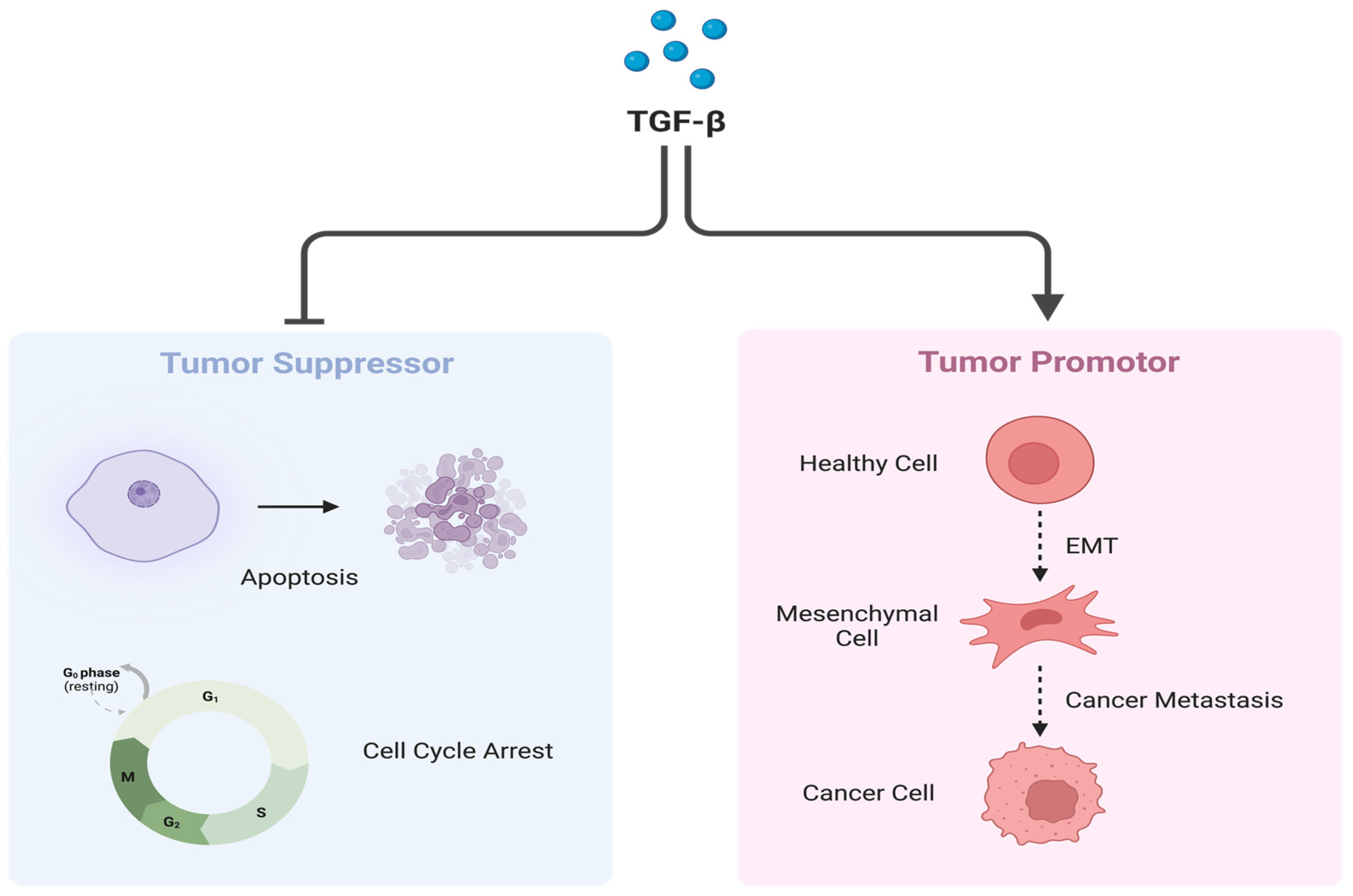
2.2. Transforming Growth Factor-β
3. ARMS Chemotherapy Drugs and Their Impact on Tumor Cell Differentiation
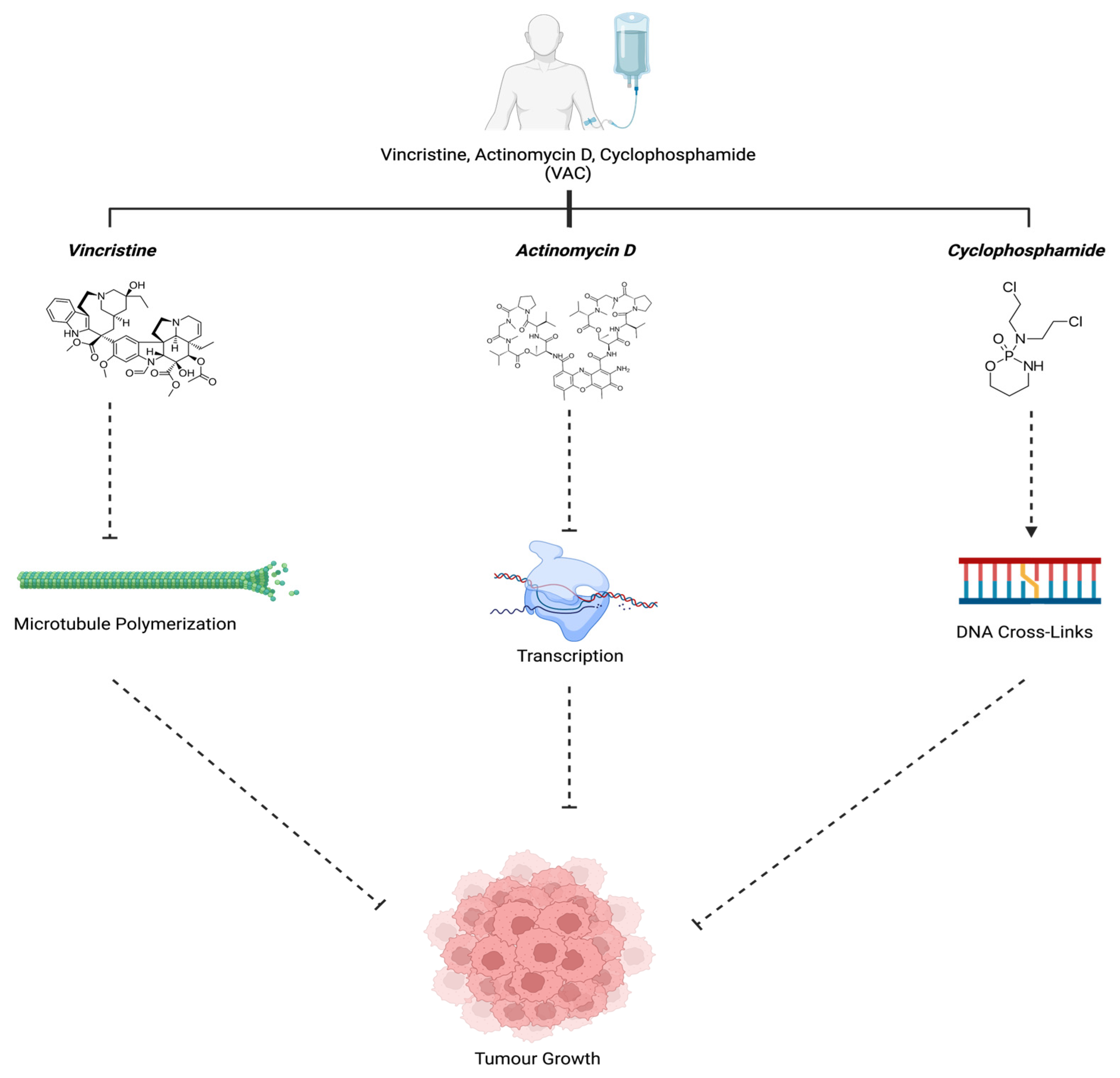
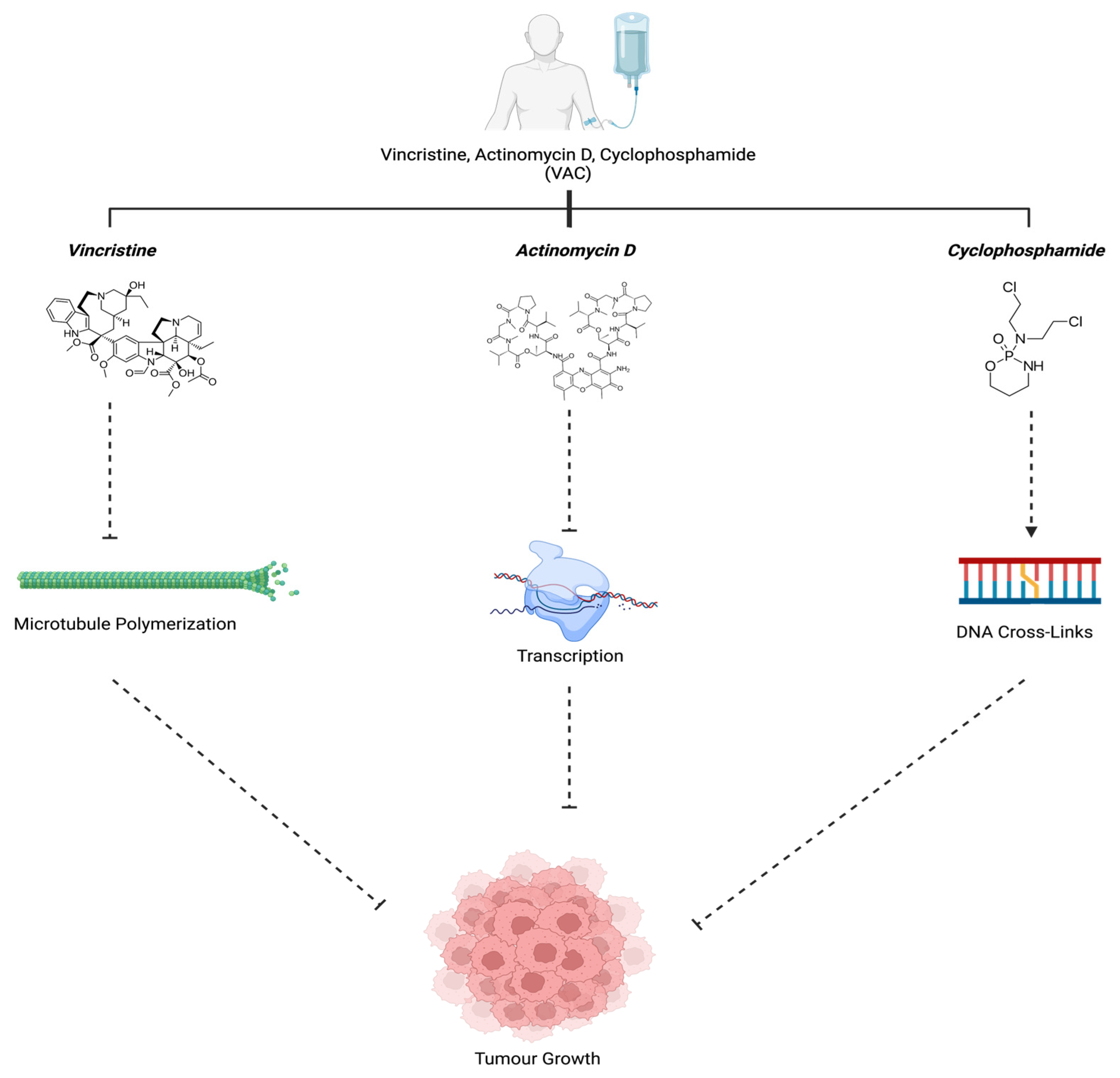
3.1. Vincristine, Actinomysin D, and Cyclophosphamide (VAC)
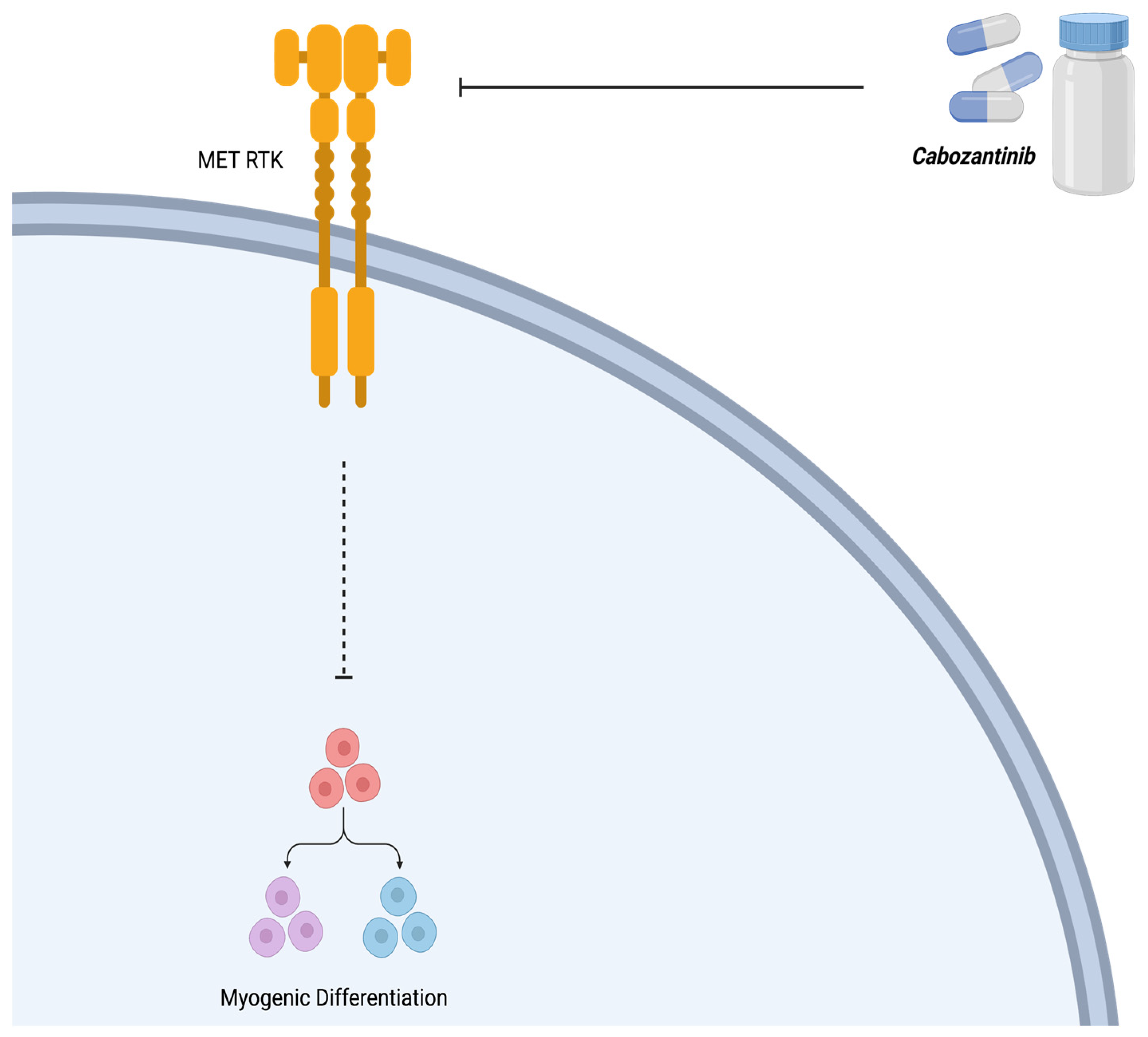
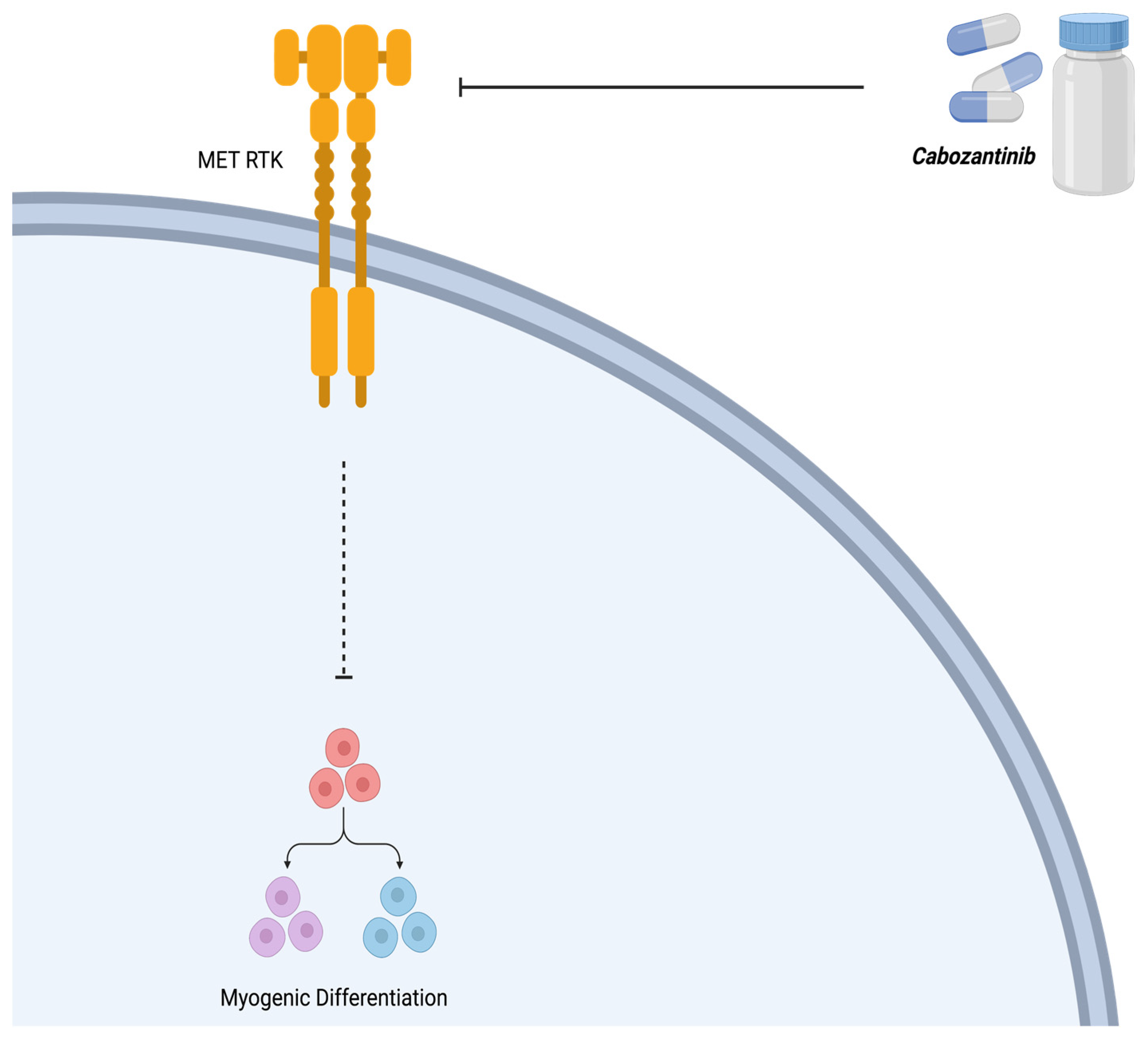
3.2. Cabozantinib (XL184)
3.3. Bortezomib
3.4. Vinorelbine
3.5. AZD1775
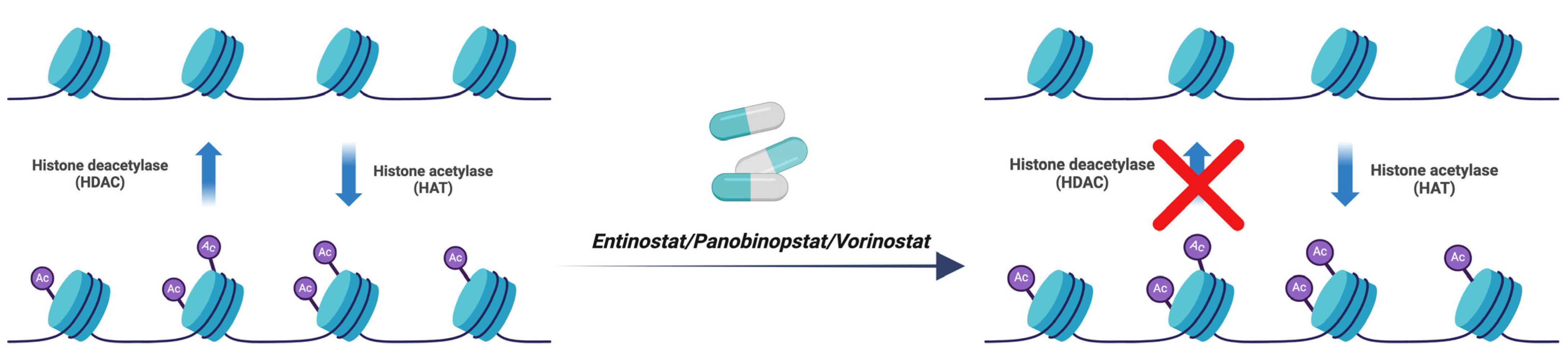
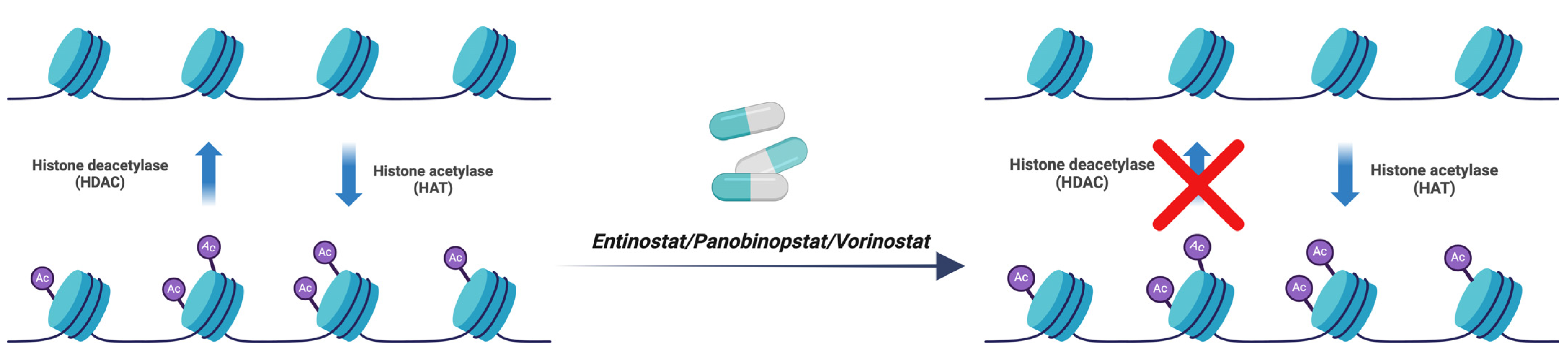
3.6. Entinostat, Panobinostat, and Vorinostat
3.7. Critotinib, Bevacizumab (mAb), and Regorafenib
3.8. All-Trans Retinoic Acid (ATRA)
3.9. Cisplatin
3.10. 5-Azacytidine
4. In Vivo Model for RMS Investigations
5. Conclusions and Future Direction
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aghaei, M.; Nasimian, A.; Rahmati, M.; Kawalec, P.; Machaj, F.; Rosik, J.; Bhushan, B.; Bathaie, S.Z.; Azarpira, N.; Los, M.J.; et al. The Role of BiP and the IRE1alpha-XBP1 Axis in Rhabdomyosarcoma Pathology. Cancers 2021, 13, 4927. [Google Scholar] [CrossRef]
- Eguia-Aguilar, P.; Lopez-Martinez, B.; Retana-Contreras, C.; Perezpena-Diazconti, M. Alveolar rhabdomyosarcoma: Origin and prognostic implications of molecular findings. Bol. Med. Hosp. Infant. Mex. 2016, 73, 405–410. [Google Scholar]
- Stefanek, E.; Samiei, E.; Kavoosi, M.; Esmaeillou, M.; Geraylow, K.R.; Emami, A.; Ashrafizadeh, M.; Perrin, D.; Gordon, J.W.; Akbari, M.; et al. A bioengineering method for modeling alveolar Rhabdomyosarcoma and assessing chemotherapy responses. MethodsX 2021, 8, 101473. [Google Scholar] [CrossRef]
