
Dualistic Effects of PRKAR1A as a Potential Anticancer Target in Cancer Cells and Cancer-Derived Stem Cells


Abstract
1. Introduction
2. Results
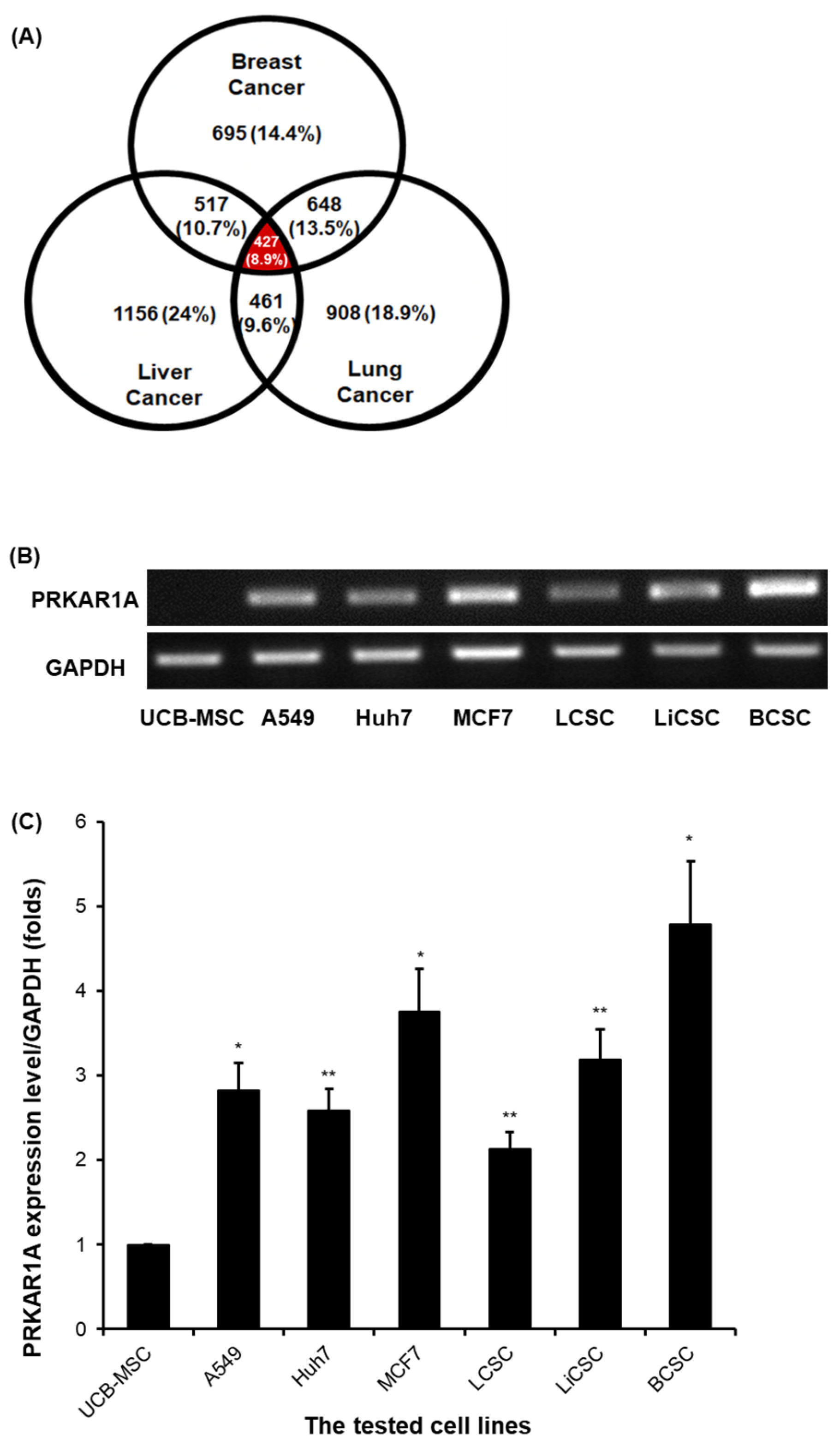
2.1. PRKAR1A Expression at the Transcriptomic Level of Various Cell Lines
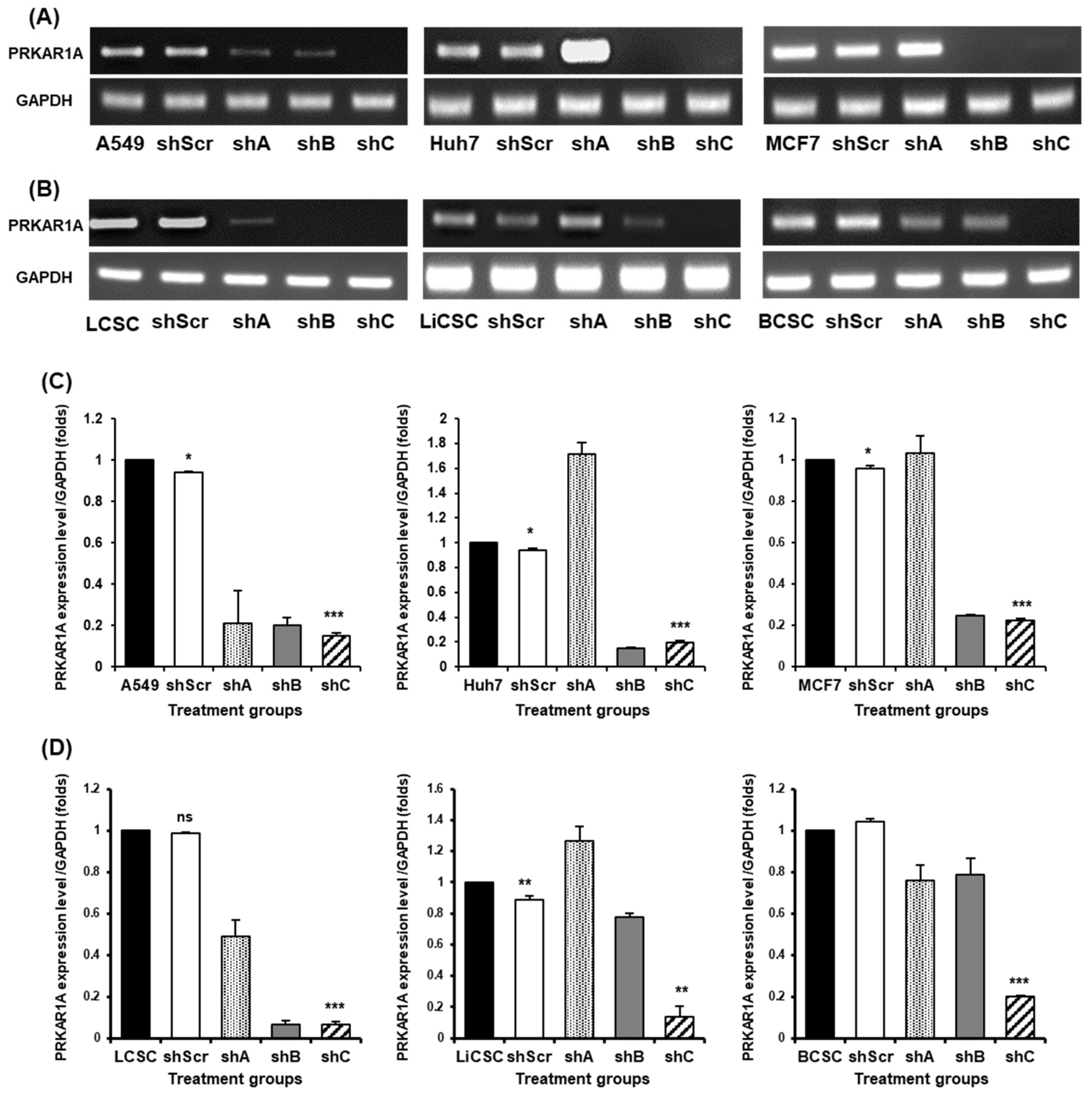
2.2. Efficient shRNA Selection for Biological Functional Assessment of PRKAR1A
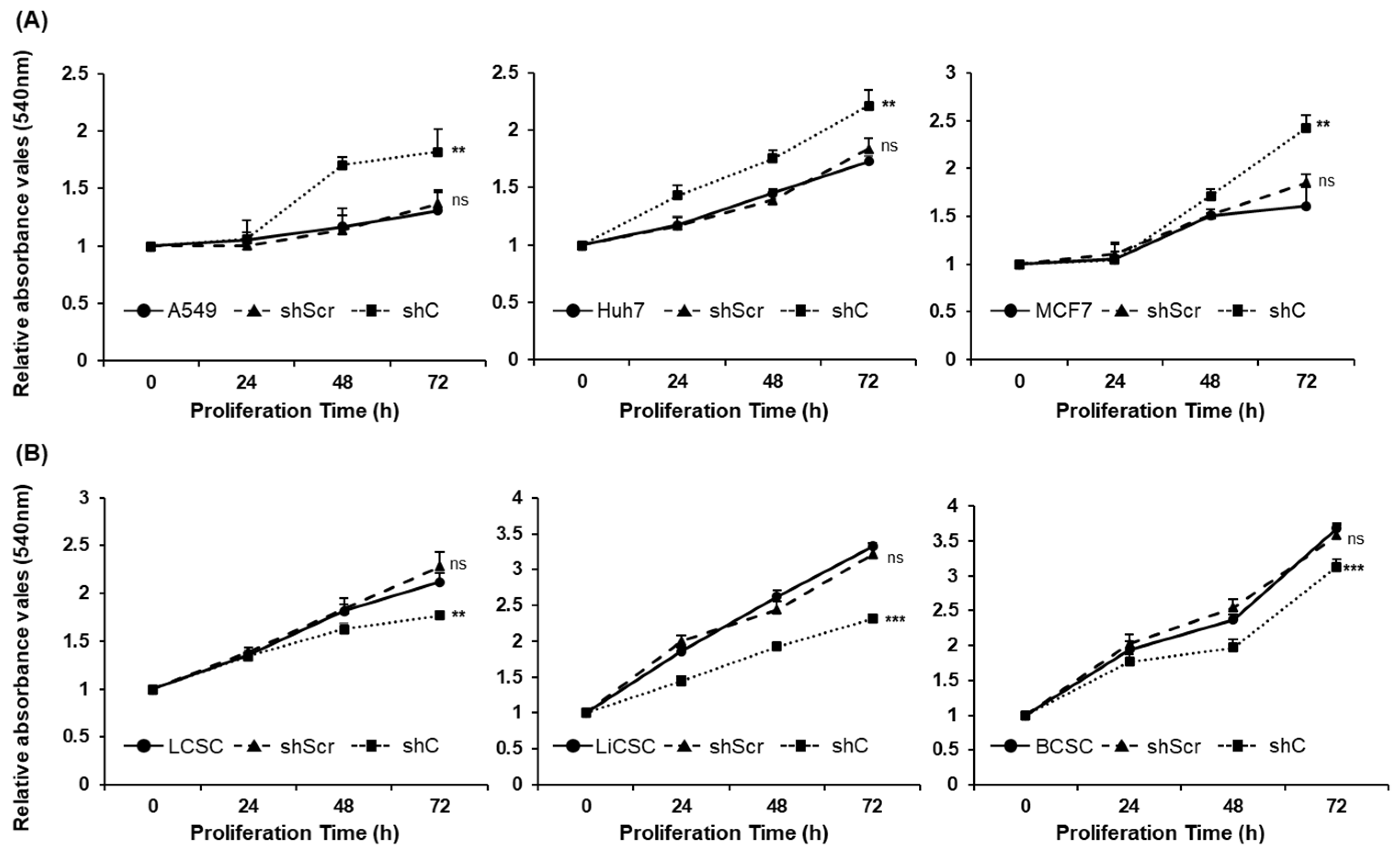
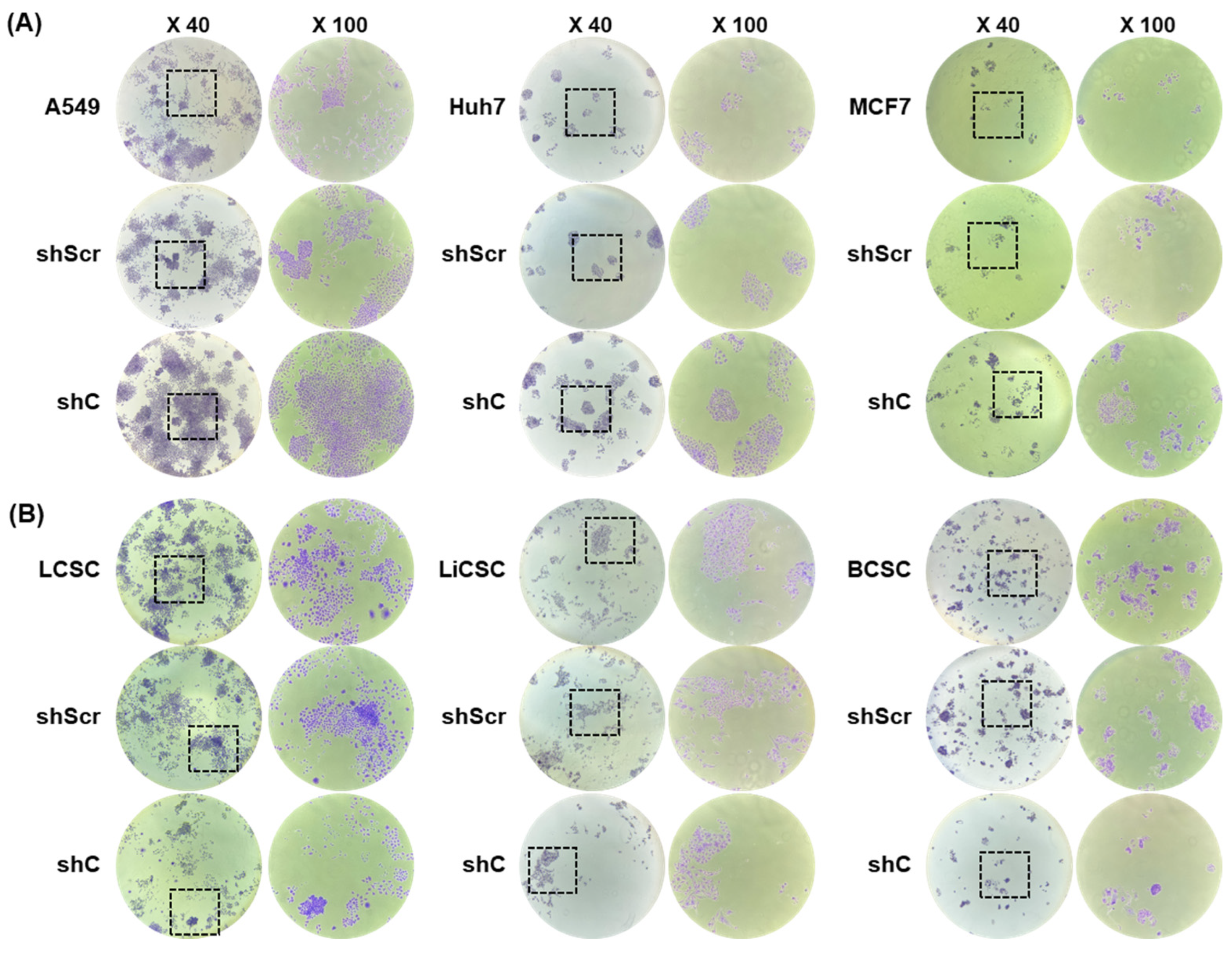
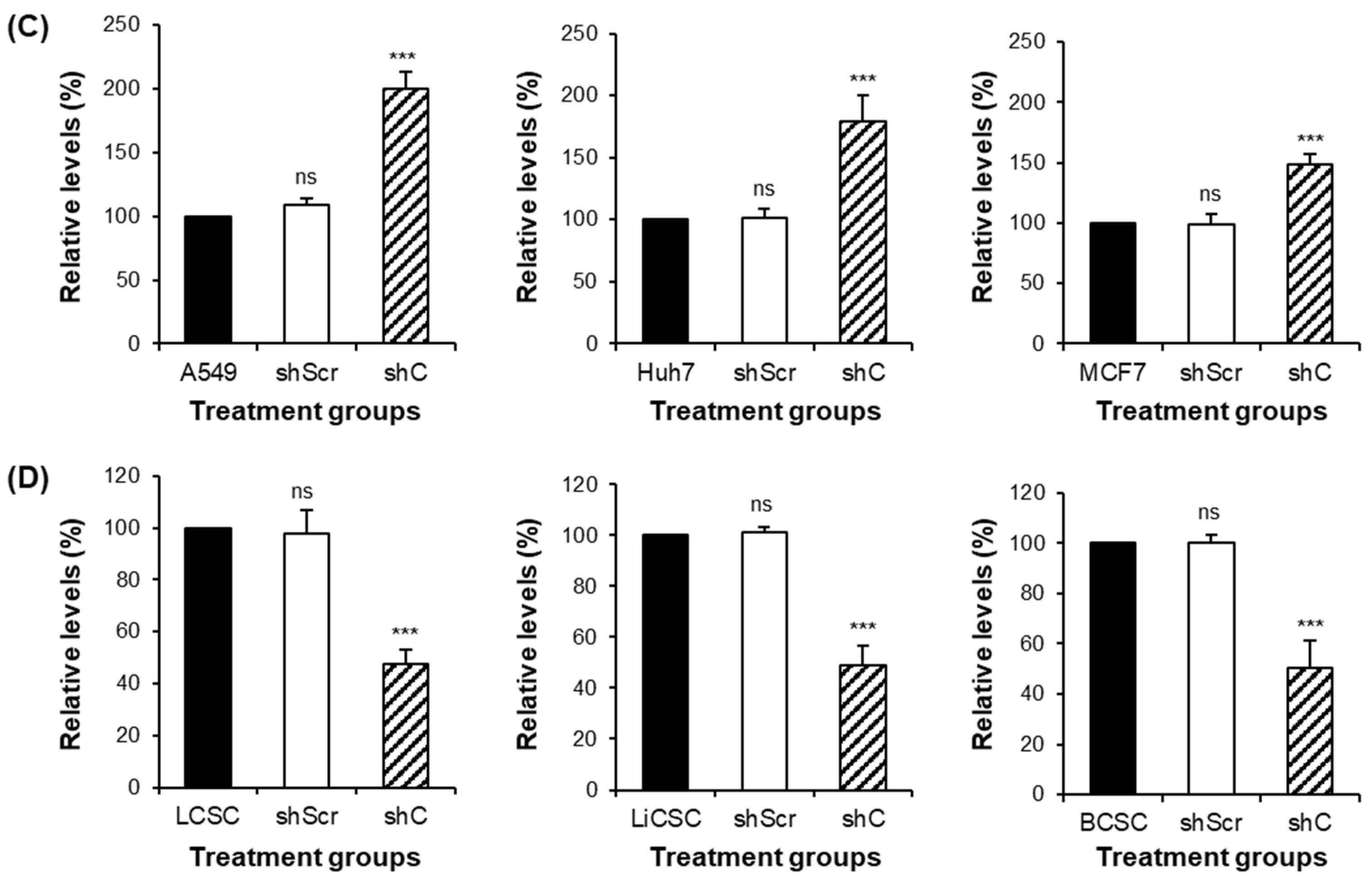
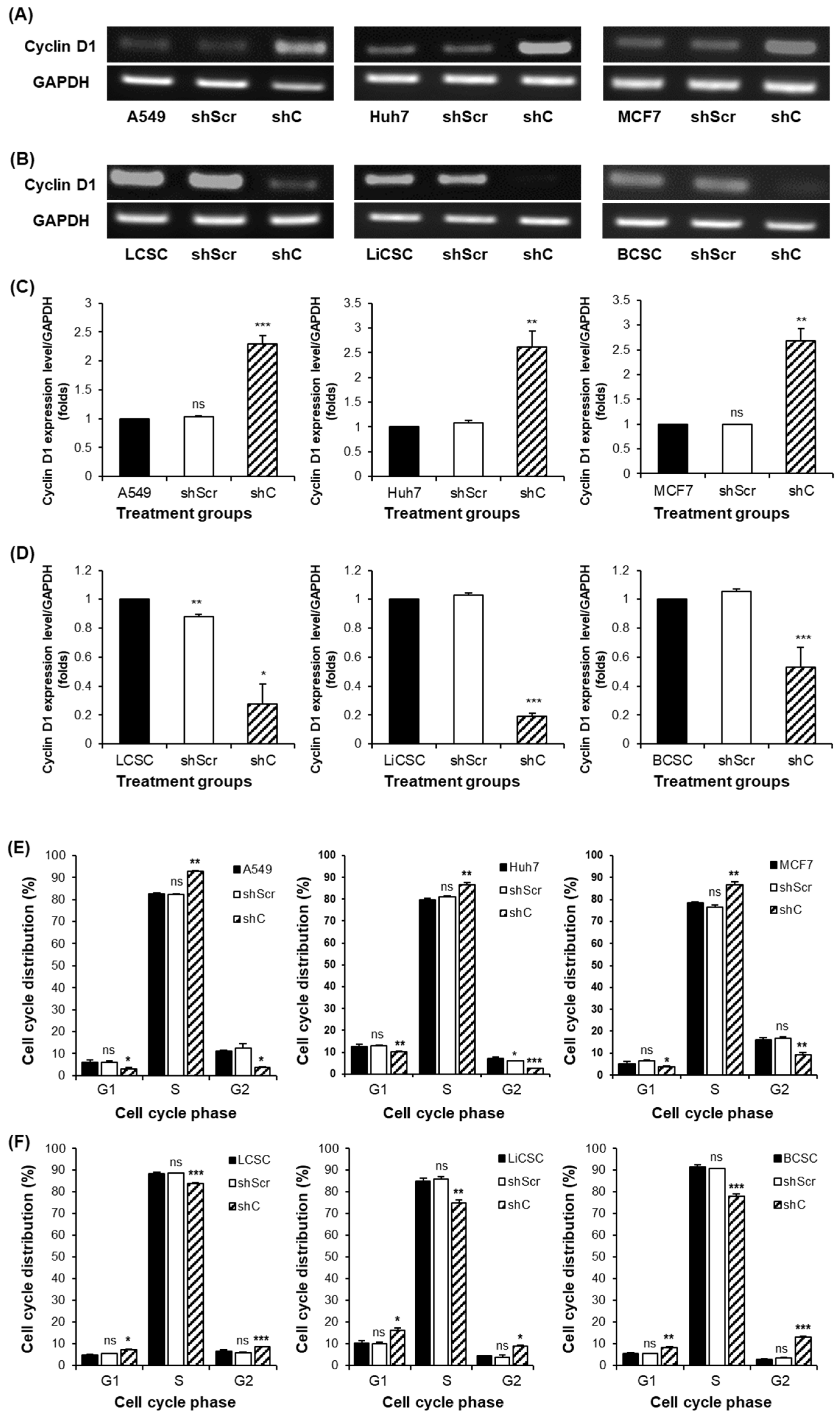
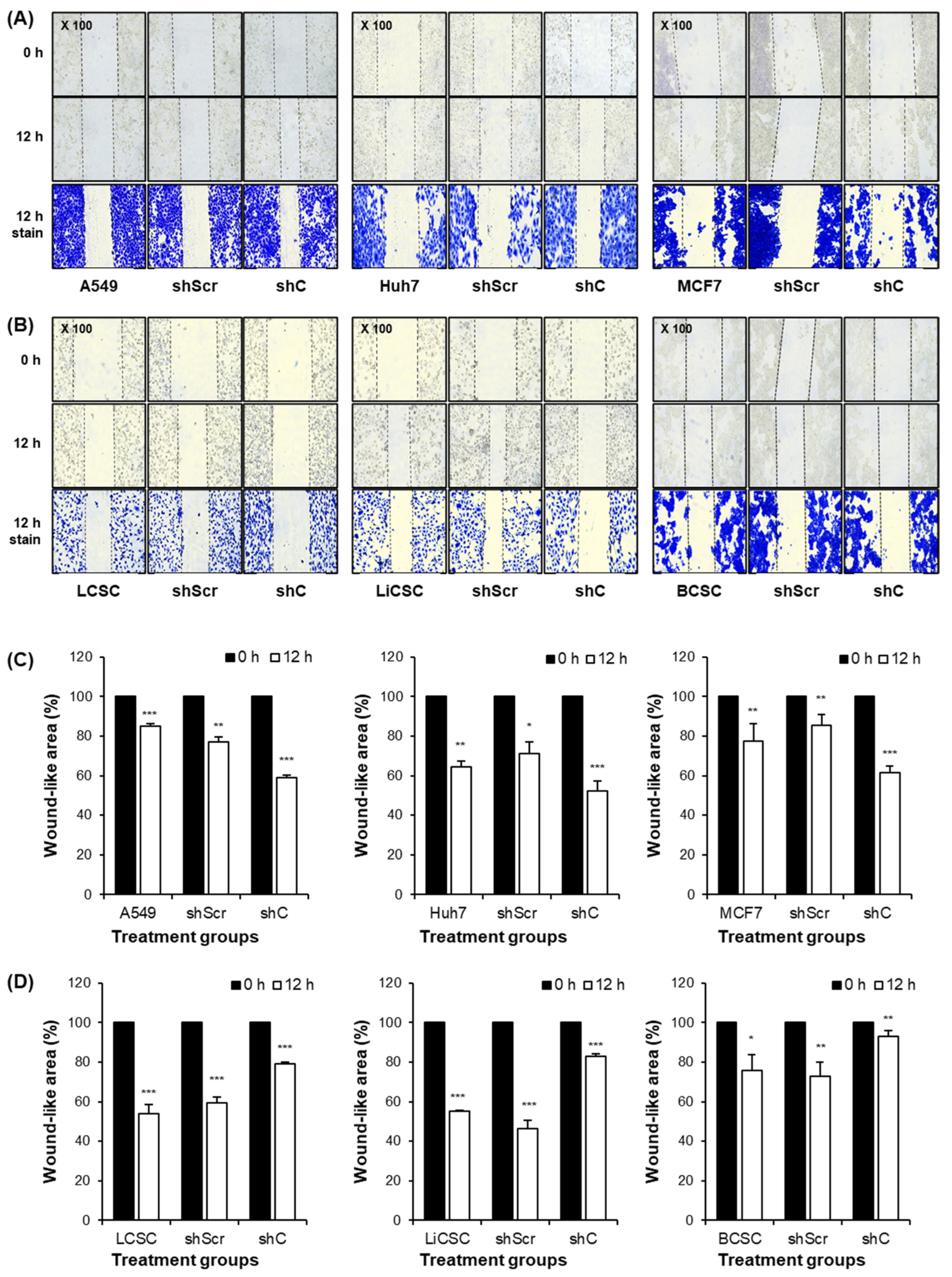
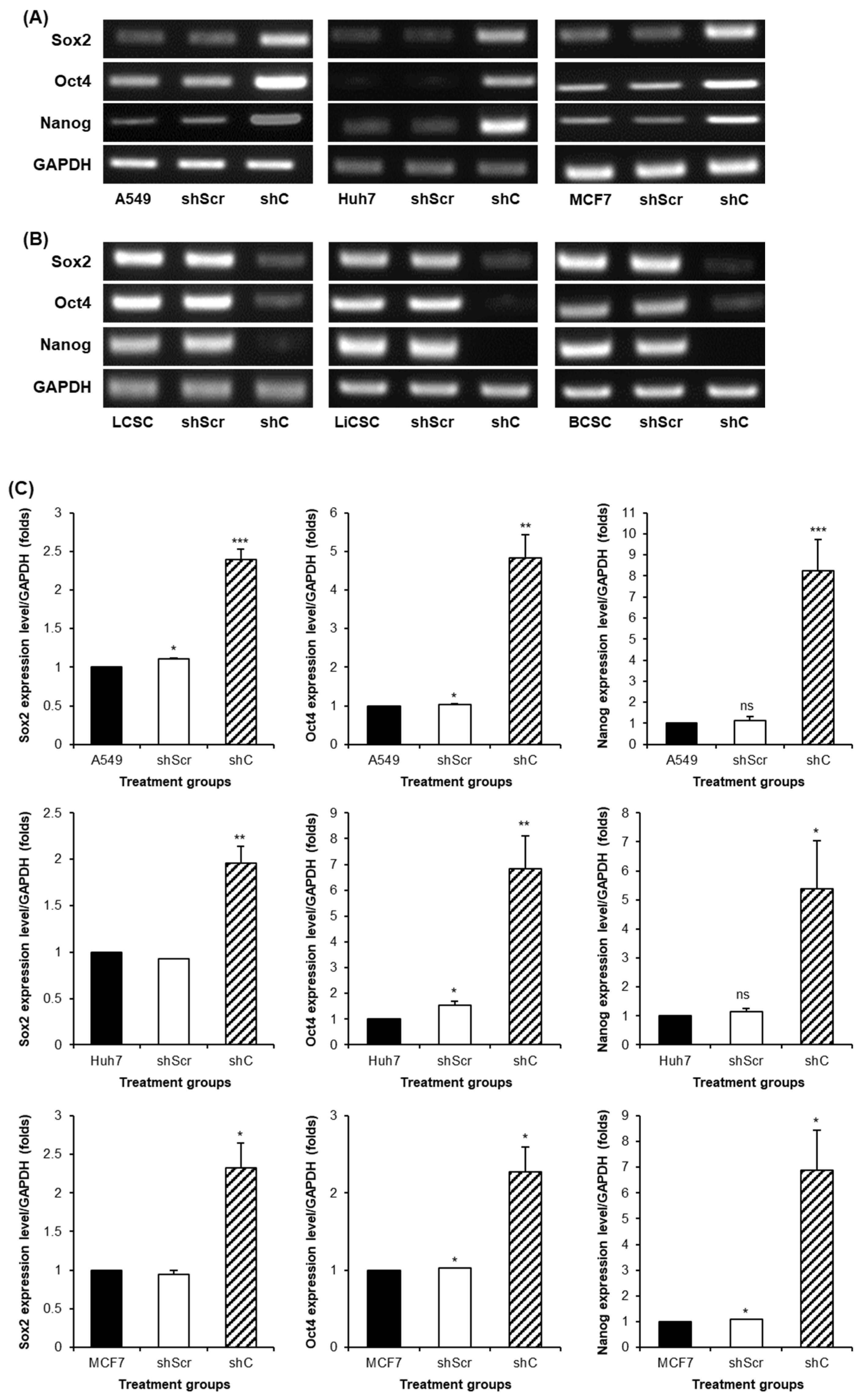
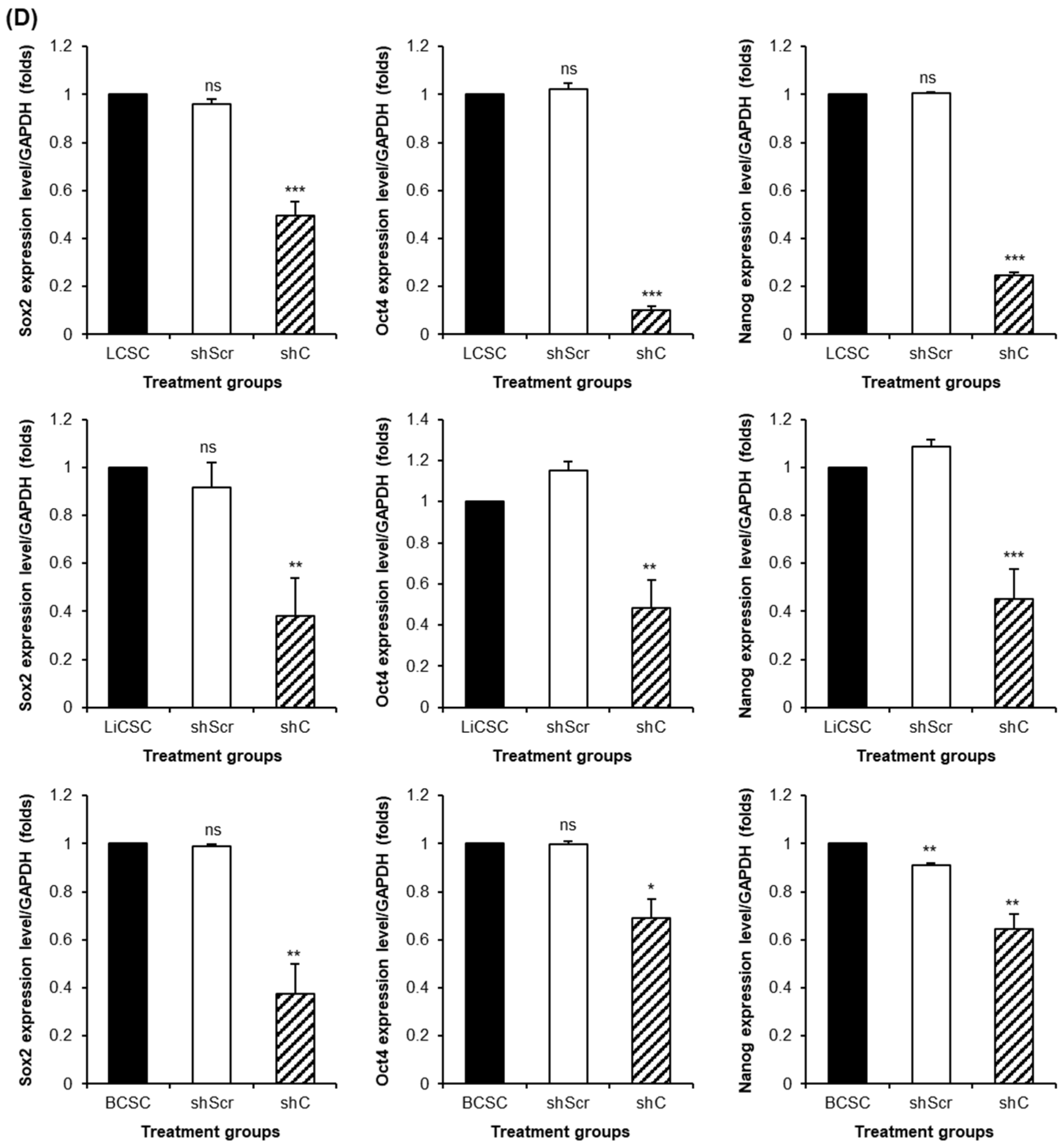
2.3. Biological Functional Assessment through PRKAR1A Knockdown Using shRNA
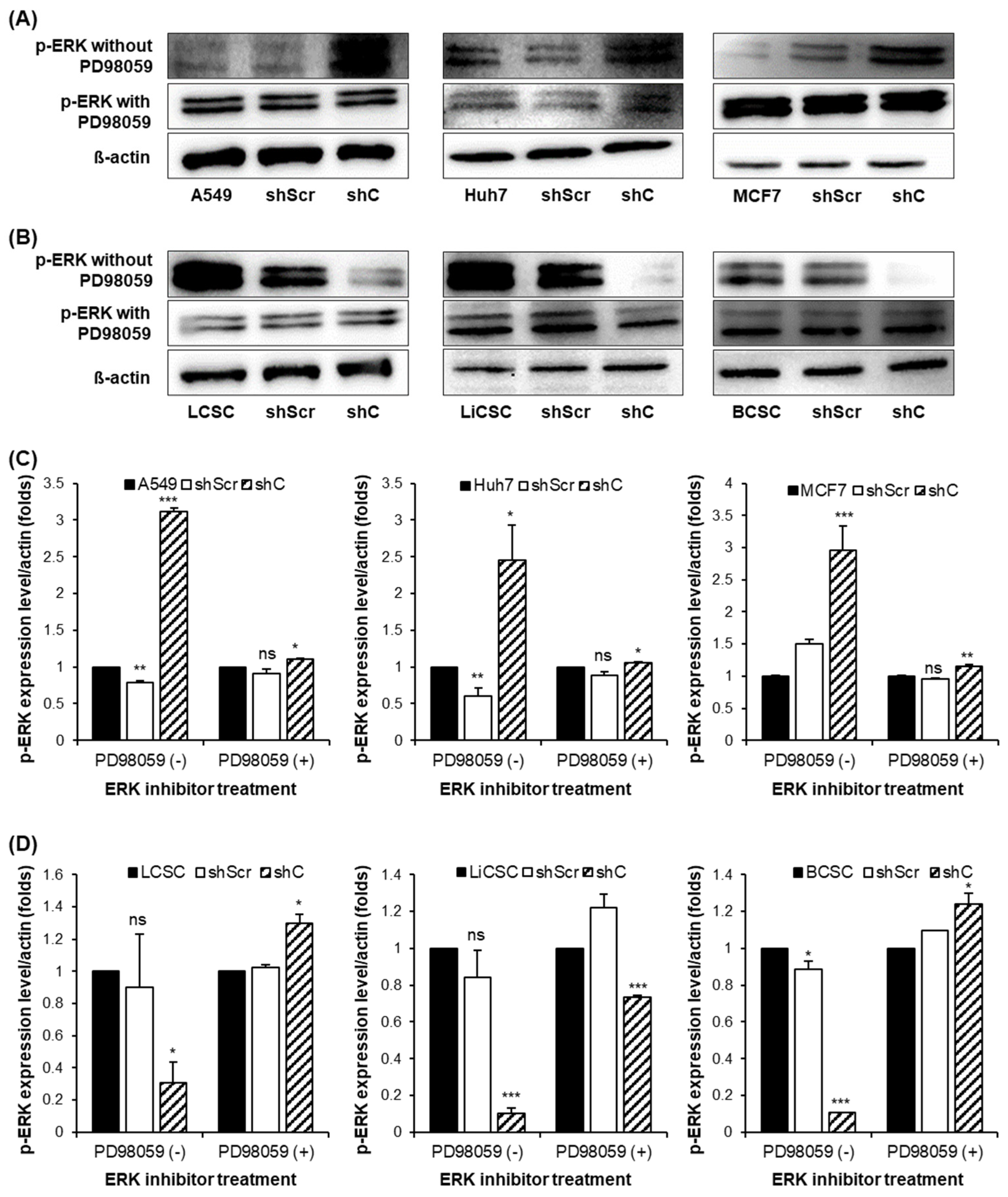
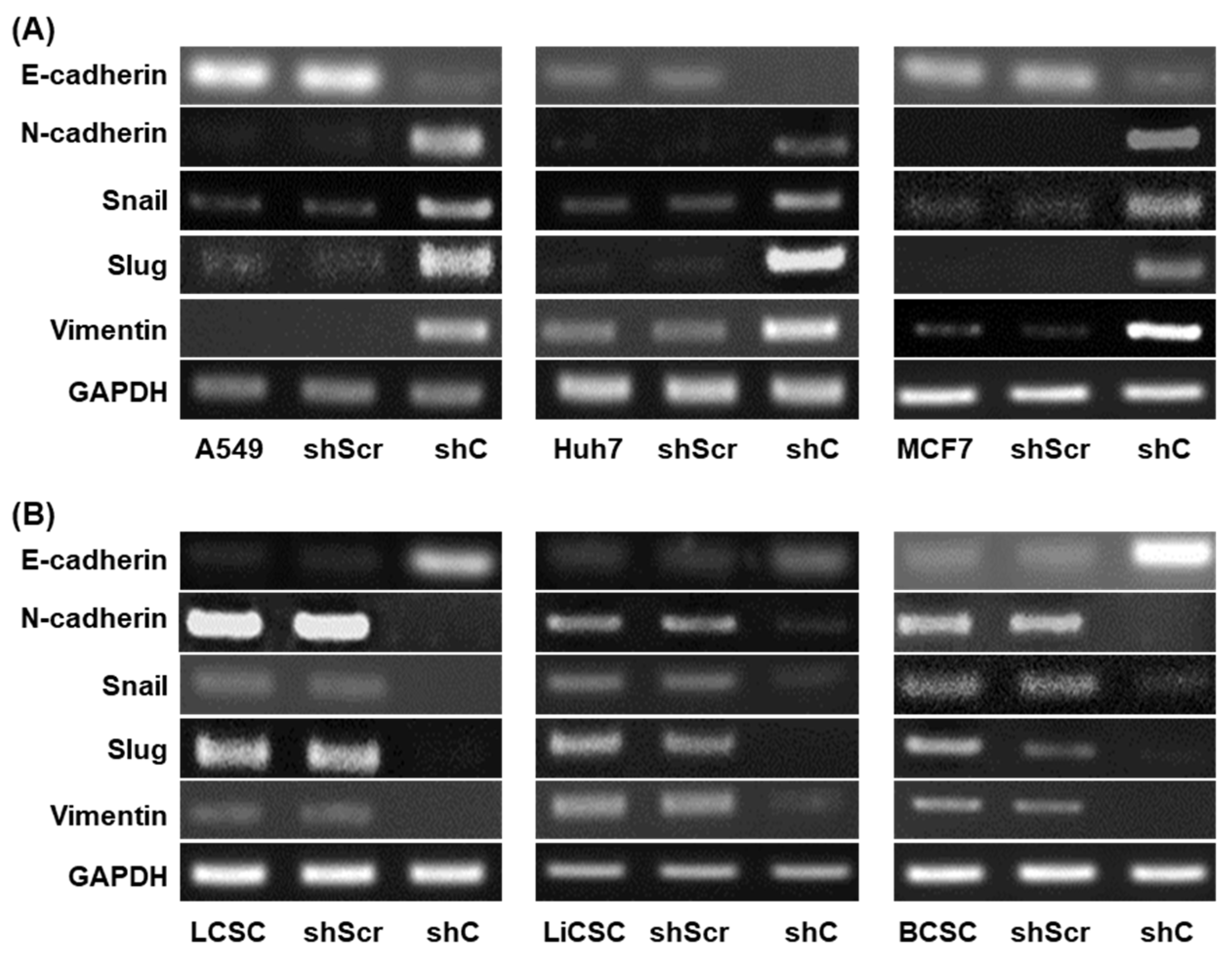
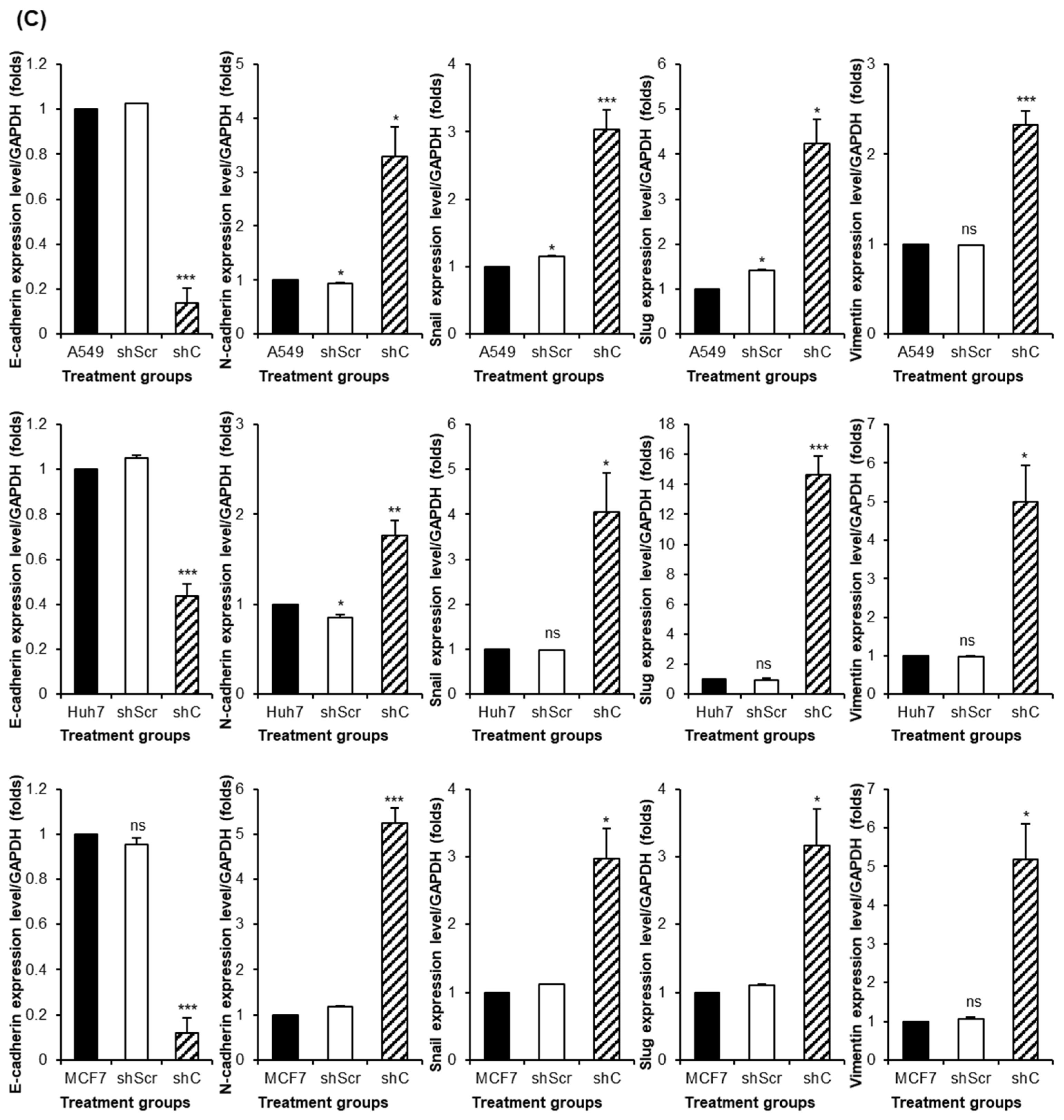
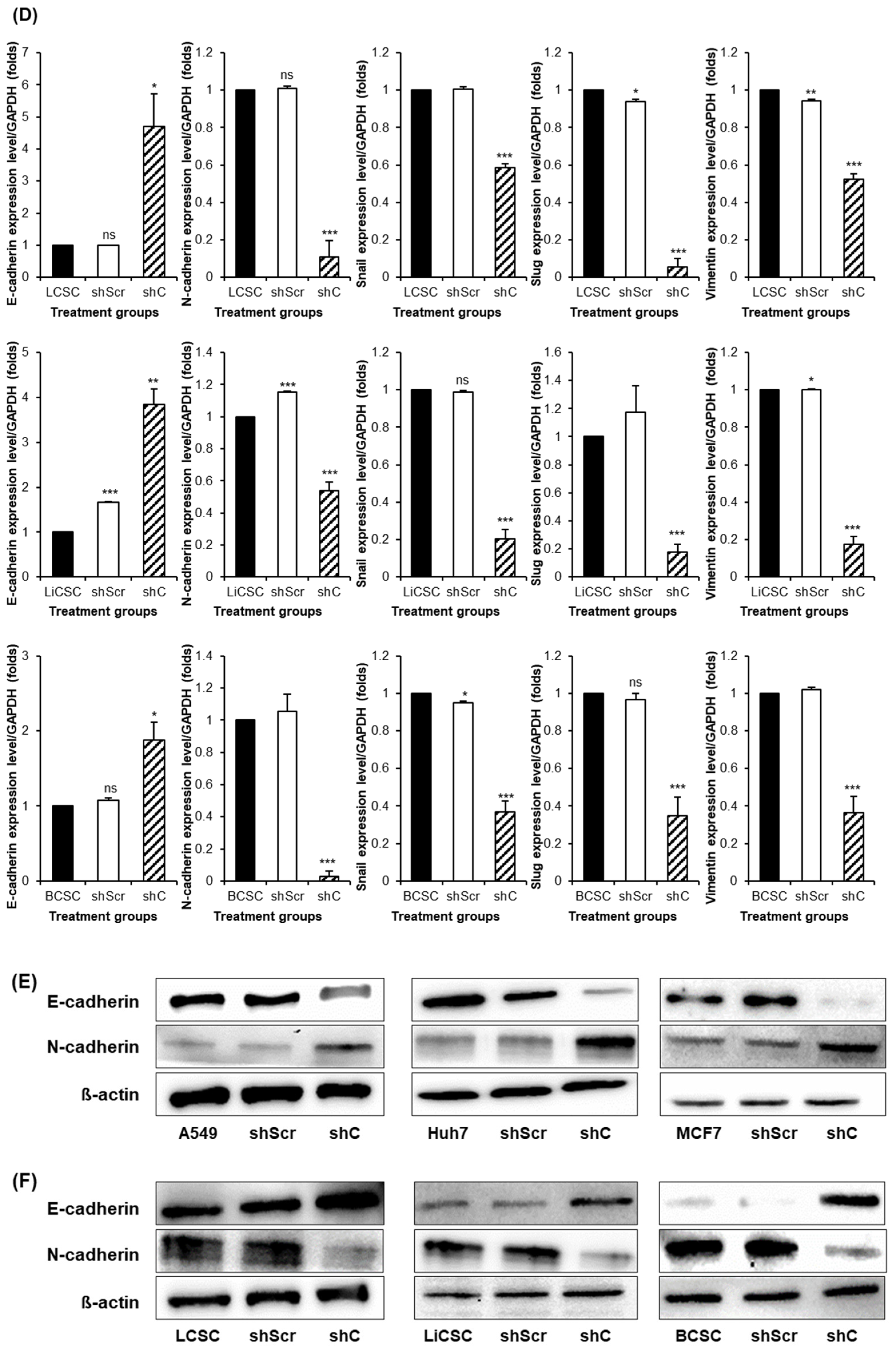
2.4. Regulation of Functionality via the ERK Signaling Pathway and EMT
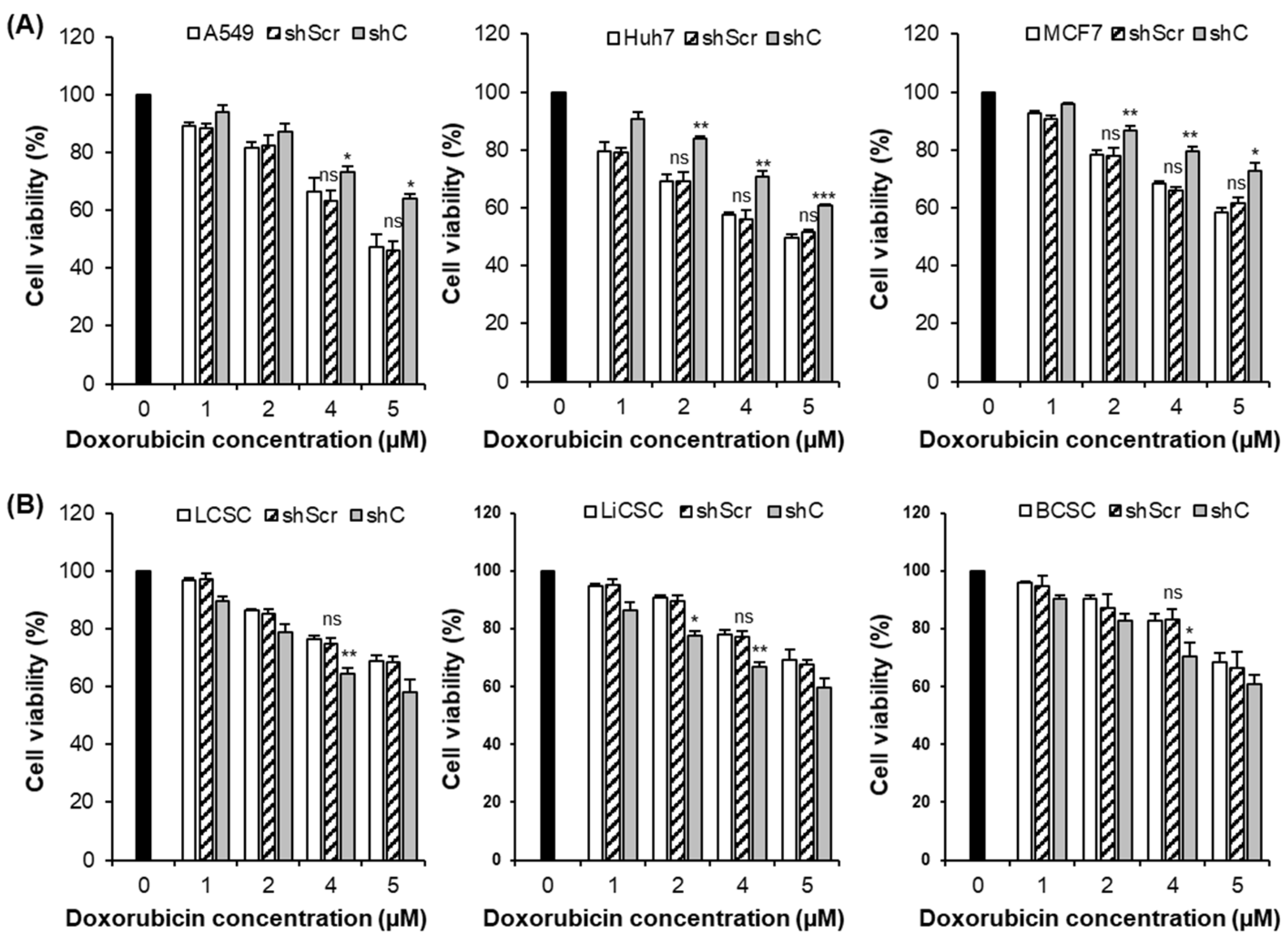
2.5. Downregulation of PRKAR1A Expression Enhanced Sensitivity to Anticancer Agents
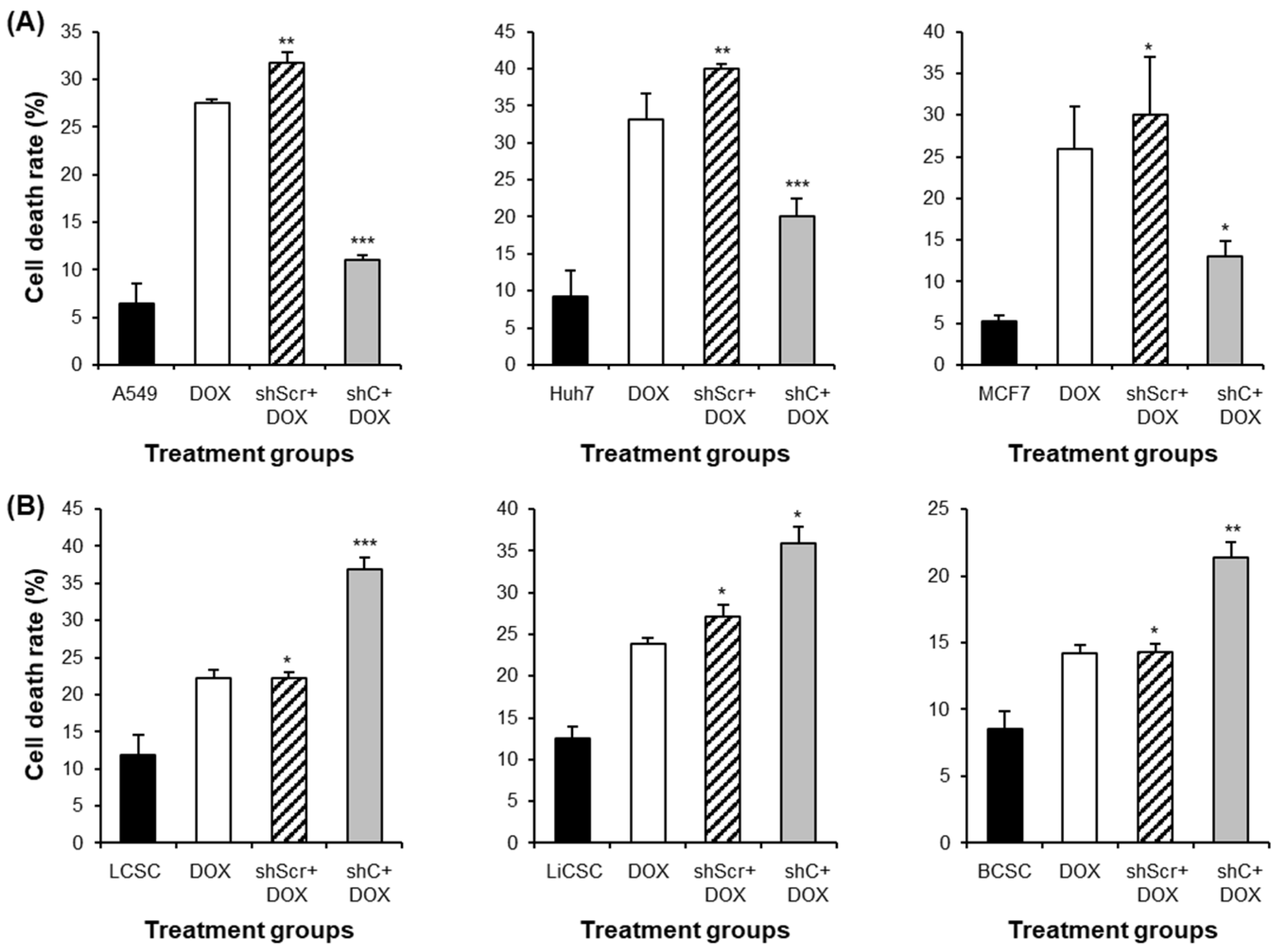
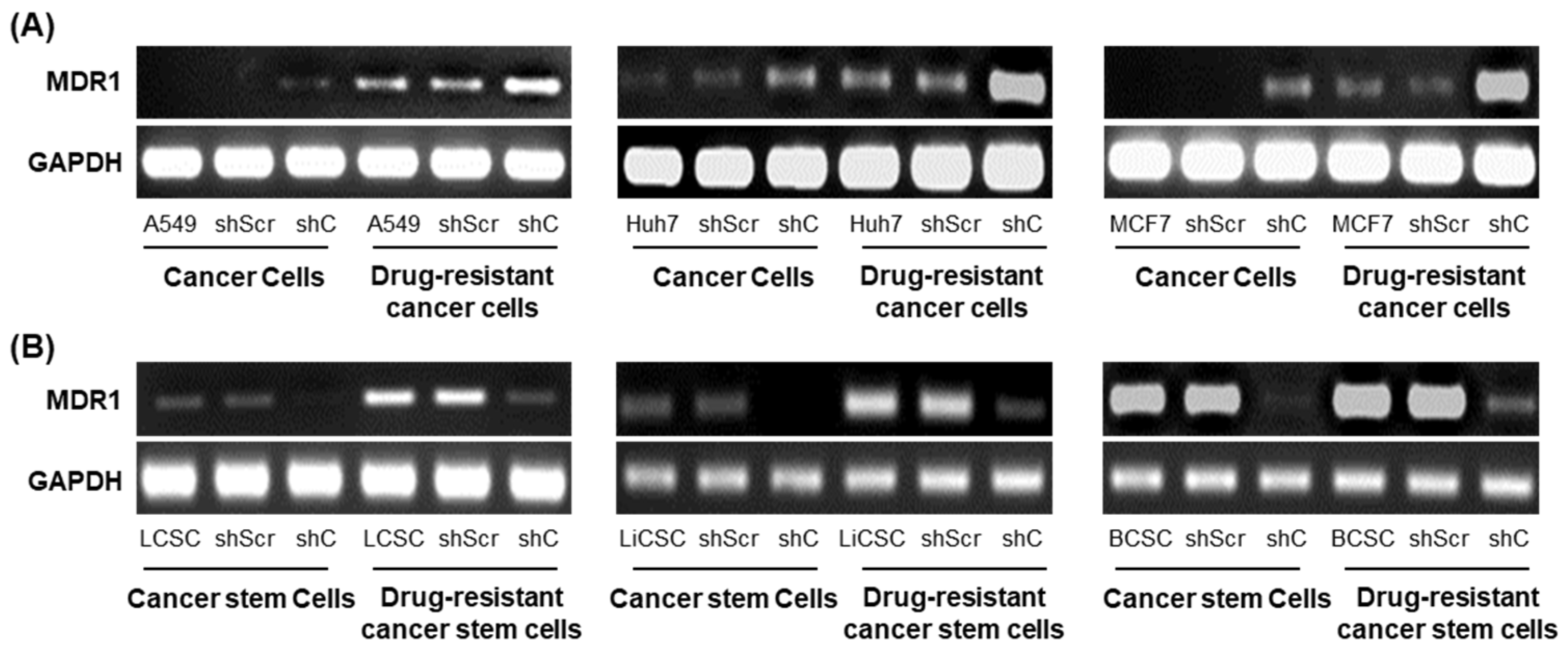
2.6. PRKAR1A shRNA Reduces Drug Resistance
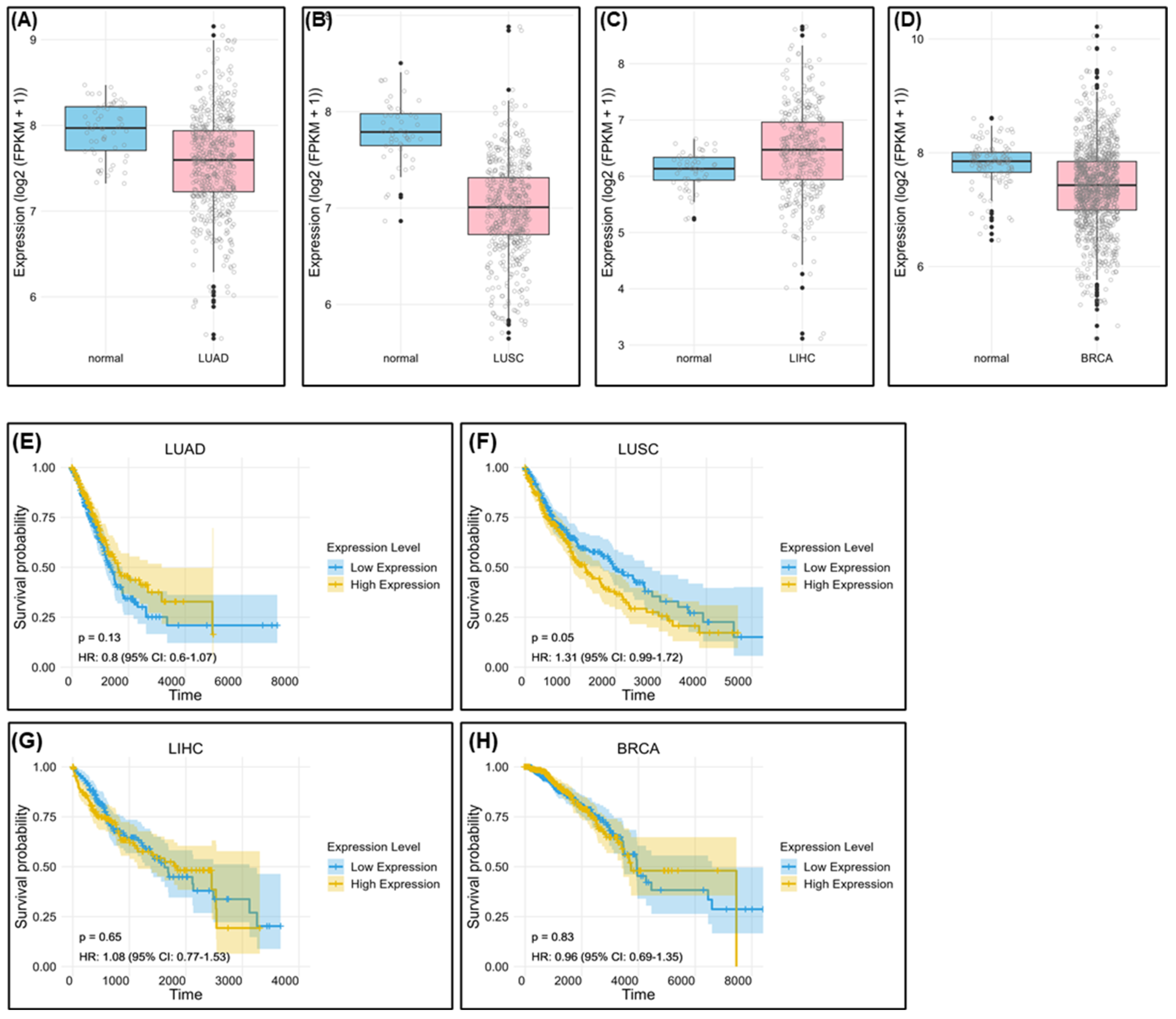
2.7. Bioinformatic Survival Data Based on PRKAR1A Expression in Patients with Cancer
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.1.1. Cancer Cells
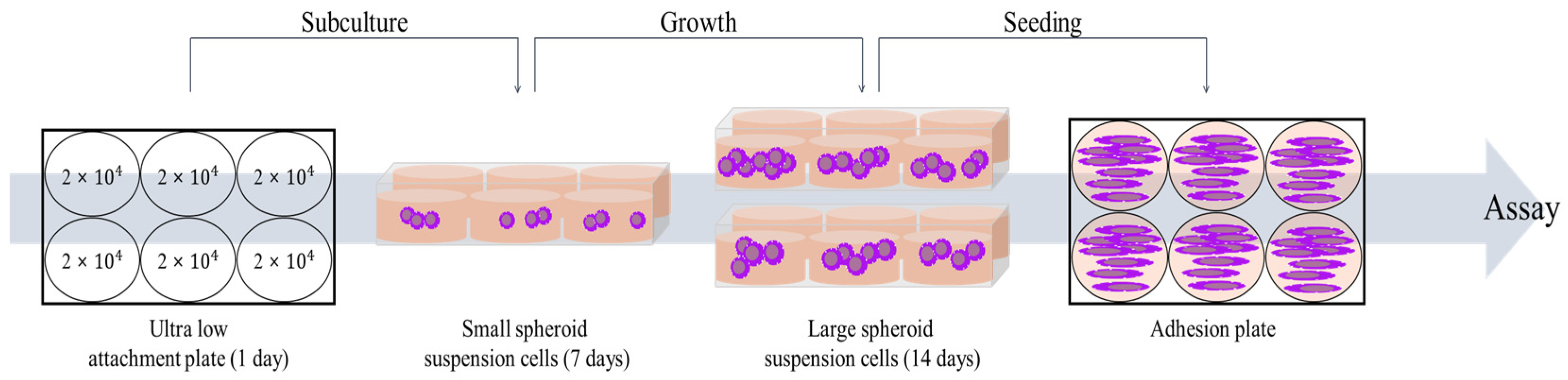
4.1.2. Cancer Stem Cells (CSCs)
