
Bulk and Single-Cell RNA Sequencing Elucidate the Etiology of Severe COVID-19


Abstract
1. Introduction
2. Functional Genomic Studies of Tissues and Cells Provide Unbiased Evidence to Understand the Etiology of COVID-19

{kind=link}
| Year of Publication and Reference | Specification of the Biological Model and Sample Type | Main Conclusions of the Study | (1) GEO ID. (2) PMID. (3) Dataset Size. (4) Number of Samples. | (1) IF. (2) Citations (According to the Elsevier Abstract and Citation Database (Scopus). Retrieved on 31 July 2023). | Platform |
|---|---|---|---|---|---|
| 2020 [54]. | Human lung cancer cell lines were infected in vitro with four common viruses: H1N1 influenza A virus (IAV), human respiratory syncytial virus (RSV), human parainfluenza virus 3 (HPIV3), and SARS-CoV-2. | Transcriptional responses did not appear to stop the replication of SARS-CoV-2. Instead, they suggested a protracted inflammatory response. | (1) GSE147507. (2) PMID 32416070. (3) 2.7 Mb. (4) 110. | (1) IF = 67. (2) 2771 citations. | Illumina NextSeq 500 (H. sapiens). |
| 2020 [24]. | Researchers used a gene expression platform to analyze the gene expression of immune cells extracted from patients with active COVID-19 infections. | A study presented a comprehensive atlas of immune modulations during COVID-19 and identified three significant immunotypes related to disease severity. | (1) GSE152418. (2) PMID 32788292. (3) 1.6 Mb. (4) 34. | (1) IF = 48. (2) 629 citations. | Illumina NovaSeq 6000 (H. sapiens). |
| 2020 [25]. | Researchers conducted a longitudinal study on PBMCs from thirteen COVID-19 patients. There were five time points profiled for each patient. In total, 358,930 cells were analyzed using scRNA-seq (single-cell RNA sequencing) technology, with an average of 10,900 cells per sample. | COVID-19 was associated with dynamic changes in expression patterns within PBMCs. Severe disease induced both hypoxic and pro-inflammatory signaling, along with impaired activation of the interferon pathway. | (1) GSE161777. (2) PMID 33296687 (3) 0.5 Mb. (4) 101. | (1) IF = 43. (2) 189 citations. | Illumina MiSeq and Illumina NovaSeq 6000 (H. sapiens). |
| 2020 [26]. | PBMCs were collected from seven hospitalized patients with COVID-19, including four with ARDS, as well as six healthy controls. | Infection with SARS-CoV-2 is characterized by strong downregulation of human leukocyte antigen (HLA) of class I and class II, and interferon-driven inflammatory reactions in monocytes. | (1) GSE150728. (2) PMID 32514174. (3) 372 Mb. (4) 13. | (1) IF = 83. (2) 896 citations. | Illumina NovaSeq 6000 (H. sapiens). |
| 2021 [55]. | Scientists studied lung cells of deceased COVID-19 patients and compared them to normal cells. | Lungs of COVID-19 patients showed inflammation and impaired T-cell response. Diseased lungs had fewer epithelial cells and more macrophages, monocytes, neuronal cells, and fibroblasts. | (1) GSE171524. (2) PMID 33915568. (3) 1601.4 Mb. (4) 27. | (1) IF = 70. (2) 213 citations. | Illumina NovaSeq 6000 (M. musculus). |
| 2021 [56]. | Blood, lungs, and airways of dead COVID-19 patients were profiled. | Results of the gene expression screening highlighted the importance of changes in immunological gene expression. Not only epithelial cell types, but also infiltrating immune cells, contributed to the signal. | (1) GSE147507. (2) PMID 33782412. (3) 1800.9 Mb. (4) 110. | (1) IF = 5. (2) 75 citations. | Illumina NextSeq 500 (H. sapiens). |
| 2021 [29]. | CSF was collected from seven men and one woman with neuro-COVID, aged from 53 to 82. | This study identified the expansion of populations of dedifferentiated monocytes and exhausted CD4+ T-cells in the CSF of neuro-COVID patients. | (1) GSE163005. (2) PMID 33382973. (3) 83 Mb. (4) 16. | (1) IF = 43. (2) 84 citations. | Illumina NextSeq 500, or Illumina NovaSeq 6000 (H. sapiens). |
| 2021 [28]. | Cell culture of human pancreatic islets. | After infecting cultured pancreatic islets with the SARS-CoV-2 virus, it was observed that the number of insulin-secretory granules in β-cells was reduced, leading to impaired glucose-dependent insulin secretion. The infection caused morphological, transcriptional, and functional changes in the β-cells of the islets, resulting in impaired glucose-stimulated insulin secretion. | (1) GSE159717. (2) PMID 33536639. (3) 3.9 Mb. (4) 4. | (1) IF = 21. (2) 318 citations. | HiSeq 4000 instrument (Illumina). |
| 2022 [27]. | The study investigated tissue samples from COVID-19 patients. Blood samples were taken for transcriptome profiling from 21 patients. In total, 57,049 single-cell transcriptomes of PBMCs were sequenced. | Endothelial injury and thrombotic events are common in COVID-19 and linked to increased myeloid cell activity. | (1) GSE208337. (2) PMID 35895716. (3) 573.4 Mb. (4) 105. | (1) IF = 13. (2) 7 citations. | Illumina NovaSeq 6000 (H. sapiens). |