- Nhung, T.H.; Le Minh, V.; Tuyet, T.T.; Cuong, T.M.; Le Lam, N.; Trang, H.T.; Quy, N.X.; Thong, P.M.; Thanh, D.K.; Duc, N.M. Orbital rhabdomyosarcoma in a 19-year-old male patient: A case report and literature review. Radiol. Case Rep. 2023, 18, 2744–2749. [Google Scholar] [CrossRef]
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Prim. 2019, 5, 1. [Google Scholar] [CrossRef]
- Emami, A.; Shojaei, S.; da Silva Rosa, S.C.; Aghaei, M.; Samiei, E.; Vosoughi, A.R.; Kalantari, F.; Kawalec, P.; Thliveris, J.; Sharma, P.; et al. Mechanisms of simvastatin myotoxicity: The role of autophagy flux inhibition. Eur. J. Pharmacol. 2019, 862, 172616. [Google Scholar] [CrossRef]
- Martin-Giacalone, B.A.; Weinstein, P.A.; Plon, S.E.; Lupo, P.J. Pediatric Rhabdomyosarcoma: Epidemiology and Genetic Susceptibility. J. Clin. Med. 2021, 10, 2028. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, A.R.; da Silva Rosa, S.C.; Samiei, E.; Alizadeh, J.; Field, J.; Kawalec, P.; Thliveris, J.; Akbari, M.; Ghavami, S.; Gordon, J.W. Autophagy modulates temozolomide-induced cell death in alveolar Rhabdomyosarcoma cells. Cell Death Discov. 2018, 4, 52. [Google Scholar] [CrossRef] [PubMed]
- Allen-Rhoades, W.; Lupo, P.J.; Scheurer, M.E.; Chi, Y.; Kuttesch, J.F.; Venkatramani, R.; Meyer, W.H.; Mascarenhas, L. Alveolar rhabdomyosarcoma has superior response rates to vinorelbine compared to embryonal rhabdomyosarcoma in patients with relapsed/refractory disease: A meta-analysis. Cancer Med. 2023, 12, 10222–10229. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, D.; Szewczyk, A.; Radzka, J.; Dubińska-Magiera, M.; Kazimierczak, W.; Daczewska, M.; Migocka-Patrzałek, M. The natural origins of cytostatic compounds used in rhabdomyosarcoma therapy. Adv. Clin. Exp. Med. 2023, 32, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.C.; Singh, C.; Pambuccian, S.E. Cytological diagnosis of metastatic alveolar rhabdomyosarcoma in the ascitic fluid: Report of a case highlighting the diagnostic difficulties. Cytojournal 2012, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Sannino, G.; Marchetto, A.; Kirchner, T.; Grünewald, T.G. Epithelial-to-Mesenchymal and Mesenchymal-to-Epithelial Transition in Mesenchymal Tumors: A Paradox in Sarcomas? Cancer Res. 2017, 77, 4556–4561. [Google Scholar] [CrossRef]
- Lagutina, I.V.; Valentine, V.; Picchione, F.; Harwood, F.; Valentine, M.B.; Villarejo-Balcells, B.; Carvajal, J.J.; Grosveld, G.C. Modeling of the human alveolar rhabdomyosarcoma Pax3-Foxo1 chromosome translocation in mouse myoblasts using CRISPR-Cas9 nuclease. PLoS Genet. 2015, 11, e1004951. [Google Scholar] [CrossRef] [PubMed]
- Azorsa, D.O.; Bode, P.K.; Wachtel, M.; Cheuk, A.T.C.; Meltzer, P.S.; Vokuhl, C.; Camenisch, U.; Khov, H.L.; Bode, B.; Schäfer, B.W.; et al. Immunohistochemical detection of PAX-FOXO1 fusion proteins in alveolar rhabdomyosarcoma using breakpoint specific monoclonal antibodies. Mod. Pathol. 2020, 34, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Skrzypek, K.; Kusienicka, A.; Trzyna, E.; Szewczyk, B.; Ulman, A.; Konieczny, P.; Adamus, T.; Badyra, B.; Kortylewski, M.; Majka, M. SNAIL is a key regulator of alveolar rhabdomyosarcoma tumor growth and differentiation through repression of MYF5 and MYOD function. Cell Death Dis. 2018, 9, 643. [Google Scholar] [CrossRef]