4.2. Mass Spectrometry Analysis
4.3. RNA Extraction and Conventional Polymerase Chain Reaction (PCR)
4.4. Transfection
4.5. Cell Proliferation Assay
4.6. Cell Colony Formation Assay and Crystal Violet Stain
4.7. Cell Cycle Assay by Flow Cytometry
4.8. Wound Healing Scratch Assay
4.9. Western Blot Analysis
4.10. Drug Resistance Assay
4.11. Generation of Chemoresistant Cell Lines
4.12. Apoptosis Assay
4.13. Data Acquisition and Preprocessing
4.14. Statistical Analysis of Expression Data
4.15. Survival Analysis
4.16. Software
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer incidence and mortality rates and trends—An update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Alieva, M.; van Rheenen, J.; Broekman, M.L. Potential impact of invasive surgical procedures on primary tumor growth and metastasis. Clin. Exp. Metastasis 2018, 35, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yan, Q.; Liu, S.; Guan, X. Cancer stem cells in hepatocellular carcinoma: Intrinsic and extrinsic molecular mechanisms in stemness regulation. Int. J. Mol. Sci. 2022, 23, 12327. [Google Scholar] [CrossRef] [PubMed]
- Aponte, P.M.; Caicedo, A. Stemness in cancer: Stem cells, cancer stem cells, and their microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef] [PubMed]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.; Lee, S.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment. Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef]
