
Molecular Mechanisms Linking Osteoarthritis and Alzheimer’s Disease: Shared Pathways, Mechanisms and Breakthrough Prospects

{kind=link}
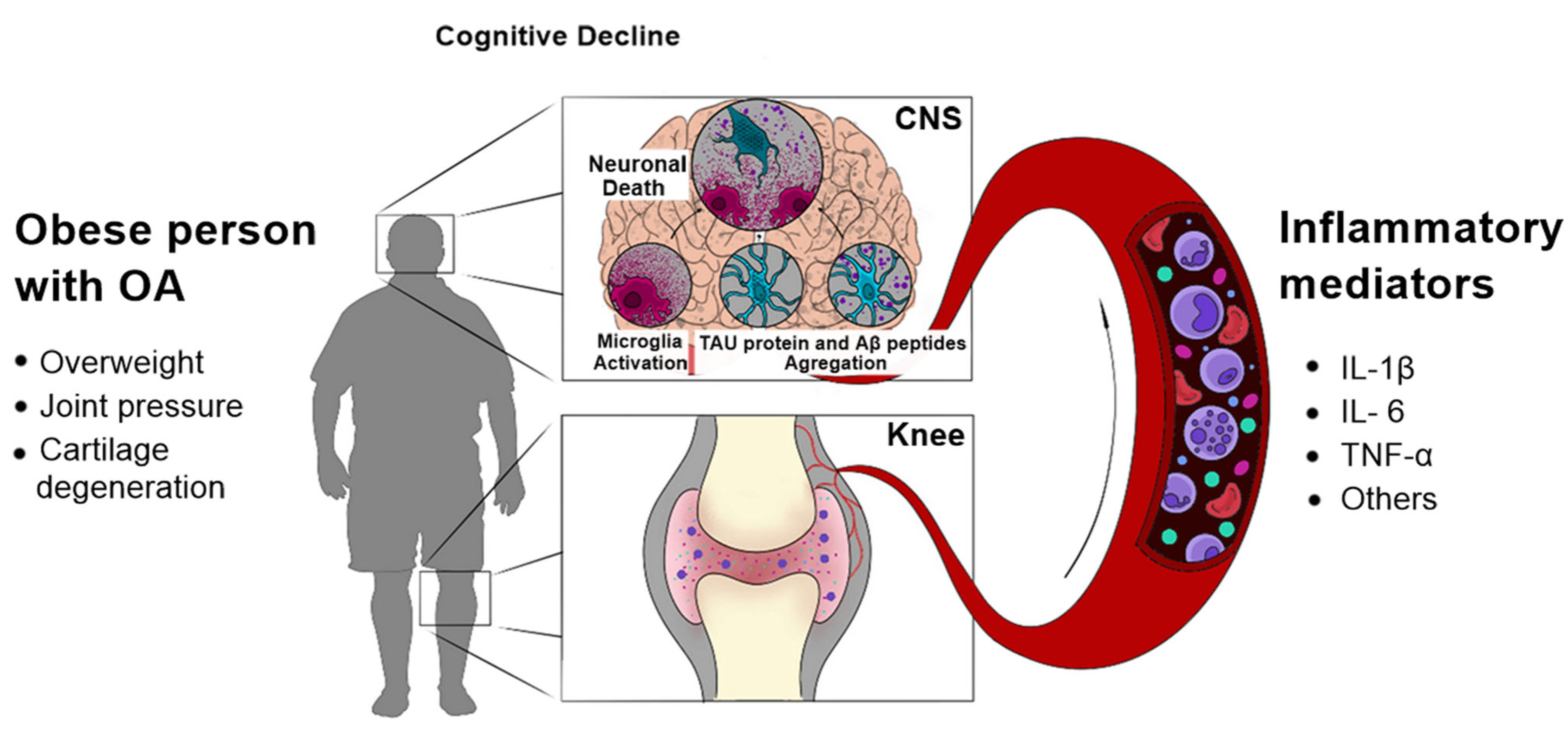
Abstract
1. Alzheimer’s Disease: Pathogenesis and Epidemiology
1.1. Pathogenesis
1.1.1. Neuroinflammation
1.1.2. Amyloid-β Hypothesis
1.1.3. TAU Pathology
1.1.4. Dysregulated miRNAs
1.1.5. Vascular Factors
1.1.6. APOE and Amyloid Metabolism in the Brain
1.1.7. Trisomy 21—Down’s Syndrome
1.1.8. Genetics in EOFAD and LOAD
1.2. Epidemiology of AD
1.2.1. Global Prevalence
1.2.2. Global Variation
1.2.3. Gender Disparities
2. Peripheral Inflammation and Neurodegenerative Diseases: Insights into AD Pathogenesis
2.1. The Role of Inflammation in AD Pathogenesis
Systemic Inflammation and Blood–Brain Barrier Dysfunction
2.2. Peripheral Inflammation and AD Risk Factors
2.2.1. Genetic Susceptibility
2.2.2. Environmental Factors
2.2.3. Activation of Microglia
2.2.4. Peripheral Inflammation and Infections
2.3. Therapeutic Implications
2.3.1. Immunomodulation
2.3.2. Anti-Microbial/Pathogenic Medications
2.3.3. Precision Medicine
3. Pathogenesis and Risk Factors of Osteoarthritis and Possible Interplay with AD
3.1. Pathogenesis
3.1.1. Definition/Introduction
3.1.2. Factors Contributing to the Pathogenesis of OA:
The Inflammatory Process in OA
Articular Cartilage Degeneration
Mechanical Stress
3.2. Modifiable and Non-Modifiable Risk Factors for Osteoarthritis Development
3.2.1. Obesity
3.2.2. Joint Overuse and Injuries
3.2.3. Hormones and Arthritis
3.2.4. Aging
3.2.5. Race
4. AD Development and OA: An Extensive Insight
4.1. The OA–Dementia Link
4.1.1. Animal Experimental Models
4.1.2. Biological Mechanisms
Inflammation as A Common Theme
The Role of APOE Variants
Common miRNA Alterations in AD and OA
4.1.3. Epidemiological Insights, Observational Studies, Systematic Reviews and Meta-Analysis
5. Perspectives on Potential Therapeutic and Preventive Strategies by Targeting Common Pathways between OA and AD
5.1. Targeting Inflammation
5.1.1. NSAIDs
5.1.2. Disease-Modifying Antirheumatic Drugs (DMARDs)—Immunosuppressive and Modulatory Agents
5.2. Disease-Modifying Osteoarthritis Drugs (DMOADs)
5.3. Statins
5.4. Glucocorticoids
5.5. Molecular Approaches
5.6. Personalized Interventions—Precision Medicine
6. Preliminary Conclusions
6.1. Unraveling the Interplay between OA and AD
6.1.1. Shared Pathogenic Mechanisms
6.1.2. Epidemiological Insights
6.1.3. Biological Insights
6.1.4. Prospective Therapeutic and Preventive Strategies
6.1.5. Precision Medicine and Genetic Approaches
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 1999, 399, A23–A31. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012, 2012, 756357. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 1–8. [Google Scholar] [CrossRef]
- Madadi, S.; Schwarzenbach, H.; Saidijam, M.; Mahjub, R.; Soleimani, M. Potential microRNA-related targets in clearance pathways of amyloid-β: Novel therapeutic approach for the treatment of Alzheimer’s disease. Cell Biosci. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Lu, H.C. microRNAs in neurodegeneration: Current findings and potential impacts. J. Alzheimer’s Dis. Park. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Brennan, S.; Keon, M.; Liu, B.; Su, Z.; Saksena, N.K. Panoramic visualization of circulating microRNAs across neurodegenerative diseases in humans. Mol. Neurobiol. 2019, 56, 7380–7407. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.; Magdy, R. MicroRNAs in central nervous system disorders: Current advances in pathogenesis and treatment. Egypt. J. Neurol. Psychiatry Neurosurg. 2021, 57, 1–11. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, F.; Guan, Y.; Meng, F.; Zhao, Z.; Su, Q.; Bao, W.; Wang, X.; Zhao, J.; Huo, Z.; et al. The biogenesis of miRNAs and their role in the development of amyotrophic lateral sclerosis. Cells 2022, 11, 572. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Jin, L.; Zhang, F.; Sarnow, P.; Kay, M.A. Biological basis for restriction of microRNA targets to the 3′ untranslated region in mammalian mRNAs. Nat. Struct. Mol. Biol. 2009, 16, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Zoltowska, K.M.; Laskowska-Kaszub, K.; Wojda, U. microRNA diagnostic panel for Alzheimer’s disease and epigenetic trade-off between neurodegeneration and cancer. Ageing Res. Rev. 2019, 49, 125–143. [Google Scholar] [CrossRef] [PubMed]