- Skrzypek, K.; Nieszporek, A.; Badyra, B.; Lasota, M.; Majka, M. Enhancement of myogenic differentiation and inhibition of rhabdomyosarcoma progression by miR-28-3p and miR-193a-5p regulated by SNAIL. Mol. Ther.-Nucleic Acids 2021, 24, 888–904. [Google Scholar] [CrossRef]
- Charytonowicz, E.; Cordon-Cardo, C.; Matushansky, I.; Ziman, M. Alveolar rhabdomyosarcoma: Is the cell of origin a mesenchymal stem cell? Cancer Lett. 2009, 279, 126–136. [Google Scholar] [CrossRef]
- Miekus, K.; Lukasiewicz, E.; Jarocha, D.; Sekula, M.; Drabik, G.; Majka, M. The decreased metastatic potential of rhabdomyosarcoma cells obtained through MET receptor downregulation and the induction of differentiation. Cell Death Dis. 2013, 4, e459. [Google Scholar] [CrossRef]
- Ramadan, F.; Saab, R.; Hussein, N.; Clézardin, P.; Cohen, P.A.; Ghayad, S.E. Non-coding RNA in rhabdomyosarcoma progression and metastasis. Front. Oncol. 2022, 12, 971174. [Google Scholar] [CrossRef]
- Skrzypek, K.; Kot, M.; Konieczny, P.; Nieszporek, A.; Kusienicka, A.; Lasota, M.; Bobela, W.; Jankowska, U.; Kędracka-Krok, S.; Majka, M. SNAIL Promotes Metastatic Behavior of Rhabdomyosarcoma by Increasing EZRIN and AKT Expression and Regulating MicroRNA Networks. Cancers 2020, 12, 1870. [Google Scholar] [CrossRef]
- Skrzypek, K.; Adamek, G.; Kot, M.; Badyra, B.; Majka, M. Progression and Differentiation of Alveolar Rhabdomyosarcoma Is Regulated by PAX7 Transcription Factor-Significance of Tumor Subclones. Cells 2021, 10, 1870. [Google Scholar] [CrossRef] [PubMed]