| 2022 [30]. | Organotypic clusters of conjunctival epithelium were isolated from deceased patients’ eyes and cultured. The transcriptomes of 15,821 cells from three infected and three uninfected cultures were processed. | There was no evidence of productive viral replication in ocular epithelial cells; however, there was evidence of the entry of the coronavirus into these cells. | (1) GSE191232. (2) PMID 35750043. (3) 78 Mb. (4) 7. | (1) IF = 7. (2) 2 citations. |
| Year of Publication and Reference | Specification of the Biological Model and Sample Type | Main Conclusions of the Study | (1) GEO ID. (2) PMID. (3) Dataset Size. (4) Number of Samples. | (1) IF. (2) Citations (According to the Scopus Database. Retrieved on 31 July 2023). | Platform |
|---|---|---|---|---|---|
| 2020 [31]. | Five COVID-19 patients and three healthy individuals donated their blood to isolate PBMCs. | A single-cell gene expression atlas was created for both COVID-19 and influenza patients. In COVID-19 patients, three signaling pathways were activated: apoptosis, signal transducer and activator of transcription 1 (STAT1), and interferon regulatory factor 3 (IRF3). COVID-19 patients had an increased number of plasma cells. | (1) CNP0001102 (at China’s National Gene Bank Database (db.cngb.org/cnsa)). (2) PMID 32783921. (3) n/a. (4) n/a. | (1) IF = 43. (2) 206 citations. | DNBelab C4 library and DIPSEQ T1 sequencer. |
| 2020 [32]. | PBMCs were obtained from five healthy donors and 13 COVID-19 patients, including those with moderate, severe, and convalescent cases. | COVID-19 induces an acute inflammatory response, with a strong induction of the interferon-alpha pathway. There is also evidence of disorganized interferon response and immune exhaustion in severe cases. | (1) HRA000150 (this is an accession identifier (ID) in the Chinese National Genomics Data Center (NGDC)). (2) PMID 32788748. (3) n/a. (4) 64. | (1) IF = 31. (2) 356 citations. | Illumina NovaSeq 6000 (H. sapiens), scRNA-seq. |
| 2020 [40]. | This study analyzed immune cells in the lungs of COVID-19 patients and healthy controls. Samples were collected from nine COVID-19 patients and three healthy controls and sequenced for analysis. | A new computational method, Viral-Track, was developed to detect infected cells and showed that SARS-CoV-2 has a detrimental effect on the host immune system. | (1) GSE145926. (2) PMID 32479746. (3) 225.1 Mb. (4) 21. | (1) IF = 67. (2) 285 citations. | Beijing Genomics Institute MGISEQ-2000 platform, scRNA-seq. |
| 2020 [33]. | PBMCs from 10 patients, either mild, severe, or control. | Subtle changes in the percentages of neutrophil or monocyte subtypes correlate with the severity of COVID-19. | (1) E-MTAB-9221. (2) PMID 32810439. (4) 10. | (1) IF = 67. (2) 496 citations. | 10× Chromium droplet-based platform. Illumina NovaSeq 6000 (H. sapiens), scRNA-seq. |
| 2020 [40]. | A total of 18 COVID-19 patients (eight with mild symptoms and 10 with severe symptoms) had their PBMCs collected between the third and twentieth day after their symptoms were first diagnosed. In total, 48,266 single-cell expression profiles of PBMCs were analyzed, along with 50,783 control PBMCs from publicly available datasets. | During a SARS-CoV-2 infection, significant changes occur in the myeloid compartment of the immune system. In mild cases, there is an increase in inflammatory monocytes. In severe cases, dysfunctional monocytes are increased, and there is a rapid generation of immature neutrophils due to excessive myelopoiesis. | (1) GSE145926. (2) PMID 32479746. (3) 225.1 Mb. (4) 21. | (1) IF = 67. (2) 805 citations. | 10× Chromium droplet-based platform. Illumina NovaSeq 6000 (H. sapiens), scRNA-seq. |
| 2020 [41]. | The study examined nasopharyngeal and bronchial samples from 19 COVID-19 patients who had a moderate or critical disease, alongside five healthy controls. In total, transcriptional profiles of 160,528 cells were analyzed from 36 samples. The study identified 22 different cell types and states within the populations of epithelial cells and immune cells. | In critical cases, there were stronger interactions between epithelial and immune cells, leading to more severe lung injury, respiratory failure, and tissue inflammation. | (1) EGAS00001004481 (in the European Genome-Phenome Archive). (2) PMID 32591762. (4) 36. | (1) IF = 59. (2) 635 citations. | Illumina NovaSeq 6000 (H. sapiens), scRNA-seq. |
| 2020 [35]. | PBMCs were collected from COVID-19 patients (hospitalized and non-hospitalized), healthy non-exposed subjects, and from subjects both before and after receiving a flu vaccination. | ScRNA-seq analysis of CD4+ T-cells from 40 COVID-19 patients demonstrated that hospitalization caused an increase in the proportion of cytotoxic follicular helper cells and cytotoxic T helper cells, while reducing the population of regulatory T-cells. | (1) GSE152522. (2) PMID 33096020. (3) 518 Mb. (4) 78. | (1) IF = 67. (2) 300 citations. | Illumina NovaSeq6000 (H. sapiens), scRNA-seq. |