- Gentile, G.; Morello, G.; La Cognata, V.; Guarnaccia, M.; Conforti, F.L.; Cavallaro, S. Dysregulated miRNAs as biomarkers and therapeutical targets in neurodegenerative diseases. J. Pers. Med. 2022, 12, 770. [Google Scholar] [CrossRef] [PubMed]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimer’s Dis. 2008, 14, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; Huang, Q.; Hu, Y.; Stromberg, A.J.; Nelson, P.T. Patterns of microRNA expression in normal and early Alzheimer’s disease human temporal cortex: White matter versus gray matter. Acta Neuropathol. 2011, 121, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Sethi, P.; Lukiw, W.J. Micro-RNA abundance and stability in human brain: Specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci. Lett. 2009, 459, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, P.B.; Scuteri, A.; Black, S.E.; DeCarli, C.; Greenberg, S.M.; Iadecola, C.; Launer, L.J.; Laurent, S.; Lopez, O.L.; Nyenhuis, D.; et al. Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011, 42, 2672–2713. [Google Scholar] [CrossRef]
- MDorrance, A.; Matin, N.; WPires, P. The effects of obesity on the cerebral vasculature. Curr. Vasc. Pharmacol. 2014, 12, 462–472. [Google Scholar] [CrossRef]
- Payabvash, S.; Souza, L.C.; Wang, Y.; Schaefer, P.W.; Furie, K.L.; Halpern, E.F.; Gonzalez, R.G.; Lev, M.H. Regional ischemic vulnerability of the brain to hypoperfusion: The need for location specific computed tomography perfusion thresholds in acute stroke patients. Stroke 2011, 42, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kåreholt, I.; Winblad, B.; Helkala, E.L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.T.; Tan, L.; Hardy, J. Apolipoprotein E in Alzheimer’s disease: An update. Annu. Rev. Neurosci. 2014, 37, 79–100. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. The genetic epidemiology of neurodegenerative disease. J. Clin. Investig. 2005, 115, 1449–1457. [Google Scholar] [CrossRef]
- Guerreiro, R.J.; Lohmann, E.; Brás, J.M.; Gibbs, J.R.; Rohrer, J.D.; Gurunlian, N.; Dursun, B.; Bilgic, B.; Hanagasi, H.; Gurvit, H.; et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia–like syndrome without bone involvement. JAMA Neurol. 2013, 70, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Cruchaga, C.; Nowotny, P.; Kauwe, J.S.; Ridge, P.G.; Mayo, K.; Bertelsen, S.; Hinrichs, A.; Fagan, A.M.; Holtzman, D.M.; Morris, J.C.; et al. Association and expression analyses with single-nucleotide polymorphisms in TOMM40 in Alzheimer disease. Arch. Neurol. 2011, 68, 1013–1019. [Google Scholar] [CrossRef]
- Rogaeva, E.; Meng, Y.; Lee, J.H.; Gu, Y.; Kawarai, T.; Zou, F.; Katayama, T.; Baldwin, C.T.; Cheng, R.; Hasegawa, H.; et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 2007, 39, 168–177. [Google Scholar] [CrossRef]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Norton, S.; Matthews, F.E.; Barnes, D.E.; Yaffe, K.; Brayne, C. Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol. 2014, 13, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, R.A.; Gunderson, E.P.; Barrett-Connor, E.; Quesenberry, C.P.; Yaffe, K. Obesity in middle age and future risk of dementia: A 27 year longitudinal population based study. BMJ 2005, 330, 1360. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Qiu, C.; Gatz, M.; Pedersen, N.L.; Johansson, B.; Fratiglioni, L. Mid-and late-life diabetes in relation to the risk of dementia: A population-based twin study. Diabetes 2009, 58, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Rusanen, M.; Kivipelto, M.; Quesenberry, C.P.; Zhou, J.; Whitmer, R.A. Heavy smoking in midlife and long-term risk of Alzheimer disease and vascular dementia. Arch. Intern. Med. 2011, 171, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, D.A. Modifiable, non-modifiable, and clinical factors associated with progression of Alzheimer’s disease. J. Alzheimer’s Dis. 2021, 80, 1–27. [Google Scholar] [CrossRef]