- Laubscher, D.; Gryder, B.E.; Sunkel, B.D.; Andresson, T.; Wachtel, M.; Das, S.; Roschitzki, B.; Wolski, W.; Wu, X.S.; Chou, H.-C.; et al. BAF complexes drive proliferation and block myogenic differentiation in fusion-positive rhabdomyosarcoma. Nat. Commun. 2021, 12, 6924. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Ney, M.; Camussi, G. The PAX3-FOXO1 fusion protein present in rhabdomyosarcoma interferes with normal FOXO activity and the TGF-β pathway. PLoS ONE 2015, 10, e0121474. [Google Scholar] [CrossRef]
- Petragnano, F.; Pietrantoni, I.; Camero, S.; Codenotti, S.; Milazzo, L.; Vulcano, F.; Macioce, G.; Giordani, I.; Tini, P.; Cheleschi, S.; et al. Clinically relevant radioresistant rhabdomyosarcoma cell lines: Functional, molecular and immune-related characterization. J. Biomed. Sci. 2020, 27, 90. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Guo, L.; Dong, L.; Guo, L.; Li, S.; Zhang, J.; Sun, M. TGF-β1 signal pathway may contribute to rhabdomyosarcoma development by inhibiting differentiation. Cancer Sci. 2010, 101, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Dalvand, A.; da Silva Rosa, S.C.; Ghavami, S.; Marzban, H. Potential role of TGFBeta and autophagy in early crebellum development. Biochem. Biophys. Rep. 2022, 32, 101358. [Google Scholar]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef]
- Siapoush, S.; Rezaei, R.; Alavifard, H.; Hatami, B.; Zali, M.R.; Vosough, M.; Lorzadeh, S.; Łos, M.J.; Baghaei, K.; Ghavami, S. Therapeutic implications of targeting autophagy and TGF-β crosstalk for the treatment of liver fibrosis. Life Sci. 2023, 329, 121894. [Google Scholar] [CrossRef]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef]