| 2021 [36]. | A total of 68 samples of peripheral blood were obtained, including 52 samples from 13 patients with severe COVID-19 and five samples from five healthy donors. NKs were isolated from these samples through cell sorting, resulting in 80,325 NKs. Single-cell transcriptomes were then generated from these NKs. The UMAP representation categorized the transcriptomes into different types, such as effector I and II, terminally differentiated, transitional, and proliferating. | Severe COVID-19 is associated with early high serum levels of TGF-beta. High levels of this cytokine early in the course of infection are associated with the inhibition of interferon-driven activation of NKs, and impaired immunological response. | (1) GSE184329. (2) PMID 34695836. (3) 174.8 Mb. (4) 13. | (1) IF = 65. (2) 104 citations. | Illumina NextSeq500 (H. sapiens), scRNA-seq. |
| 2021 [43]. | Single-cell atlases of 24 lung, 19 heart, 16 liver, and 16 kidney tissue samples from COVID-19 autopsies were generated. | COVID-19 is characterized by failed tissue regeneration and pathological remodeling of diseased tissues. In COVID-19 samples of the lungs, there were more fibroblasts and fewer epithelial cells. The RNA of SARS-CoV-2 was found to be enriched in phagocytic and ECs. In the heart, a reduction in the number of cardiomyocytes and pericytes was observed, with an increase in the number of vascular ECs. | (1) GSE163530. (2) PMID 33915569. (3) 40.4 Mb. (4) 1194. | (1) IF = 65. (2) 320 citations. | NextSeq 550, Illumina NovaSeq 6000, NextSeq 550, Nanostring GeoMx 2020 Broad COVID Platform, scRNA-seq. |
| 2021 [42]. | Brain and choroid plexus samples isolated from dead patients with severe COVID-19 were profiled at a single-cell level. A total of 65,309 high-quality nuclei were analyzed. | There is significant evidence to suggest that both COVID-19 and neurodegenerative diseases involve long-term inflammation. | (1) GSE159812. (2) PMID 34153974. (3) 944 Mb. (4) 30. | (1) IF = 70. (2) 270 citations. | Illumina NovaSeq 6000 (M. musculus), scRNA-seq. |
| 2021 [37]. | The study used scRNA-seq to profile COVID-19, MIS-C, healthy pediatric, and adult individuals. The scRNA-seq results were correlated with disease severity, flow cytometry, antigen receptor repertoire analysis, and serum proteomics. | The research has identified gene expression signatures that are linked to MIS-C. These signatures could be useful in diagnosing inflammatory complications related to COVID-19. The study found that MIS-C tissues showed higher levels of S100A family alarmins, which is a sign of reduced antigen presentation, and increased cytotoxicity in NK and CD8+ T-cells. | (1) GSE166489. (2) PMID 33891889. (3) 826.1 Mb. (4) 54. | (1) IF = 43. (2) 118 citations. | Illumina NovaSeq 6000 (H. sapiens), scRNA-seq. |
| 2021 [38]. | PBMCs were isolated from a total of 15 donors, including five healthy individuals, seven who had recovered from moderate COVID-19, and three who had recovered from severe COVID-19. The 97,315 resulting PBMC epigenomes were categorized into different subtypes such as monocytes, effector, memory, naive, plasma cells, or NKs using UMAP representation. | This study documented the global chromatin accessibility landscape remodeling in COVID-19 recovered patients. The remodeling patterns indicated the development of immunity through immunological memory against SARS-CoV-2. | (1) HRA000562 (this is an accession ID in the Chinese National Genomics Data Center (NGDC)), PRJNA718009 (this is an accession ID in the Sequence Read Archive (SRA)). (2) PMID 34108657. (3) n/a. (4) n/a. | (1) IF = 28. (2) 43 citations. | Libraries (these are libraries for single-cell T-cell-receptor (TCR) sequencing (scTCR-seq), and TCR-FACS-index-ATAC sequencing (Ti-ATAC-seq)). were processed on HiSeq X Ten platform, Illumina. |
| 2021 [57]. | To investigate the role of neutrophils in COVID-19 and determine the effects of dexamethasone. | A COVID-19 single-cell atlas of neutrophil states and the molecular mechanisms of dexamethasone action were created. For more information, visit http://biernaskielab.ca/COVID_neutrophil. | (1) GSE157789. (2) PMID 34782790. (3) 360 Mb. (4) 31. | (1) IF = 53. (2) 90 citations. | Sequencing was performed using Illumina NovaSeq S2 and SP 100. |
| 2022 [39]. | PBMCs available from seven children diagnosed with MIS-C, as well as six healthy control samples. | Researchers have found that long non-coding RNAs (lncRNAs) play a crucial role in regulating immune responses to SARS-CoV-2. One example is lncRNA PIRAT, which forms a negative feedback loop with the PU.1 transcription factor. This loop helps promote transcription of alarmins, which are proteins that trigger inflammation in response to the virus. However, COVID-19 promotes inflammation by downregulating PIRAT and upregulating lung cancer-associated transcript 1 (LUCAT1), which is another lncRNA that promotes inflammation. | (1) GSE142503. (2) PMID 35998224. (3) 16.7 Mb. (4) 15. | (1) IF = 10. (2) 6 citations. | Illumina NovaSeq 6000 (H. sapiens), scRNA-seq. |