- World Alzheimer Report. Alzheimer’s Disease International. 2019. Available online: https://www.alzint.org/u/WorldAlzheimerReport2019.pdf (accessed on 18 December 2023).
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.C.; Wu, Y.T.; Prina, M. World Alzheimer Report 2015. The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends. Doctoral Dissertation, Alzheimer’s Disease International, London, UK, 2015. [Google Scholar]
- Prince, M.; Ali, G.C.; Guerchet, M.; Prina, A.M.; Albanese, E.; Wu, Y.T. Recent global trends in the prevalence and incidence of dementia, and survival with dementia. Alzheimer’s Res. Ther. 2016, 8, 1–13. [Google Scholar] [CrossRef]
- Alzheimer’s Disease International. World Alzheimer Report 2021: Journey through the Diagnosis of Dementia. 2021. Available online: https://www.alzint.org/u/World-Alzheimer-Report-2021.pdf (accessed on 18 December 2023).
- Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 18 December 2023).
- Scarmeas, N.; Stern, Y.; Mayeux, R.; Luchsinger, J.A. Mediterranean diet, Alzheimer disease, and vascular mediation. Arch. Neurol. 2006, 63, 1709–1717. [Google Scholar] [CrossRef]
- Knight, A.; Bryan, J.; Wilson, C.; Hodgson, J.M.; Davis, C.R.; Murphy, K.J. The Mediterranean diet and cognitive function among healthy older adults in a 6-month randomised controlled trial: The MedLey Study. Nutrients 2016, 8, 579. [Google Scholar] [CrossRef] [PubMed]
- Laws, K.R.; Irvine, K.; Gale, T.M. Sex differences in Alzheimer’s disease. Curr. Opin. Psychiatry 2018, 31, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef]
- Sudduth, T.L.; Schmitt, F.A.; Nelson, P.T.; Wilcock, D.M. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Zheng, H. Peripheral immune system in aging and Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 1–17. [Google Scholar]
- Banks, W.A. The blood-brain barrier in neuroimmunology: Tales of separation and assimilation. Brain Behav. Immun. 2015, 44, 1–8. [Google Scholar] [CrossRef]
- Rochfort, K.D.; Collins, L.E.; McLoughlin, A.; Cummins, P.M. Shear-dependent attenuation of cellular ROS levels can suppress proinflammatory cytokine injury to human brain microvascular endothelial barrier properties. J. Cereb. Blood Flow Metab. 2015, 35, 1648–1656. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and dysfunction of the blood-brain barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Prendecki, M.; Kowalska, M.; Łagan-Jędrzejczyk, U.; Piekut, T.; Krokos, A.; Kozubski, W.; Dorszewska, J. Genetic factors related to the immune system in subjects at risk of developing Alzheimer’s disease. J. Integr. Neurosci. 2020, 19, 359–371. [Google Scholar]
- Kleinberger, G.; Yamanishi, Y.; Suárez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 2014, 6, 243ra86. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.; Simpson, J.F.; Parikh, I.; Wilfred, B.R.; Fardo, D.W.; Nelson, P.T.; Estus, S. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J. Neurosci. 2013, 33, 13320–13325. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Jeng, A.T.; Nowotny, P.; Cady, J.; Cruchaga, C.; Goate, A.M. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS ONE 2012, 7, e50976. [Google Scholar] [CrossRef] [PubMed]
- Sindi, S.; Mangialasche, F.; Kivipelto, M. Advances in the prevention of Alzheimer’s Disease. F1000prime Rep. 2015, 7, 50. [Google Scholar] [CrossRef]
- Rahman, M.A.; Rahman, M.S.; Uddin, M.J.; Mamum-Or-Rashid, A.N.; Pang, M.G.; Rhim, H. Emerging risk of environmental factors: Insight mechanisms of Alzheimer’s diseases. Environ. Sci. Pollut. Res. 2020, 27, 44659–44672. [Google Scholar] [CrossRef]
- Abuznait, A.H.; Qosa, H.; Busnena, B.A.; El Sayed, K.A.; Kaddoumi, A. Olive-oil-derived oleocanthal enhances β-amyloid clearance as a potential neuroprotective mechanism against Alzheimer’s disease: In vitro and in vivo studies. ACS Chem. Neurosci. 2013, 4, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Omar, S.H.; Scott, C.J.; Hamlin, A.S.; Obied, H.K. Olive biophenols reduces alzheimer’s pathology in SH-SY5Y cells and APPswe mice. Int. J. Mol. Sci. 2018, 20, 125. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xu, Z.; Tang, J.; Sun, J.; Gao, J.; Wu, T.; Xiao, M. Voluntary exercise counteracts Aβ25-35-induced memory impairment in mice. Behav. Brain Res. 2013, 256, 618–625. [Google Scholar] [CrossRef]