- Alizadeh, J.; Glogowska, A.; Thliveris, J.; Kalantari, F.; Shojaei, S.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Autophagy modulates transforming growth factor β 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2018, 1865, 749–768. [Google Scholar]
- Esmaeilzadeh, A.; Mohammadi, V.; Elahi, R. Transforming growth factor β (TGF-β) pathway in the immunopathogenesis of multiple sclerosis (MS); molecular approaches. Mol. Biol. Rep. 2023, 50, 6121–6131. [Google Scholar] [CrossRef]
- Peng, D.; Fu, M.; Wang, M.; Wei, Y.; Wei, X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol. Cancer 2022, 21, 104. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef]
- Chen, C.; Garcia, H.D.; Scheer, M.; Henssen, A.G. Current and Future Treatment Strategies for Rhabdomyosarcoma. Front. Oncol. 2019, 9, 1458. [Google Scholar] [CrossRef] [PubMed]
- Kahen, E.; Yu, D.; Harrison, D.J.; Clark, J.; Hingorani, P.; Cubitt, C.L.; Reed, D.R. Identification of clinically achievable combination therapies in childhood rhabdomyosarcoma. Cancer Chemother. Pharmacol. 2016, 78, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Makimoto, A. Optimizing Rhabdomyosarcoma Treatment in Adolescents and Young Adults. Cancers 2022, 14, 2270. [Google Scholar] [CrossRef]
- George, P.; Journey, L.J.; Goldstein, M.N. Effect of vincristine on the fine structure of HeLa cells during mitosis. JNCI J. Natl. Cancer Inst. 1965, 35, 355–375. [Google Scholar]
- Gidding, C.E.; Kellie, S.J.; Kamps, W.A.; de Graaf, S.S. Vincristine revisited. Crit. Rev. Oncol. Hematol. 1999, 29, 267–287. [Google Scholar] [CrossRef] [PubMed]
- Awosika, A.O.; Below, J.; MD, J. Vincristine. StatPearls. Available online: https://www.ncbi.nlm.nih.gov/books/NBK537122/.2023 (accessed on 22 June 2023).