- Ranji, P.; Salmani Kesejini, T.; Saeedikhoo, S.; Alizadeh, A.M. Targeting cancer stem cell-specific markers and/or associated signaling pathways for overcoming cancer drug resistance. Tumor Biol. 2016, 37, 13059–13075. [Google Scholar] [CrossRef]
- Hiom, S.C. Diagnosing cancer earlier: Reviewing the evidence for improving cancer survival. Br. J. Cancer 2015, 112, S1–S5. [Google Scholar] [CrossRef]
- Bradley, C.J.; Given, C.W.; Roberts, C. Disparities in cancer diagnosis and survival. Cancer 2001, 91, 178–188. [Google Scholar] [CrossRef]
- Raymond, A.C.; Gao, B.; Girard, L.; Minna, J.D.; Gomika Udugamasooriya, D. Unbiased peptoid combinatorial cell screen identifies plectin protein as a potential biomarker for lung cancer stem cells. Sci. Rep. 2019, 9, 14954. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Li, Z.; Jiang, X.; Lum, Y.L.; Khin, E.; Lee, N.P.; Wu, G.; Luk, J.M. Osteopontin as potential biomarker and therapeutic target in gastric and liver cancers. World J. Gastroenterol. WJG 2012, 18, 3923. [Google Scholar] [CrossRef] [PubMed]
- Bensalah, K.; Montorsi, F.; Shariat, S.F. Challenges of cancer biomarker profiling. Eur. Urol. 2007, 52, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Rizvi, H.; Preeshagul, I.R.; Egger, J.V.; Hoyos, D.; Bandlamudi, C.; McCarthy, C.G.; Falcon, C.J.; Schoenfeld, A.J.; Arbour, K.C. COVID-19 in patients with lung cancer. Ann. Oncol. 2020, 31, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Zelber-Sagi, S.; Noureddin, M.; Shibolet, O. Lifestyle and hepatocellular carcinoma what is the evidence and prevention recommendations. Cancers 2021, 14, 103. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.; Allan, A.L. Molecular mechanisms of breast cancer metastasis to the lung: Clinical and experimental perspectives. Int. J. Mol. Sci. 2019, 20, 2272. [Google Scholar] [CrossRef] [PubMed]
- Bossis, I.; Stratakis, C.A. Minireview: PRKAR1A: Normal and abnormal functions. Endocrinology 2004, 145, 5452–5458. [Google Scholar] [CrossRef] [PubMed]
- Pitsava, G.; Stratakis, C.A.; Faucz, F.R. PRKAR1A and thyroid tumors. Cancers 2021, 13, 3834. [Google Scholar] [CrossRef]
- Nadella, K.S.; Jones, G.N.; Trimboli, A.; Stratakis, C.A.; Leone, G.; Kirschner, L.S. Targeted deletion of Prkar1a reveals a role for protein kinase A in mesenchymal-to-epithelial transition. Cancer Res. 2008, 68, 2671–2677. [Google Scholar] [CrossRef]
- Loilome, W.; Juntana, S.; Namwat, N.; Bhudhisawasdi, V.; Puapairoj, A.; Sripa, B.; Miwa, M.; Saya, H.; Riggins, G.J.; Yongvanit, P. PRKAR1A is overexpressed and represents a possible therapeutic target in human cholangiocarcinoma. Int. J. Cancer 2011, 129, 34–44. [Google Scholar] [CrossRef]
- Wang, S.; Cheng, Y.; Zheng, Y.; He, Z.; Chen, W.; Zhou, W.; Duan, C.; Zhang, C. PRKAR1A is a functional tumor suppressor inhibiting ERK/Snail/E-cadherin pathway in lung adenocarcinoma. Sci. Rep. 2016, 6, 39630. [Google Scholar] [CrossRef]