- Koo, J.H.; Kang, E.B.; Oh, Y.S.; Yang, D.S.; Cho, J.Y. Treadmill exercise decreases amyloid-β burden possibly via activation of SIRT-1 signaling in a mouse model of Alzheimer’s disease. Exp. Neurol. 2017, 288, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Cacciottolo, M.; Wang, X.; Driscoll, I.; Woodward, N.; Saffari, A.; Reyes, J.; Serre, M.L.; Vizuete, W.; Sioutas, C.; Morgan, T.E.; et al. Particulate air pollutants, APOE alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models. Transl. Psychiatry 2017, 7, e1022. [Google Scholar] [CrossRef]
- Bihaqi, S.W.; Zawia, N.H. Enhanced taupathy and AD-like pathology in aged primate brains decades after infantile exposure to lead (Pb). Neurotoxicology 2013, 39, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Park, J.D.; Choi, B.S. Mercury-induced amyloid-beta (Aβ) accumulation in the brain is mediated by disruption of Aβ transport. J. Toxicol. Sci. 2014, 39, 625–635. [Google Scholar] [CrossRef]
- Li, Z.; Wang, H.; Yin, Y. Peripheral inflammation is a potential etiological factor in Alzheimer’s disease. Rev. Neurosci. 2024, 35, 99–120. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T. Inflammasome activation and innate immunity in Alzheimer’s disease. Brain Pathol. 2017, 27, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Vigasova, D.; Nemergut, M.; Liskova, B.; Damborsky, J. Multi-pathogen infections and Alzheimer’s disease. Microb. Cell Factories 2021, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, T.; Munawara, U.; Larbi, A.; Desroches, M.; Rodrigues, S.; Catanzaro, M.; Guidolin, A.; Khalil, A.; Bernier, F.; Barron, A.E.; et al. Targeting infectious agents as a therapeutic strategy in Alzheimer’s disease. CNS Drugs 2020, 34, 673–695. [Google Scholar] [CrossRef] [PubMed]
- Vojtechova, I.; Machacek, T.; Kristofikova, Z.; Stuchlik, A.; Petrasek, T. Infectious origin of Alzheimer’s disease: Amyloid beta as a component of brain antimicrobial immunity. PLoS Pathog. 2022, 18, e1010929. [Google Scholar] [CrossRef]
- Fernandes, B.; Enduru, N.; Bahrami, S.; Dai, Y.; Andreassen, O.; Zhao, Z. Genetic overlap between Alzheimer’s disease and immune-mediated diseases: An atlas of shared genetic determinants and biological convergence. Res. Sq. 2023; preprint . [Google Scholar] [CrossRef]
- Scanzello, C.R.; Goldring, S.R. The role of synovitis in osteoarthritis pathogenesis. Bone 2012, 51, 249–257. [Google Scholar] [CrossRef]
- Cyr, B.; Hadad, R.; Keane, R.W.; de Rivero Vaccari, J.P. The role of non-canonical and canonical inflammasomes in inflammaging. Front. Mol. Neurosci. 2022, 15, 774014. [Google Scholar] [CrossRef]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697. [Google Scholar] [CrossRef]
- Goldring, S.R.; Goldring, M.B. Eating bone or adding it: The Wnt pathway decides. Nat. Med. 2007, 13, 133–134. [Google Scholar] [CrossRef] [PubMed]
- Felson, D.; Niu, J.; Sack, B.; Aliabadi, P.; McCullough, C.; Nevitt, M.C. Progression of osteoarthritis as a state of inertia. Ann. Rheum. Dis. 2012, 72, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Ngandu, T.; Lehtisalo, J.; Solomon, A.; Levälahti, E.; Ahtiluoto, S.; Antikainen, R.; Bäckman, L.; Hänninen, T.; Jula, A.; Laatikainen, T.; et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomised controlled trial. Lancet 2015, 385, 2255–2263. [Google Scholar] [CrossRef]
- Fraenkel, L.; Zhang, Y.; Chaisson, C.E.; Evans, S.R.; Wilson, P.W.; Felson, D.T. The association of estrogen replacement therapy and the Raynaud phenomenon in postmenopausal women. Ann. Intern. Med. 1998, 129, 208–211. [Google Scholar] [CrossRef]
- Maki, P.M. The critical window hypothesis of hormone therapy and cognition: A scientific update on clinical studies. Menopause 2013, 20, 695. [Google Scholar] [CrossRef] [PubMed]