| 2022 [58]. | During a period of 2 to 3 months, a total of 209 patients with COVID-19 (in addition to 100 patients with post-acute COVID-19) and 457 healthy individuals were studied. The patients were examined at the time of their initial diagnosis, during the acute phase of the disease, as well as during the recovery phase. | The diagnosis of PASC has been found to be associated with certain risk factors, such as diabetes, viremia, and autoimmune conditions. | (1) E-MTAB-10129. (2) PMID 35216672. (3) 5.2 Mb. (4) 309. | (1) IF = 67. (2) 802 citations. | Illumina NovaSeq 6000. |
| 2023 [45]. | The research study conducted single-nucleus RNA sequencing on frozen lung samples from seven patients who died from COVID-19, six pairs of lungs from patients with idiopathic pulmonary fibrosis (IPF), and 12 individuals from a control group. The study identified 38,794 cell nuclei from a vascular fraction, which were further divided into 14 subtypes of ECs. Additionally, there were 38,794 non-vascular nuclei. The primary focus of the study was on ECs and a comparison between IPF and COVID-19. | Genes involved in cellular stress were enriched, alongside a signature of reduced immunomodulation and impaired vessel integrity. There was also specific receptor–ligand interactions that were either enriched or depleted in COVID-19 or IPF. | (1) GSE159585. (2) PMID 35998078. (3) 488 Mb. (4) 53. | (1) IF = 13. (2) 7 citations. | Illumina HiSeq 4000/NovaSeq 6000 (H. sapiens), scRNA-seq |
| Year of Publication and Reference | Goal | Conclusions | Datasets | Computational Methods | (1) IF. (2) Citations. (3) PMID. |
|---|---|---|---|---|---|
| 2020 [46]. | A comprehensive meta-analysis of scRNA-seq datasets to identify cell subsets expressing ACE2 and, therefore, targeted by SARS-CoV-2. | ACE2 and TMPRSS2 promote cellular entry of SARS-CoV-2. Type I interferons, and to a lesser extent type II interferons, upregulate ACE2. Cells vulnerable to infection were identified in the lungs. | GSE148829, GSE135069, GSE19190, GSE22147. | Re-analysis with Drop-Seq Computational Protocol v2.0, Seurat. Meta-analysis with UMAP, PCA, gene set enrichment, etc. | (1) IF = 65. (2) 1574 citations. (3) PMID 32413319. |
| 2020 [47]. | To investigate the impact of smoking on COVID-19 utilizing scRNA-seq gene expression data from lung and airway epithelial samples from humans, mice, or rats. | For ACE2, the levels of protein and mRNA were highly correlated (r = 0.82, p-value < 0.0001) across 53 cell lines. Smokers had higher levels of gene expression of ACE2 in the lungs. ACE2 expression was uncorrelated with age or sex. However, inflammation in the lungs was linked to the induction of expression of ACE2. ACE2 was also stimulated in expression by interferon signaling. | GSE132040, GSE53960, GSE53960, GSE34378, GSE53960, GSE44555, GSE132040, GSE6591, GSE80680, GSE1643, GSE18344, GSE13933, GSE22047, GSE64614, GSE76925, GSE79209, GSE121611, GSE122960, GSE134174, GSE75715, GSE39059, GSE135188, GSE57148, GSE103174, GSE2052, GSE47460, GSE43696, GSE16538, GSE3100, GSE11056, GSE86623, GSE41789, GSE32138, GSE32138, GSE47963, GSE100504, GSE51392, and GSE19392, plus some samples from TCGA, the GTEx portal, the Human Cell Atlas, the Human Protein Atlas, and the Single-Cell Expression Atlas. | The meta-analysis was performed using Python, Excel, and GraphPad Prism. Regressions were performed using Python, using ordinary least squares and the statsmodels package. Analysis of single-cell expression data was performed using Python, Scanpy, and Multicore-TSNE packages. Variable genes were prioritized utilizing the Seurat function in Scanpy. The variable genes were then analyzed using PCA. | (1) IF = 14. (2) 275 citations. (3) PMID 32425701. |
| 2020 [48]. | To determine expression patterns for ACE2 or other receptors for SARS-CoV-2 in the respiratory mucosa using scRNA-seq data. | The levels of mRNA and protein of ACE2 are very low in the upper airways and in the lungs. However, there is a mechanism dynamically regulating ACE2 expression in response to SARS-CoV-2. | GSE19190, GSE11906, GSE4302, GSE67472, GSE37147, GSE108134, GSE135893, the FANTOM5 dataset, and a few proteomics datasets, including the Human Proteome Map. | The Cell Ranger pipeline, UMAP, Zenbu genome browser, R packages: pheatmap, Seurat, ggplot2. | (1) IF = 25. (2) 107 citations. (3) PMID 32675206. |
| 2020 [50]. | In this study, scRNA-seq data were used to investigate how kidney diseases or medications may alter ACE2 expression in kidneys. | ACE2 expression in proximal tubular epithelial cells of the kidney facilitated infection with SARS-CoV-2. | Nephrocell data were archived at http://nephrocell.miktmc.org. COVID-19 kidney data were archived at https://hb.flatironinstitute.org/covid-kidney. | The scRNA-seq data were analyzed according to protocols of the Kidney Precision Medicine Project (see https://www.kpmp.org/for-researchers#protocols). | (1) IF = 8. (2) 52 citations. (3) PMID 33038424. |