- Lu, D.-F.; Wang, Y.-S.; Li, C.; Wei, G.-J.; Chen, R.; Dong, D.-M.; Yao, M. Actinomycin D inhibits cell proliferations and promotes apoptosis in osteosarcoma cells. Int. J. Clin. Exp. Med. 2015, 8, 1904–1911. [Google Scholar]
- Marchal, J.A.; Prados, J.; Melguizo, C.; Fernández, J.E.; Velez, C.; Alvarez, L.; Aránega, A. Actinomycin D treatment leads to differentiation and inhibits proliferation in rhabdomyosarcoma cells. J. Lab. Clin. Med. 1997, 130, 42–50. [Google Scholar] [CrossRef]
- Ogino, M.H.; Tadi, P. Cyclophosphamide. StatPearls. Available online: https://www.ncbi.nlm.nih.gov/books/NBK553087/.2023 (accessed on 22 June 2023).
- Chuk, M.K.; Widemann, B.C.; Minard, C.G.; Liu, X.; Kim, A.; Bernhardt, M.B.; Kudgus, R.A.; Reid, J.M.; Voss, S.D.; Blaney, S.; et al. A phase 1 study of cabozantinib in children and adolescents with recurrent or refractory solid tumors, including CNS tumors: Trial ADVL1211, a report from the Children’s Oncology Group. Pediatr. Blood Cancer 2018, 65, e27077. [Google Scholar] [CrossRef]
- Casanova, M.; Ferrari, A.; Bisogno, G.; Merks, J.H.; De Salvo, G.L.; Meazza, C.; Tettoni, K.; Provenzi, M.; Mazzarino, I.; Carli, M. Vinorelbine and low-dose cyclophosphamide in the treatment of pediatric sarcomas: Pilot study for the upcoming European Rhabdomyosarcoma Protocol. Cancer 2004, 101, 1664–1671. [Google Scholar] [CrossRef]
- Liang, C.; Qiao, G.; Liu, Y.; Tian, L.; Hui, N.; Li, J.; Ma, Y.; Li, H.; Zhao, Q.; Cao, W.; et al. Overview of all-trans-retinoic acid (ATRA) and its analogues: Structures, activities, and mechanisms in acute promyelocytic leukaemia. Eur. J. Med. Chem. 2021, 220, 113451. [Google Scholar] [CrossRef]
- Ni, X.; Hu, G.; Cai, X. The success and the challenge of all-trans retinoic acid in the treatment of cancer. Crit. Rev. Food Sci. Nutr. 2019, 59 (Suppl. S1), S71–S80. [Google Scholar] [CrossRef] [PubMed]
- Szymański, Ł.; Skopek, R.; Palusińska, M.; Schenk, T.; Stengel, S.; Lewicki, S.; Kraj, L.; Kamiński, P.; Zelent, A. Retinoic Acid and Its Derivatives in Skin. Cells 2020, 9, 2660. [Google Scholar] [CrossRef]
- le Maire, A.; Teyssier, C.; Balaguer, P.; Bourguet, W.; Germain, P. Regulation of RXR-RAR Heterodimers by RXR- and RAR-Specific Ligands and Their Combinations. Cells 2019, 8, 1392. [Google Scholar] [CrossRef]
- O’brien, E.; Tse, C.; Tracy, I.; Reddin, I.; Selfe, J.; Gibson, J.; Tapper, W.; Pengelly, R.J.; Gao, J.; Aladowicz, E.; et al. Pharmacological EZH2 inhibition combined with retinoic acid treatment promotes differentiation and apoptosis in rhabdomyosarcoma cells. Clin. Epigenetics 2023, 15, 167. [Google Scholar] [CrossRef]
- Williams, A.P.; Waters, A.M.; Stewart, J.E.; Atigadda, V.R.; Mroczek-Musulman, E.; Muccio, D.D.; Grubbs, C.J.; Beierle, E.A. A novel retinoid X receptor agonist, UAB30, inhibits rhabdomyosarcoma cells in vitro. J. Surg. Res. 2018, 228, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Q. Implication of retinoic acid receptor selective signaling in myogenic differentiation. Sci. Rep. 2016, 6, 18856. [Google Scholar] [CrossRef] [PubMed]
- Gudas, L.J. Retinoids induce stem cell differentiation via epigenetic changes. Semin. Cell Dev. Biol. 2013, 24, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Luo, Q.; Zhang, Y.; Jia, F.; Zhao, Y.; Wang, F. Advances in Toxicological Research of the Anticancer Drug Cisplatin. Chem. Res. Toxicol. 2019, 32, 1469–1486. [Google Scholar] [CrossRef] [PubMed]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Zarrabi, A.; Perrin, D.; Kavoosi, M.; Sommer, M.; Sezen, S.; Mehrbod, P.; Bhushan, B.; Machaj, F.; Rosik, J.; Kawalec, P.; et al. Rhabdomyosarcoma: Current Therapy, Challenges, and Future Approaches to Treatment Strategies. Cancers 2023, 15, 5269. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Cojocari, D.; Purkal, J.J.; Popovic, R.; Talaty, N.N.; Xiao, Y.; Solomon, L.R.; Boghaert, E.R.; Leverson, J.D.; Phillips, D.C. 5-Azacitidine Induces NOXA to Prime AML Cells for Venetoclax-Mediated Apoptosis. Clin. Cancer Res. 2020, 26, 3371–3383. [Google Scholar] [CrossRef]
- Mahoney, S.E.; Yao, Z.; Keyes, C.C.; Tapscott, S.J.; Diede, S.J. Genome-wide DNA methylation studies suggest distinct DNA methylation patterns in pediatric embryonal and alveolar rhabdomyosarcomas. Epigenetics 2012, 7, 400–408. [Google Scholar] [CrossRef]
- Zhu, Q.; Liang, F.; Cai, S.; Luo, X.; Duo, T.; Liang, Z.; He, Z.; Chen, Y.; Mo, D. KDM4A regulates myogenesis by demethylating H3K9me3 of myogenic regulatory factors. Cell Death Dis. 2021, 12, 514. [Google Scholar] [CrossRef]