- Henderson, V.W. Alzheimer’s disease: Review of hormone therapy trials and implications for treatment and prevention after menopause. J. Steroid Biochem. Mol. Biol. 2014, 142, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Pike, C.J.; Carroll, J.C.; Rosario, E.R.; Barron, A.M. Protective actions of sex steroid hormones in Alzheimer’s disease. Front. Neuroendocrinol. 2009, 30, 239–258. [Google Scholar] [CrossRef]
- Lawrence, R.C.; Felson, D.T.; Helmick, C.G.; Arnold, L.M.; Choi, H.; Deyo, R.A.; Gabriel, S.; Hirsch, R.; Hochberg, M.C.; Hunder, G.G.; et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: Part II. Arthritis Rheum. 2008, 58, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.; Schwartz, T.A.; Helmick, C.G.; Renner, J.B.; Tudor, G.; Koch, G.; Dragomir, A.; Kalsbeek, W.D.; Luta, G.; Jordan, J.M. Lifetime risk of symptomatic knee osteoarthritis. Arthritis Care Res. Off. J. Am. Coll. Rheumatol. 2008, 59, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement. 2013, 9, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Wolters, F.J.; Chibnik, L.B.; Waziry, R.; Anderson, R.; Berr, C.; Beiser, A.; Bis, J.C.; Blacker, D.; Bos, D.; Brayne, C.; et al. Twenty-seven-year time trends in dementia incidence in Europe and the United States: The Alzheimer Cohorts Consortium. Neurology 2020, 95, e519–e531. [Google Scholar] [CrossRef]
- Jordan, J.M.; Helmick, C.G.; Renner, J.B.; Luta, G.; Dragomir, A.D.; Woodard, J.; Fang, F.; Schwartz, T.A.; Abbate, L.M.; Callahan, L.F.; et al. Prevalence of knee symptoms and radiographic and symptomatic knee osteoarthritis in African Americans and Caucasians: The Johnston County Osteoarthritis Project. J. Rheumatol. 2007, 34, 172–180. [Google Scholar]
- Loeser, R.F.; Collins, J.A.; Diekman, B.O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420. [Google Scholar] [CrossRef]
- Valdes, A.M.; Spector, T.D. The contribution of genes to osteoarthritis. Rheum. Dis. Clin. N. Am. 2008, 34, 581–603. [Google Scholar] [CrossRef]
- Wang, T.; Liang, Y.; Li, H.; Li, H.; He, Q.; Xue, Y.; Shen, C.; Zhang, C.; Xiang, J.; Ding, J.; et al. Single nucleotide polymorphisms and osteoarthritis: An overview and a meta-analysis. Medicine 2016, 95, e2811. [Google Scholar] [CrossRef]
- Loughlin, J. Genetic contribution to osteoarthritis development: Current state of evidence. Curr. Opin. Rheumatol. 2015, 27, 284. [Google Scholar] [CrossRef] [PubMed]
- Grillet, B.; Pereira, R.V.; Van Damme, J.; Abu El-Asrar, A.; Proost, P.; Opdenakker, G. Matrix metalloproteinases in arthritis: Towards precision medicine. Nat. Rev. Rheumatol. 2023, 19, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-β/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Reynard, L.N.; Loughlin, J. Insights from human genetic studies into the pathways involved in osteoarthritis. Nat. Rev. Rheumatol. 2013, 9, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A. Osteoarthritis year 2013 in review: Genetics and genomics. Osteoarthr. Cartil. 2013, 21, 1443–1451. [Google Scholar] [CrossRef] [PubMed][Green Version]
- van den Bosch, M.; Kruisbergen, N.; de Munter, W.; Sloetjes, A.; van den Kraan, P.; Blom, A.; van Lent, P. More severe OA joint pathology in human APOE-ε4 as compared to APOE-ε3 transgenic mice: APOE-isoforms as possible risk factor for inflammatory osteoarthritis development? Osteoarthr. Cartil. 2018, 26, S123. [Google Scholar] [CrossRef]
- Gupta, D.P.; Lee, Y.S.; Choe, Y.; Kim, K.T.; Song, G.J.; Hwang, S.C. Knee osteoarthritis accelerates amyloid beta deposition and neurodegeneration in a mouse model of Alzheimer’s disease. Mol. Brain 2023, 16, 1–10. [Google Scholar] [CrossRef]
- Kyrkanides, S.; Tallents, R.H.; Miller, J.N.; Olschowka, M.E.; Johnson, R.; Yang, M.; Olschowka, J.A.; Brouxhon, S.M.; O’Banion, M.K. Osteoarthritis accelerates and exacerbates Alzheimer’s disease pathology in mice. J. Neuroinflamm. 2011, 8, 112. [Google Scholar] [CrossRef]
- Wojdasiewicz, P.; Poniatowski, Ł.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2014, 2014, 561459. [Google Scholar] [CrossRef]