| 2021 [51]. | In this study, scRNA-seq data were used to identify cellular phenotypes shared across disparate inflammatory diseases. | Similarities in gene expression and signaling between COVID-19 and other inflammatory diseases are uncovered. | GSE134809, GSE122960, GSE145926, GSE155249, GSE47189, GSE147507, GSE168710, phs001457.v1.p1, phs001529.v1.p1, phs001457.v1.p1, SCP259. | A meta-analysis and integration pipeline was constructed, which models and removes the effects of the technology, the tissue of origin, and the donor. The meta-analysis built a reference library for immune cells in a normal body and in different diseases. | (1) IF = 12. (2) 83 citations. (3) PMID 33879239. |
| 2021 [49]. | Muus et al. performed a meta-analysis of receptor genes for SARS-CoV-2, by looking at gene expression in a meta-analysis of 31 lung scRNA-seq studies. | An atlas of cell type-specific associations of age, sex, and smoking with expression levels of ACE2 (and other co-receptors for SARS-CoV-2). | SCP865, SCP895, SCP891, SCP903, SCP871, SCP870, SCP874, SCP878, SCP887, SCP899, SCP898, SCP902, SCP894, SCP869, SCP872, SCP866, SCP1240, SCP1241, SCP868, SCP877, SCP867, SCP897, SCP886, SCP879, SCP860, SCP881, SCP875, SCP876, SCP900, SCP892, SCP890, SCP889, SCP880, SCP882, SCP882, GSE102592, GSE103918, GSE104600, GSE107747, GSE108571, GSE109037, GSE110973, GSE111014, GSE111360, GSE112570, GSE112845, GSE113036, GSE114530, GSE114724, GSE114802, GSE115149, GSE115189, GSE117211, GSE117403, GSE117824, GSE118127, GSE119212, GSE119506, GSE119507, GSE119561, GSE119594, GSE120446, GSE121267, GSE121600, GSE122342, GSE122703, GSE122960, GSE123926, GSE124263, GSE124334, GSE124472, GSE124494, GSE124898, GSE125680, GSE127472, GSE128066, GSE128169, GSE128518, GSE128889, GSE129845, GSE130073, GSE130117, GSE130151, GSE130238, GSE130318, GSE130430, GSE130888, GSE131685, GSE132802, GSE133704, GSE134809, GSE135618, GSE135929, GSE136103, GSE136314, GSE136394, GSE139249, GSE139324, GSE98201. | Data generated by Chromium instrument 10× were integrated using the Cell Ranger pipeline. Datasets were post-processed using Python harmony-pytorch (for batch correction and Leiden clustering) in Scanpy. | (1) IF = 87. (2) 183 citations. (3) PMID 33654293. |
| 2021 [52]. | Chen et al. used cell-line and bulk-tissue RNA-seq to detect overlaps between the host transcriptional responses observed in cancer and those observed in COVID-19 in response to SARS-CoV-2. | There were many similarities in host–disease interactions between cancer and COVID-19. In particular, immune infiltration and inflammation were characteristic of both the diseases. | GSE147507, GSE148729, GSE36969, GSE59185, GSE68820, GSE119856, GSE115770, GSE147507, GSE146507, GSE156063. | All data were public and derived either from GEO or from TCGA. Pathway analysis was performed using GO, wikiPathways, SigTerms software. Standard statistical methods were used, for example two-sided p-values, log-transformed gene expression values, false discovery rate correction for multiple testing, and heat maps. | (1) IF = 4.3. (2) 12 citations. (3) PMID 33510359. |
| 2021 [53]. | Garg et al. set up a multi-dataset that included samples from nine scRNA-seq studies. | The multi-dataset consisted of 159 samples, deriving from seven medical procedures focused on PBMCs and two focused on BALF. Eight out of 20 evaluated hypotheses were confirmed. | PRJCA002413, PRJCA002564, GSE149689, PRJCA002579, GSE150728, GSE145926, GSE147143, and EGAS00001004481. | The Scanpy protocol was used for the meta-analysis. The following steps were included: normalization of the datasets, log transformation of the data, selection of genes variable in expression levels, and PCA. Harmony was used to integrate data from different samples. UMAP was then used to cluster, visualize, and annotate gene expression data by cell type. | (1) IF = 4.3. (2) 12 citations. (3) PMID 34675242. |
2.1. RNA Sequencing Can Be Performed on Cell Lines or Whole Tissues to Investigate the Underlying Causes of COVID-19
2.2. Examples of RNA-seq Studies of Individual Cells in COVID-19
2.2.1. Expression Profiling of Single Cells in the Immune System
2.2.2. Expression Profiling of the Lung Tissue
2.2.3. Expression Profiling of the Brain, Ocular Epithelia, and the Vasculature
3. A Review of Selected COVID-19 Meta-Analyses
4. The Etiology of Severe COVID-19 in the Light of Gene Expression Data
| Tissue Type | Positive Cell Types | Type of Evidence | Computational Method | Reference |
|---|---|---|---|---|
| If detected, transcription of the viral genome in swabs or brush specimens was used to confirm the diagnosis of COVID-19. | n/a | The entire SARS-CoV-2 genome sequence was annotated as one viral ‘gene’. The viral gene was appended to the hg19 annotation gtf file. All reads aligning to the SARS-CoV-2 genome per sample were aggregated and divided by the total number of reads in that sample. | Transcripts were aligned to a customized reference genome in which the SARS-CoV-2 genome (RefSeq ID: NC_045512) was added as an additional chromosome to the human reference genome hg19. Viral load was calculated on the raw data matrices output by Cell Ranger. | [41]. |
| BALF from severe and mild COVID-19 patients. | Epithelial cells and macrophages. | Viral mRNAs were identified among scRNA-seq reads that did not map to the human genome. | Viral-Track was an R-based pipeline that utilized the STAR algorithm to align reads to the SARS-CoV-2 genome. | [40]. |