- Filip, K.; Lewińska, A.; Adamczyk-Grochala, J.; Marino Gammazza, A.; Cappello, F.; Lauricella, M.; Wnuk, M. 5-Azacytidine Inhibits the Activation of Senescence Program and Promotes Cytotoxic Autophagy during Trdmt1-Mediated Oxidative Stress Response in Insulinoma β-TC-6 Cells. Cells 2022, 11, 1213. [Google Scholar] [CrossRef]
- Chen, E.Y.; Langenau, D.M. Zebrafish models of rhabdomyosarcoma. Methods Cell Biol. 2011, 105, 383–402. [Google Scholar]
- Kahsay, A.; Rodriguez-Marquez, E.; López-Pérez, A.; Hörnblad, A.; von Hofsten, J. Pax3 loss of function delays tumour progression in kRAS-induced zebrafish rhabdomyosarcoma models. Sci. Rep. 2022, 12, 17149. [Google Scholar] [CrossRef]
- Kahsay, A.; Rodriguez-Marquez, E.; López-Pérez, A.; Hörnblad, A.; von Hofsten, J. PAX3-FOXO1 transgenic zebrafish models identify HES3 as a mediator of rhabdomyosarcoma tumorigenesis. eLife 2018, 7, 17149. [Google Scholar]
- Krukemeyer, M.G.; Krenn, V.; Jakobs, M.; Wagner, W. Magnetic drug targeting in a rhabdomyosarcoma rat model using magnetite-dextran composite nanoparticle-bound mitoxantrone and 0.6 tesla extracorporeal magnets—Sarcoma treatment in progress. J. Drug Target. 2012, 20, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.-Y.; Cheng, R.; Yang, Z.; Tian, Z.-M. Nanotechnology for Cancer Therapy Based on Chemotherapy. Molecules 2018, 23, 826. [Google Scholar] [CrossRef] [PubMed]
- Mercatali, L.; Vanni, S.; Miserocchi, G.; Liverani, C.; Spadazzi, C.; Cocchi, C.; Calabrese, C.; Gurrieri, L.; Fausti, V.; Riva, N.; et al. The emerging role of cancer nanotechnology in the panorama of sarcoma. Front. Bioeng. Biotechnol. 2022, 10, 953555. [Google Scholar] [CrossRef] [PubMed]
- Chramiec, A.; Teles, D.; Yeager, K.; Marturano-Kruik, A.; Pak, J.; Chen, T.; Hao, L.; Wang, M.; Lock, R.; Tavakol, D.N.; et al. Integrated human organ-on-a-chip model for predictive studies of anti-tumor drug efficacy and cardiac safety. Lab A Chip 2020, 20, 4357–4372. [Google Scholar] [CrossRef]
- Rengaswamy, V.; Zimmer, D.; Suss, R.; Rossler, J. RGD liposome-protamine-siRNA (LPR) nanoparticles targeting PAX3-FOXO1 for alveolar rhabdomyosarcoma therapy. J. Control. Release 2016, 235, 319–327. [Google Scholar] [CrossRef]
- Mirani, B.; Pagan, E.; Shojaei, S.; Duchscherer, J.; Toyota, B.D.; Ghavami, S.; Akbari, M. A 3D bioprinted hydrogel mesh loaded with all-trans retinoic acid for treatment of glioblastoma. Eur. J. Pharmacol. 2019, 854, 201–212. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug/Compound | Molecular Target | References |
|---|---|---|
| Vincristine, Actinomycin D, and Cyclophosphamide (VAC) | Microtubule Polymerization, Guanine Nucleotide in DNA, and Cross-Linkages with Guanine N-7, respectively | [40,42] |
| Cabozantinib (XL184) | Tyrosine Kinase (MET) | [35] |
| Bortezomib | 26s Proteosome | [35] |
| Vinorelbin | Microtubular Proteins | [9] |
| AZD1775 | Wee1 | [35] |
| Entinostat, Panobinostat, and Vorinostat | Histone Deacetylase (HDAC) | [34] |
| Critotinib, Bevacizumab (mAb), and Regorafenib | Receptor Tyrosine Kinae (RTK) | [34] |
| All-Trans Retinoic Acid (ATRA) | Retinoic Acid Receptors (RARs) | [20] |
| Cisplatin | DNA | [53] |
| 5-Azacytidine | DNA Methyltransferase | [56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhushan, B.; Iranpour, R.; Eshtiaghi, A.; da Silva Rosa, S.C.; Lindsey, B.W.; Gordon, J.W.; Ghavami, S. Transforming Growth Factor Beta and Alveolar Rhabdomyosarcoma: A Challenge of Tumor Differentiation and Chemotherapy Response. Int. J. Mol. Sci. 2024, 25, 2791. https://doi.org/10.3390/ijms25052791
Bhushan B, Iranpour R, Eshtiaghi A, da Silva Rosa SC, Lindsey BW, Gordon JW, Ghavami S. Transforming Growth Factor Beta and Alveolar Rhabdomyosarcoma: A Challenge of Tumor Differentiation and Chemotherapy Response. International Journal of Molecular Sciences. 2024; 25(5):2791. https://doi.org/10.3390/ijms25052791
Chicago/Turabian StyleBhushan, Bhavya, Rosa Iranpour, Amirmohammad Eshtiaghi, Simone C. da Silva Rosa, Benjamin W. Lindsey, Joseph W. Gordon, and Saeid Ghavami. 2024. "Transforming Growth Factor Beta and Alveolar Rhabdomyosarcoma: A Challenge of Tumor Differentiation and Chemotherapy Response" International Journal of Molecular Sciences 25, no. 5: 2791. https://doi.org/10.3390/ijms25052791
APA StyleBhushan, B., Iranpour, R., Eshtiaghi, A., da Silva Rosa, S. C., Lindsey, B. W., Gordon, J. W., & Ghavami, S. (2024). Transforming Growth Factor Beta and Alveolar Rhabdomyosarcoma: A Challenge of Tumor Differentiation and Chemotherapy Response. International Journal of Molecular Sciences, 25(5), 2791. https://doi.org/10.3390/ijms25052791