- Westacott, C.I.; Barakat, A.F.; Wood, L.; Perry, M.J.; Neison, P.; Bisbinas, I.; Armstrong, L.; Millar, A.B.; Elson, C.J. Tumor necrosis factor alpha can contribute to focal loss of cartilage in osteoarthritis. Osteoarthr. Cartil. 2000, 8, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Chen, Y.; Zhang, R.; Dai, H.; Zeng, C.; Zeng, H.; Feng, H.; Du, G.; Fang, H.; Cai, D. c-Jun N-terminal kinase–c-Jun pathway transactivates Bim to promote osteoarthritis. Can. J. Physiol. Pharmacol. 2014, 92, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Armada, M.J.; Carames, B.; Lires-Dean, M.; Cillero-Pastor, B.; Ruiz-Romero, C.; Galdo, F.; Blanco, F.J. Cytokines, tumor necrosis factor-α and interleukin-1β, differentially regulate apoptosis in osteoarthritis cultured human chondrocytes. Osteoarthr. Cartil. 2006, 14, 660–669. [Google Scholar] [CrossRef]
- Heraud, F.; Heraud, A.; Harmand, M. Apoptosis in normal and osteoarthritic human articular cartilage. Ann. Rheum. Dis. 2000, 59, 959. [Google Scholar] [CrossRef] [PubMed]
- Joos, H.; Wildner, A.; Hogrefe, C.; Reichel, H.; Brenner, R.E. Interleukin-1 beta and tumor necrosis factor alpha inhibit migration activity of chondrogenic progenitor cells from non-fibrillated osteoarthritic cartilage. Arthritis Res. Ther. 2013, 15, R119. [Google Scholar] [CrossRef] [PubMed]
- Séguin, C.A.; Bernier, S.M. TNFα suppresses link protein and type II collagen expression in chondrocytes: Role of MEK1/2 and NF-κB signaling pathways. J. Cell. Physiol. 2003, 197, 356–369. [Google Scholar] [CrossRef]
- Xue, J.; Wang, J.; Liu, Q.; Luo, A. Tumor necrosis factor-α induces ADAMTS-4 expression in human osteoarthritis chondrocytes. Mol. Med. Rep. 2013, 8, 1755–1760. [Google Scholar] [CrossRef]
- Henderson, B.; Pettipher, E.R. Arthritogenic actions of recombinant IL-1 and tumour necrosis factor α in the rabbit: Evidence for synergistic interactions between cytokines in vivo. Clin. Exp. Immunol. 1989, 75, 306–310. [Google Scholar]
- Jian, J.; Konopka, J.; Liu, C. Insights into the role of progranulin in immunity, infection, and inflammation. J. Leukoc. Biol. 2013, 93, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Konopka, J.; Richbourgh, B.; Liu, C. The role of PGRN in musculoskeletal development and disease. Front. Biosci. 2014, 19, 662. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guo, F.; Lai, Y.; Tian, Q.; Lin, E.A.; Kong, L.; Liu, C. Granulin-epithelin precursor binds directly to ADAMTS-7 and ADAMTS-12 and inhibits their degradation of cartilage oligomeric matrix protein. Arthritis Rheum. 2010, 62, 2023–2036. [Google Scholar] [CrossRef]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef]
- Von Bernhardi, R.; Tichauer, J.E.; Eugenín, J. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J. Neurochem. 2010, 112, 1099–1114. [Google Scholar] [CrossRef] [PubMed]
- Olianas, M.C.; Dedoni, S.; Onali, P. Inhibition of TNF-α-induced neuronal apoptosis by antidepressants acting through the lysophosphatidic acid receptor LPA1. Apoptosis 2019, 24, 478–498. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar]
- Koenigsknecht-Talboo, J.; Landreth, G.E. Microglial Phagocytosis Induced by Fibrillar β-Amyloid and IgGs Are Differentially Regulated by Proinflammatory Cytokines. J. Neurosci. 2005, 25, 8240–8249. [Google Scholar] [CrossRef]
- Walker, K.A.; Le Page, L.M.; Terrando, N.; Duggan, M.R.; Heneka, M.T.; Bettcher, B.M. The role of peripheral inflammatory insults in Alzheimer’s disease: A review and research roadmap. Mol. Neurodegener. 2023, 18, 1–9. [Google Scholar] [CrossRef]
- Du, J.; Li, A.; Shi, D.; Chen, X.; Wang, Q.; Liu, Z.; Sun, K.; Guo, T. Alzheimer’s Disease Neuroimaging Initiative. Association of APOE-ε4, Osteoarthritis, β-Amyloid, and Tau Accumulation in Primary Motor and Somatosensory Regions in Alzheimer Disease. Neurology 2023, 101, e40–e49. [Google Scholar] [CrossRef]
- Chen, M.; Fu, W.; Xu, H.; Liu, C.J. Tau deficiency inhibits classically activated macrophage polarization and protects against collagen-induced arthritis in mice. Arthritis Res. Ther. 2023, 25, 146. [Google Scholar] [CrossRef]