- Gentili, C.; Sanfilippo, O.; Silvestrini, R. Cell proliferation and its relationship to clinical features and relapse in breast cancers. Cancer 1981, 48, 974–979. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yu, H.; Kong, Q.; Liu, C.; Tian, Y.; Zeng, X.; Li, D. Effect of ERβ-regulated ERK1/2 signaling on biological behaviors of prostate cancer cells. Am. J. Transl. Res. 2017, 9, 2775. [Google Scholar] [PubMed]
- Hu, X.; Zhai, Y.; Kong, P.; Cui, H.; Yan, T.; Yang, J.; Qian, Y.; Ma, Y.; Wang, F.; Li, H. FAT1 prevents epithelial mesenchymal transition (EMT) via MAPK/ERK signaling pathway in esophageal squamous cell cancer. Cancer Lett. 2017, 397, 83–93. [Google Scholar] [CrossRef]
- Singh, M.; Yelle, N.; Venugopal, C.; Singh, S.K. EMT: Mechanisms and therapeutic implications. Pharmacol. Ther. 2018, 182, 80–94. [Google Scholar] [CrossRef]
- Pradella, D.; Naro, C.; Sette, C.; Ghigna, C. EMT and stemness: Flexible processes tuned by alternative splicing in development and cancer progression. Mol. Cancer 2017, 16, 8. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Malfettone, A.; Soukupova, J. New insights into the crossroads between EMT and stemness in the context of cancer. J. Clin. Med. 2016, 5, 37. [Google Scholar] [CrossRef]
- Manfioletti, G.; Fedele, M. Epithelial–Mesenchymal Transition (EMT) 2021. Int. J. Mol. Sci. 2022, 23, 5848. [Google Scholar] [CrossRef]
- Roy, S.; Sunkara, R.R.; Parmar, M.Y.; Shaikh, S.; Waghmare, S.K. EMT imparts cancer stemness and plasticity: New perspectives and therapeutic potential. Front. Biosci.-Landmark 2020, 26, 238–265. [Google Scholar] [CrossRef]