| BALF from two severe COVID-19 patients [62]. | Epithelial cells, lymphocytes, macrophages, neutrophils. | Yeskit integrates host gene expression profiles with virus detection. | R and Python-based packages utilizing the STAR algorithm. Yeskit is an R package for data integration, clustering, identification of DEGs, functional annotation, and visualization. | [66]. |
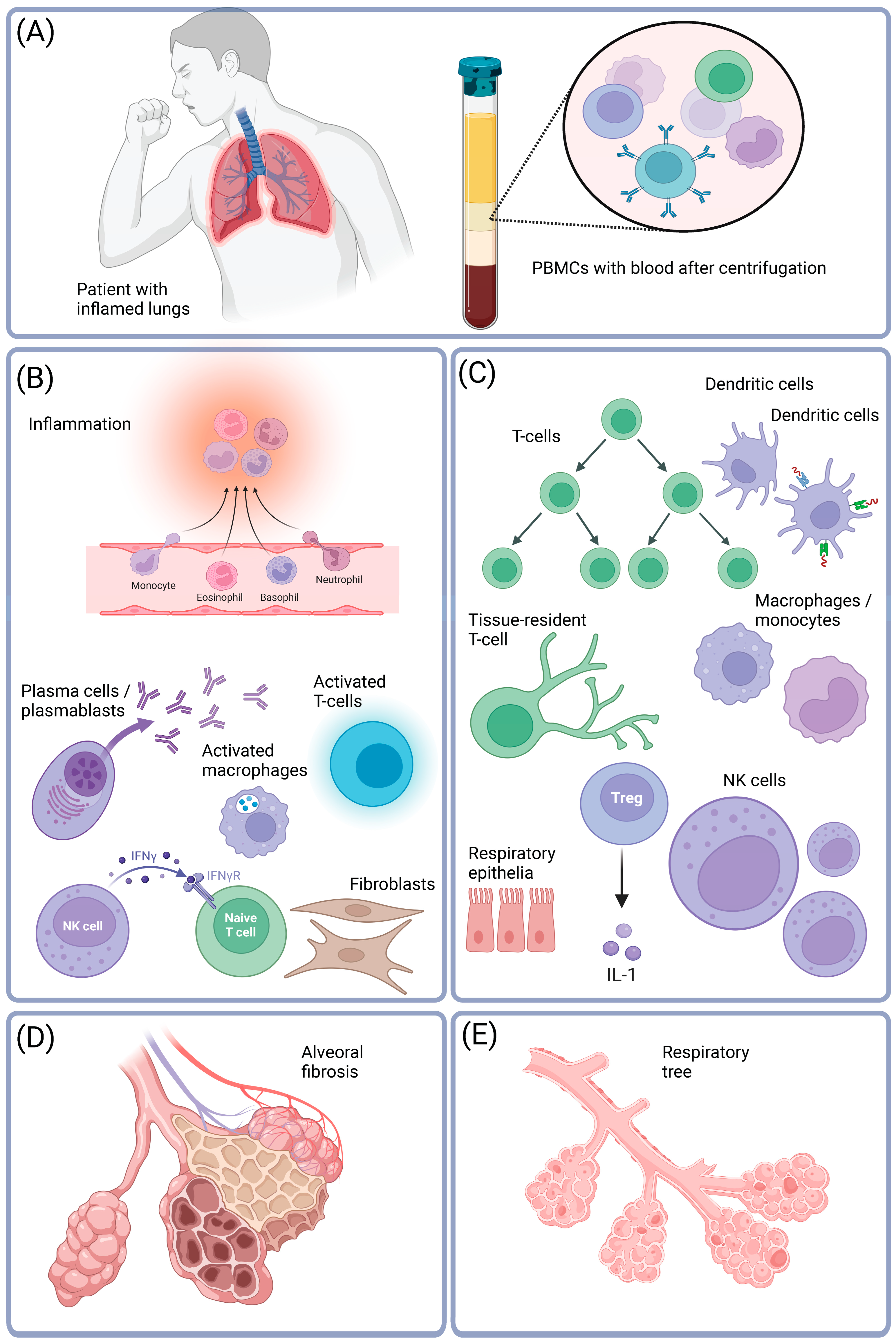
5. Methods
6. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE2 | Angiotensin-converting enzyme 2. |
| ARDS | Acute respiratory distress syndrome. |
| AT1 | Alveolar type I. |
| AT2 | Alveolar type II. |
| BALF | Broncho-alveolar lavage fluid. |
| BioC | Bioconductor. |
| CD4 | Cluster of differentiation 4. |
| CD8 | Cluster of differentiation 8. |
| COVID-19 | Coronavirus disease 2019. |
| CSF | Cerebrospinal fluid. |
| DEG | Differentially expressed gene. |
| EC | Endothelial cell. |
| GEO | Gene Expression Omnibus. |
| GSEA | Gene set enrichment. |
| GZMA | Granzyme A. |
| GZMB | Granzyme B. |
| HPIV3 | Human parainfluenza virus 3. |
| HUVEC | Human umbilical vein EC. |
| IAV | H1N1 influenza A virus. |
| ID | Identifier. |
| IPF | Idiopathic pulmonary fibrosis. |
| ISG15 | Interferon-stimulated gene 15. |
| LUCAT1 | Lung cancer-associated transcript 1. |
| MIS-C | Multisystem inflammatory syndrome. |
| NGS | Next-generation sequencing. |
| NK | Natural killer. |
| ORFs | Open reading frames. |
| PBMCs | Peripheral blood mononuclear cells. |
| PMID | PubMed unique identifier. |
| RNA-seq | RNA sequencing. |
| SARS | Severe acute respiratory syndrome. |
| SARS-CoV-2 | SARS coronavirus 2. |
| STRING | Protein–protein interaction networks functional enrichment analysis. |
| Scopus | Elsevier abstract and citation database. |
| TCR | T-cell receptor. |
| TCGA | The Cancer Genome Atlas. |
| UMAP | Uniform manifold approximation and projection. |
| hACE2 | Human angiotensin-converting enzyme 2. |
| pDCs | Plasmacytoid dendritic cells. |
| scRNA-seq | Single-cell RNA sequencing. |
References
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Gostin, L.O.; Gronvall, G.K. The Origins of COVID-19—Why It Matters (and Why It Doesn’t). N. Engl. J. Med. 2023, 388, 2305–2308. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C.; Goldstein, S.A.; Rasmussen, A.L.; Robertson, D.L.; Crits-Christoph, A.; Wertheim, J.O.; Anthony, S.J.; Barclay, W.S.; Boni, M.F.; Doherty, P.C.; et al. The origins of SARS-CoV-2: A critical review. Cell 2021, 184, 4848–4856. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72‚314 Cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef]
- Sherif, Z.A.; Gomez, C.R.; Connors, T.J.; Henrich, T.J.; Reeves, W.B. Pathogenic mechanisms of post-acute sequelae of SARS-CoV-2 infection (PASC). eLife 2023, 12, e86002. [Google Scholar] [CrossRef]
- Konstantinos, E.; Dimitris, V.; Koralia, P.; Periklis, G.F.; Nefeli, L.; Marios, D.; Angelos, P.; Bindu, K.; Orsalia, H.; Aikaterini, P.; et al. Pulmonary infection by SARS-CoV-2 induces senescence accompanied by an inflammatory phenotype in severe COVID-19: Possible implications for viral mutagenesis. Eur. Respir. J. 2022, 60, 2102951. [Google Scholar]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Costa, T.J.; Potje, S.R.; Fraga-Silva, T.F.C.; da Silva-Neto, J.A.; Barros, P.R.; Rodrigues, D.; Machado, M.R.; Martins, R.B.; Santos-Eichler, R.A.; Benatti, M.N.; et al. Mitochondrial DNA and TLR9 activation contribute to SARS-CoV-2-induced endothelial cell damage. Vasc. Pharmacol. 2022, 142, 106946. [Google Scholar] [CrossRef]
- Liu, F.; Han, K.; Blair, R.; Kenst, K.; Qin, Z.; Upcin, B.; Wrsdrfer, P.; Midkiff, C.C.; Mudd, J.; Belyaeva, E.; et al. SARS-CoV-2 Infects Endothelial Cells In Vivo and In Vitro. Front. Cell. Infect. Microbiol. 2021, 11, 701278. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Connors, J.M.; Levy, J.H. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflamm. Res. 2020, 69, 1181–1189. [Google Scholar] [CrossRef]
- Trypsteen, W.; Van Cleemput, J.; Snippenberg, W.V.; Gerlo, S.; Vandekerckhove, L. On the whereabouts of SARS-CoV-2 in the human body: A systematic review. PLoS Pathog. 2020, 16, e1009037. [Google Scholar] [CrossRef] [PubMed]
- Stjepanovic, M.I.; Stojanovic, M.R.; Stankovic, S.; Cvejic, J.; Dimic-Janjic, S.; Popevic, S.; Buha, I.; Belic, S.; Djurdjevic, N.; Stjepanovic, M.M.; et al. Autoimmune and immunoserological markers of COVID-19 pneumonia: Can they help in the assessment of disease severity. Front. Med. 2022, 9, 934270. [Google Scholar] [CrossRef] [PubMed]
- Dotan, A.; Muller, S.; Kanduc, D.; David, P.; Halpert, G.; Shoenfeld, Y. The SARS-CoV-2 as an instrumental trigger of autoimmunity. Autoimmun. Rev. 2021, 20, 102792. [Google Scholar] [CrossRef] [PubMed]
- Damoiseaux, J.; Dotan, A.; Fritzler, M.J.; Bogdanos, D.P.; Meroni, P.L.; Roggenbuck, D.; Goldman, M.; Landegren, N.; Bastard, P.; Shoenfeld, Y.; et al. Autoantibodies and SARS-CoV2 infection: The spectrum from association to clinical implication: Report of the 15th Dresden Symposium on Autoantibodies. Autoimmun. Rev. 2022, 21, 103012. [Google Scholar] [CrossRef]