- Alasmari, F.; Alshammari, M.A.; Alasmari, A.F.; Alanazi, W.A.; Alhazzani, K. Neuroinflammatory cytokines induce amyloid beta neurotoxicity through modulating amyloid precursor protein levels/metabolism. Biomed. Res. Int. 2018, 78, 3087475. [Google Scholar] [CrossRef] [PubMed]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, T.J.; Cavalier, A.N.; Roberts, C.M.; Lemieux, M.R.; Ramesh, P.; Garcia, M.A.; Link, C.D. Amyloid beta acts synergistically as a pro-inflammatory cytokine. Neurobiol. Dis. 2021, 159, 105493. [Google Scholar] [CrossRef] [PubMed]
- Stanciugelu, S.I.; Homorogan, C.; Selaru, C.; Patrascu, J.M.; Patrascu, J.M., Jr.; Stoica, R.; Nitusca, D.; Marian, C. Osteoarthritis and microRNAs: Do They Provide Novel Insights into the Pathophysiology of This Degenerative Disorder? Life 2022, 12, 1914. [Google Scholar]
- Jiang, L.; Sun, X.; Kong, H. microRNA-9 might be a novel protective factor for osteoarthritis patients. Hereditas 2020, 157, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, C.; Zhao, J.; Xu, J.; Geng, Y.; Dai, L.; Huang, Y.; Fu, S.C.; Dai, K.; Zhang, X. miR-146a facilitates osteoarthritis by regulating cartilage homeostasis via targeting Camk2d and Ppp3r2. Cell Death Dis. 2017, 8, e2734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hu, C.; Zhang, C.; Luo, C.; Zhong, B.; Yu, X. MiRNA-132 regulates the development of osteoarthritis in correlation with the modulation of PTEN/PI3K/AKT signaling. BMC Geriatr. 2021, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.W.; Wang, W.T.; Chou, L.C.; Liao, C.D.; Liou, T.H.; Lin, H.W. Osteoarthritis increases the risk of dementia: A nationwide cohort study in Taiwan. Sci. Rep. 2015, 5, 10145. [Google Scholar] [CrossRef]
- Ikram, M.; Innes, K.; Sambamoorthi, U. Association of osteoarthritis and pain with Alzheimer’s diseases and related dementias among older adults in the United States. Osteoarthr. Cartil. 2019, 27, 1470–1480. [Google Scholar] [CrossRef]
- Weber, A.; hung Mak, S.; Berenbaum, F.; Sellam, J.; Zheng, Y.P.; Han, Y.; Wen, C. Association between osteoarthritis and increased risk of dementia: A systemic review and meta-analysis. Medicine 2019, 98, e14355. [Google Scholar] [CrossRef]
- Yamada, K.; Kubota, Y.; Tabuchi, T.; Shirai, K.; Iso, H.; Kondo, N.; Kondo, K. A prospective study of knee pain, low back pain, and risk of dementia: The JAGES project. Sci. Rep. 2019, 9, 10690. [Google Scholar] [CrossRef]
- Siviero, P.; Veronese, N.; Smith, T.; Stubbs, B.; Limongi, F.; Zambon, S.; Dennison, E.M.; Edwards, M.; Cooper, C.; Timmermans, E.J.; et al. Association between osteoarthritis and social isolation: Data from the EPOSA study. J. Am. Geriatr. Soc. 2020, 68, 87–95. [Google Scholar] [CrossRef]
- Innes, K.E.; Sambamoorthi, U. The association of osteoarthritis and related pain burden to incident Alzheimer’s disease and related dementias: A retrospective cohort study of US Medicare beneficiaries. J. Alzheimer’s Dis. 2020, 75, 789–805. [Google Scholar] [CrossRef]
- Gregori, D.; Giacovelli, G.; Minto, C.; Barbetta, B.; Gualtieri, F.; Azzolina, D.; Vaghi, P.; Rovati, L.C. Association of pharmacological treatments with long-term pain control in patients with knee osteoarthritis: A systematic review and meta-analysis. JAMA 2018, 320, 2564–2579. [Google Scholar] [CrossRef] [PubMed]
- Lyseng-Williamson, K.A. Anakinra in Still’s disease: A profile of its use. Drugs Ther. Perspect. 2018, 34, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.; Mousavi, M.J.; Jamalzehi, S.; Alimohammadi, R.; Bezvan, M.H.; Mohammadi, H.; Aslani, S. Strategies toward rheumatoid arthritis therapy; the old and the new. J. Cell. Physiol. 2019, 234, 10018–10031. [Google Scholar] [CrossRef]
- Benjamin , O.; Goyal , A.; Lappin, S.L. Disease-Modifying Antirheumatic Drugs (DMARD). 2023. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Rodriguez-Merchan, E.C. The current role of disease-modifying osteoarthritis drugs. Arch. Bone Jt. Surg. 2023, 11, 11–22. [Google Scholar] [PubMed]
- Wandel, S.; Jüni, P.; Tendal, B.; Nüesch, E.; Villiger, P.M.; Welton, N.J.; Reichenbach, S.; Trelle, S. Effects of glucosamine, chondroitin, or placebo in patients with osteoarthritis of hip or knee: Network meta-analysis. BMJ 2010, 16, 341. [Google Scholar] [CrossRef]