- Dave, B.; Mittal, V.; Tan, N.M.; Chang, J.C. Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res. 2012, 14, 202. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Kohno, M.; Pouyssegur, J. Targeting the ERK signaling pathway in cancer therapy. Ann. Med. 2006, 38, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Li, Y.; Zhang, X. Stemness-related markers in cancer. Cancer Transl. Med. 2017, 3, 87. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S.; Wang, M.; Chou, Y.; Chen, C.; Hong, C.; Hsieh, W.; Chang, H.; Chen, Y.; Lin, T.; Hsu, H. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell–like properties and epithelial–mesenchymal transdifferentiation. Cancer Res. 2010, 70, 10433–10444. [Google Scholar] [CrossRef]
- Sun, C.; Sun, L.; Li, Y.; Kang, X.; Zhang, S.; Liu, Y. Sox2 expression predicts poor survival of hepatocellular carcinoma patients and it promotes liver cancer cell invasion by activating Slug. Med. Oncol. 2013, 30, 503. [Google Scholar] [CrossRef]
- Chen, Y.; Shi, L.; Zhang, L.; Li, R.; Liang, J.; Yu, W.; Sun, L.; Yang, X.; Wang, Y.; Zhang, Y. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J. Biol. Chem. 2008, 283, 17969–17978. [Google Scholar] [CrossRef]
- Rasti, A.; Mehrazma, M.; Madjd, Z.; Abolhasani, M.; Saeednejad Zanjani, L.; Asgari, M. Co-expression of cancer stem cell markers OCT4 and NANOG predicts poor prognosis in renal cell carcinomas. Sci. Rep. 2018, 8, 11739. [Google Scholar] [CrossRef]