- Hikmet, F.; Mear, L.; Edvinsson, A.; Micke, P.; Uhlen, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef]
- Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; Morgenstern, D.; Yahalom-Ronen, Y.; Tamir, H.; Achdout, H.; Stein, D.; Israeli, O.; et al. The coding capacity of SARS-CoV-2. Nature 2020, 589, 125–130. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef] [PubMed]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortola, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, P.S.; Wimmers, F.; Mok, C.K.P.; Perera, R.; Scott, M.; Hagan, T.; Sigal, N.; Feng, Y.; Bristow, L.; Tak-Yin Tsang, O.; et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 2020, 369, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, J.P.; Mishra, N.; Tran, F.; Bahmer, T.; Best, L.; Blase, J.I.; Bordoni, D.; Franzenburg, J.; Geisen, U.; Josephs-Spaulding, J.; et al. Longitudinal Multi-omics Analyses Identify Responses of Megakaryocytes, Erythroid Cells, and Plasmablasts as Hallmarks of Severe COVID-19. Immunity 2020, 53, 1296–1314.e9. [Google Scholar] [CrossRef] [PubMed]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martínez-Colón, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef]
- Iwamura, C.; Hirahara, K.; Kiuchi, M.; Ikehara, S.; Azuma, K.; Shimada, T.; Kuriyama, S.; Ohki, S.; Yamamoto, E.; Inaba, Y.; et al. Elevated Myl9 reflects the Myl9-containing microthrombi in SARS-CoV-2-induced lung exudative vasculitis and predicts COVID-19 severity. Proc. Natl. Acad. Sci. USA 2022, 119, e2203437119. [Google Scholar] [CrossRef]
- Muller, J.A.; Gross, R.; Conzelmann, C.; Kruger, J.; Merle, U.; Steinhart, J.; Weil, T.; Koepke, L.; Bozzo, C.P.; Read, C.; et al. SARS-CoV-2 infects and replicates in cells of the human endocrine and exocrine pancreas. Nat. Metab. 2021, 3, 149–165. [Google Scholar] [CrossRef]
- Heming, M.; Li, X.; Ruber, S.; Mausberg, A.K.; Brsch, A.L.; Hartlehnert, M.; Singhal, A.; Lu, I.N.; Fleischer, M.; Szepanowski, F.; et al. Neurological Manifestations of COVID-19 Feature T Cell Exhaustion and Dedifferentiated Monocytes in Cerebrospinal Fluid. Immunity 2021, 54, 164–175.e6. [Google Scholar] [CrossRef]
- Jackson, R.M.; Hatton, C.F.; Spegarova, J.S.; Georgiou, M.; Collin, J.; Stephenson, E.; Verdon, B.; Haq, I.J.; Hussain, R.; Coxhead, J.M.; et al. Conjunctival epithelial cells resist productive SARS-CoV-2 infection. Stem Cell Rep. 2022, 17, 1699–1713. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yang, P.; Zhao, Y.; Zhuang, Z.; Wang, Z.; Song, R.; Zhang, J.; Liu, C.; Gao, Q.; Xu, Q.; et al. Single-Cell Sequencing of Peripheral Mononuclear Cells Reveals Distinct Immune Response Landscapes of COVID-19 and Influenza Patients. Immunity 2020, 53, 685–696.e3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Wang, X.M.; Xing, X.; Xu, Z.; Zhang, C.; Song, J.W.; Fan, X.; Xia, P.; Fu, J.L.; Wang, S.Y.; et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat. Immunol. 2020, 21, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A.G.; Dubuisson, A.; Derosa, L.; Almire, C.; Hénon, C.; Kosmider, O.; Droin, N.; et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 2020, 182, 1401–1418.e18. [Google Scholar] [CrossRef]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Baßler, K.; Schlickeiser, S.; Zhang, B.; Krämer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440.e23. [Google Scholar] [CrossRef]
- Meckiff, B.J.; Ramirez-Sustegui, C.; Fajardo, V.; Chee, S.J.; Kusnadi, A.; Simon, H.; Eschweiler, S.; Grifoni, A.; Pelosi, E.; Weiskopf, D.; et al. Imbalance of Regulatory and Cytotoxic SARS-CoV-2-Reactive CD4(+) T-cells in COVID-19. Cell 2020, 183, 1340–1353.e16. [Google Scholar] [CrossRef]
- Witkowski, M.; Tizian, C.; Ferreira-Gomes, M.; Niemeyer, D.; Jones, T.C.; Heinrich, F.; Frischbutter, S.; Angermair, S.; Hohnstein, T.; Mattiola, I.; et al. Untimely TGF-beta responses in COVID-19 limit antiviral functions of NK cells. Nature 2021, 600, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, A.; Brodsky, N.N.; Sumida, T.S.; Comi, M.; Asashima, H.; Hoehn, K.B.; Li, N.; Liu, Y.; Shah, A.; Ravindra, N.G.; et al. Immune dysregulation and autoreactivity correlate with disease severity in SARS-CoV-2-associated multisystem inflammatory syndrome in children. Immunity 2021, 54, 1083–1095.e7. [Google Scholar] [CrossRef]
- You, M.; Chen, L.; Zhang, D.; Zhao, P.; Chen, Z.; Qin, E.Q.; Gao, Y.; Davis, M.M.; Yang, P. Single-cell epigenomic landscape of peripheral immune cells reveals establishment of trained immunity in individuals convalescing from COVID-19. Nat. Cell Biol. 2021, 23, 620–630. [Google Scholar] [CrossRef]
- Aznaourova, M.; Schmerer, N.; Janga, H.; Zhang, Z.; Pauck, K.; Bushe, J.; Volkers, S.M.; Wendisch, D.; Georg, P.; Ntini, E.; et al. Single-cell RNA sequencing uncovers the nuclear decoy lincRNA PIRAT as a regulator of systemic monocyte immunity during COVID-19. Proc. Natl. Acad. Sci. USA 2022, 119, e2120680119. [Google Scholar] [CrossRef]