- Richard, E.; Ligthart, S.A.; Moll van Charante, E.P.; Van Gool, W.A. Vascular risk factors and dementia–towards prevention strategies. Neth. J. Med. 2010, 68, 284–290. [Google Scholar] [PubMed]
- Anstey, K.J.; Ashby-Mitchell, K.; Peters, R. Updating the evidence on the association between serum cholesterol and risk of late-life dementia: Review and meta-analysis. J. Alzheimer’s Dis. 2017, 56, 215–228. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Sundahl, N.; Bridelance, J.; Libert, C.; De Bosscher, K.; Beck, I.M. Selective glucocorticoid receptor modulation: New directions with non-steroidal scaffolds. Pharmacol. Ther. 2015, 152, 28–41. [Google Scholar] [CrossRef]
- Palomer, E.; Buechler, J.; Salinas, P.C. Wnt signaling deregulation in the aging and Alzheimer’s brain. Front. Cell. Neurosci. 2019, 13, 227. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/β-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- McLeod, F.; Salinas, P.C. Wnt proteins as modulators of synaptic plasticity. Curr. Opin. Neurobiol. 2018, 53, 90–95. [Google Scholar] [CrossRef] [PubMed]
- June, R.K.; Liu-Bryan, R.; Long, F.; Griffin, T.M. Emerging role of metabolic signaling in synovial joint remodeling and osteoarthritis. J. Orthop. Res. 2016, 34, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt signaling in the nervous system and in Alzheimer’s disease. J. Mol. Cell Biol. 2014, 6, 64–74. [Google Scholar] [CrossRef]
- Marzo, A.; Galli, S.; Lopes, D.; McLeod, F.; Podpolny, M.; Segovia-Roldan, M.; Ciani, L.; Purro, S.; Cacucci, F.; Gibb, A.; et al. Reversal of synapse degeneration by restoring Wnt signaling in the adult hippocampus. Curr. Biol. 2016, 26, 2551–2561. [Google Scholar] [CrossRef] [PubMed]
- Rosso, S.B.; Inestrosa, N.C. WNT signaling in neuronal maturation and synaptogenesis. Front. Cell. Neurosci. 2013, 7, 103. [Google Scholar] [CrossRef]
- Available online: https://www.fda.gov/medical-devices/in-vitro-diagnostics/precision-medicine#:~:text=Precision%20medicine%2C%20sometimes%20known%20as,genes%2C%20environments%2C%20and%20lifestyles (accessed on 18 December 2023).
- Akhoon, N. Precision Medicine: A New Paradigm in Therapeutics. Int. J. Prev. Med. 2021, 12, 12. [Google Scholar]
- Arafah, A.; Khatoon, S.; Rasool, I.; Khan, A.; Rather, M.A.; Abujabal, K.A.; Faqih, Y.A.; Rashid, H.; Rashid, S.M.; Bilal Ahmad, S.; et al. The future of precision medicine in the cure of Alzheimer’s disease. Biomedicines 2023, 11, 335. [Google Scholar] [CrossRef] [PubMed]
- Dagenais, G.R.; Leong, D.P.; Rangarajan, S.; Lanas, F.; Lopez-Jaramillo, P.; Gupta, R.; Diaz, R.; Avezum, A.; Oliveira, G.B.; Wielgosz, A.; et al. Variations in common diseases, hospital admissions, and deaths in middle-aged adults in 21 countries from five continents (PURE): A prospective cohort study. Lancet 2020, 395, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef]
- Liu-Ambrose, T.; Barha, C.K.; Best, J.R. Physical activity for brain health in older adults. Appl. Physiol. Nutr. Metab. 2018, 43, 1105–1112. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umoh, I.O.; dos Reis, H.J.; de Oliveira, A.C.P. Molecular Mechanisms Linking Osteoarthritis and Alzheimer’s Disease: Shared Pathways, Mechanisms and Breakthrough Prospects. Int. J. Mol. Sci. 2024, 25, 3044. https://doi.org/10.3390/ijms25053044
Umoh IO, dos Reis HJ, de Oliveira ACP. Molecular Mechanisms Linking Osteoarthritis and Alzheimer’s Disease: Shared Pathways, Mechanisms and Breakthrough Prospects. International Journal of Molecular Sciences. 2024; 25(5):3044. https://doi.org/10.3390/ijms25053044
Chicago/Turabian StyleUmoh, Idiongo Okon, Helton Jose dos Reis, and Antonio Carlos Pinheiro de Oliveira. 2024. "Molecular Mechanisms Linking Osteoarthritis and Alzheimer’s Disease: Shared Pathways, Mechanisms and Breakthrough Prospects" International Journal of Molecular Sciences 25, no. 5: 3044. https://doi.org/10.3390/ijms25053044
APA StyleUmoh, I. O., dos Reis, H. J., & de Oliveira, A. C. P. (2024). Molecular Mechanisms Linking Osteoarthritis and Alzheimer’s Disease: Shared Pathways, Mechanisms and Breakthrough Prospects. International Journal of Molecular Sciences, 25(5), 3044. https://doi.org/10.3390/ijms25053044