- Smith, J.; Field, M.; Sugaya, K. Suppression of NANOG Expression Reduces Drug Resistance of Cancer Stem Cells in Glioblastoma. Genes 2023, 14, 1276. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, M.; Li, L.; Wang, X.; Han, S.; Zhao, J.; Dong, Y.; Ahmad, M.; Li, L.; Zhang, X. EMT and cancer cell stemness associated with chemotherapeutic resistance in esophageal cancer. Front. Oncol. 2021, 11, 672222. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Ajani, J.A.; Song, S. Drug resistance and Cancer stem cells. Cell Commun. Signal. 2021, 19, 19. [Google Scholar] [CrossRef]
- Elliott, A.; Adams, J.; Al-Hajj, M. The ABCs of cancer stem cell drug resistance. IDrugs Investig. Drugs J. 2010, 13, 632–635. [Google Scholar]
- Dean, M. ABC transporters, drug resistance, and cancer stem cells. J. Mammary Gland Biol. Neoplasia 2009, 14, 3–9. [Google Scholar] [CrossRef]
- Koh, E.; Kim, K.; Park, H.; Kim, J.; Kim, P. Active targeting of versatile nanocomplex using the novel biomarker of breast cancer stem cells. Int. J. Mol. Sci. 2022, 24, 685. [Google Scholar] [CrossRef]
- Koh, E.; You, J.; Jung, S.; Kim, P. Biological functions and identification of novel biomarker expressed on the surface of breast cancer-derived cancer stem cells via proteomic analysis. Mol. Cells 2020, 43, 384. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| GAPDH | AGGGCTGCTTTTAACTCTGGT | CCCCACTTGATTTTGGAGGGA |
| PRKAR1A | GCAGCCACTGTCAAAGCAAA | GGTTCTCCCTGCACCACAAT |
| E-cadherin | GCTTTGACGCCGAGAGCTA | CTTTGTCGACCGGTGCAATC |
| N-cadherin | AGGCTTCTGGTGAAATCGCA | TGGAAAGCTTCTCACGGCAT |
| Snail | GCTGCAGGACTCTAATCCAGAGTT | GACAGAGTCCCAGATGAGCATTG |
| Slug | AGATGCATATTCGGACCCAC | CCTCATGTTTGTGCAGGAGA |
| Vimentin | CGAAAACACCCTGCAATCTT | TCCAGCTTCCTGTAGGT |
| Sox2 | GCTACAGCATGATGCAGGACCA | TCTGCGAGCTGGTCATGGAGTT |
| Oct4 | CCTGAAGCAGAAGAGGATCACC | AAAGCGGCAGATGGTCGTTTGG |
| Nanog | CTCCAACATCCTGAACCTCAGC | CGTCACACCATTGCTATTCTTCG |
| Cyclin D1 | AGCTGTGCATCTACACCGAC | GAAATCGTGCGGGGTCATTG |
| MDR1 | CCCATCATTGCAATAGCAGG | GTTCAAACTTCTGCTCCTGA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baek, J.-W.; Nam, A.-R.; Kim, K.; Kim, P.-H. Dualistic Effects of PRKAR1A as a Potential Anticancer Target in Cancer Cells and Cancer-Derived Stem Cells. Int. J. Mol. Sci. 2024, 25, 2876. https://doi.org/10.3390/ijms25052876
Baek J-W, Nam A-R, Kim K, Kim P-H. Dualistic Effects of PRKAR1A as a Potential Anticancer Target in Cancer Cells and Cancer-Derived Stem Cells. International Journal of Molecular Sciences. 2024; 25(5):2876. https://doi.org/10.3390/ijms25052876
Chicago/Turabian StyleBaek, Joong-Won, A-Reum Nam, Kyunggon Kim, and Pyung-Hwan Kim. 2024. "Dualistic Effects of PRKAR1A as a Potential Anticancer Target in Cancer Cells and Cancer-Derived Stem Cells" International Journal of Molecular Sciences 25, no. 5: 2876. https://doi.org/10.3390/ijms25052876
APA StyleBaek, J.-W., Nam, A.-R., Kim, K., & Kim, P.-H. (2024). Dualistic Effects of PRKAR1A as a Potential Anticancer Target in Cancer Cells and Cancer-Derived Stem Cells. International Journal of Molecular Sciences, 25(5), 2876. https://doi.org/10.3390/ijms25052876