- Bost, P.; Giladi, A.; Liu, Y.; Bendjelal, Y.; Xu, G.; David, E.; Blecher-Gonen, R.; Cohen, M.; Medaglia, C.; Li, H.; et al. Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients. Cell 2020, 181, 1475–1488.e12. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thurmann, L.; Kurth, F.; Volker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.C.; Kern, F.; Losada, P.M.; Agam, M.R.; Maat, C.A.; Schmartz, G.P.; Fehlmann, T.; Stein, J.A.; Schaum, N.; Lee, D.P.; et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 2021, 595, 565–571. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Ö.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef]
- Assou, S.; Ahmed, E.; Morichon, L.; Nasri, A.; Foisset, F.; Bourdais, C.; Gros, N.; Tieo, S.; Petit, A.; Vachier, I.; et al. The Transcriptome Landscape of the In Vitro Human Airway Epithelium Response to SARS-CoV-2. Int. J. Mol. Sci. 2023, 24, 12017. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, L.; Becker, L.M.; Teuwen, L.A.; Boeckx, B.; Jansen, S.; Feys, S.; Verleden, S.; Liesenborghs, L.; Stalder, A.K.; Libbrecht, S.; et al. The pulmonary vasculature in lethal COVID-19 and idiopathic pulmonary fibrosis at single-cell resolution. Cardiovasc. Res. 2023, 119, 520–535. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef]
- Smith, J.C.; Sausville, E.L.; Girish, V.; Yuan, M.L.; Vasudevan, A.; John, K.M.; Sheltzer, J.M. Cigarette Smoke Exposure and Inflammatory Signaling Increase the Expression of the SARS-CoV-2 Receptor ACE2 in the Respiratory Tract. Dev. Cell 2020, 53, 514–529.e3. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, J.A.; Tremblay, B.J.; Mansfield, M.J.; Woody, O.; Lobb, B.; Banerjee, A.; Chandiramohan, A.; Tiessen, N.; Cao, Q.; Dvorkin-Gheva, A.; et al. Gene expression and in situ protein profiling of candidate SARS-CoV-2 receptors in human airway epithelial cells and lung tissue. Eur. Respir. J. 2020, 56, 2001123. [Google Scholar] [CrossRef]
- Muus, C.; Luecken, M.D.; Eraslan, G.; Sikkema, L.; Waghray, A.; Heimberg, G.; Kobayashi, Y.; Vaishnav, E.D.; Subramanian, A.; Smillie, C.; et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat. Med. 2021, 27, 546–559. [Google Scholar] [CrossRef]
- Menon, R.; Otto, E.A.; Sealfon, R.; Nair, V.; Wong, A.K.; Theesfeld, C.L.; Chen, X.; Wang, Y.; Boppana, A.S.; Luo, J.; et al. SARS-CoV-2 receptor networks in diabetic and COVID-19-associated kidney disease. Kidney Int. 2020, 98, 1502–1518. [Google Scholar] [CrossRef]
- Zhang, F.; Mears, J.R.; Shakib, L.; Beynor, J.I.; Shanaj, S.; Korsunsky, I.; Nathan, A.; Donlin, L.T.; Raychaudhuri, S. IFN-γ and TNF-α drive a CXCL10+ CCL2+ macrophage phenotype expanded in severe COVID-19 lungs and inflammatory diseases with tissue inflammation. Genome Med. 2021, 13, 64. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, Y.; Sucgang, R.; Ramani, S.; Corry, D.; Kheradmand, F.; Creighton, C.J. Meta-analysis of host transcriptional responses to SARS-CoV-2 infection reveals their manifestation in human tumors. Sci. Rep. 2021, 11, 2459. [Google Scholar] [CrossRef]
- Garg, M.; Li, X.; Moreno, P.; Papatheodorou, I.; Shu, Y.; Brazma, A.; Miao, Z. Meta-analysis of COVID-19 single-cell studies confirms eight key immune responses. Sci. Rep. 2021, 11, 20833. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Muller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Melms, J.C.; Biermann, J.; Huang, H.; Wang, Y.; Nair, A.; Tagore, S.; Katsyv, I.; Rendeiro, A.F.; Amin, A.D.; Schapiro, D.; et al. A molecular single-cell lung atlas of lethal COVID-19. Nature 2021, 595, 114–119. [Google Scholar] [CrossRef]
- Daamen, A.R.; Bachali, P.; Owen, K.A.; Kingsmore, K.M.; Hubbard, E.L.; Labonte, A.C.; Robl, R.; Shrotri, S.; Grammer, A.C.; Lipsky, P.E. Comprehensive transcriptomic analysis of COVID-19 blood, lung, and airway. Sci. Rep. 2021, 11, 7052. [Google Scholar] [CrossRef]
- Sinha, S.; Rosin, N.L.; Arora, R.; Labit, E.; Jaffer, A.; Cao, L.; Farias, R.; Nguyen, A.P.; de Almeida, L.G.N.; Dufour, A.; et al. Dexamethasone modulates immature neutrophils and interferon programming in severe COVID-19. Nat. Med. 2022, 28, 201–211. [Google Scholar] [CrossRef]
- Su, Y.; Yuan, D.; Chen, D.G.; Ng, R.H.; Wang, K.; Choi, J.; Li, S.; Hong, S.; Zhang, R.; Xie, J.; et al. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell 2022, 185, 881–895.e20. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Wen, W.; Su, W.; Tang, H.; Le, W.; Zhang, X.; Zheng, Y.; Liu, X.; Xie, L.; Li, J.; Ye, J.; et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov. 2020, 6, 31. [Google Scholar] [CrossRef]
- Lee, J.S.; Park, S.; Jeong, H.W.; Ahn, J.Y.; Choi, S.J.; Lee, H.; Choi, B.; Nam, S.K.; Sa, M.; Kwon, J.S.; et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. 2020, 5, eabd1554. [Google Scholar] [CrossRef]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, X.; Liu, Z.; Yao, T.; Zheng, D.; Gan, J.; Yu, S.; Li, L.; Chen, P.; Sun, J. Single-cell RNA sequencing reveals the sustained immune cell dysfunction in the pathogenesis of sepsis secondary to bacterial pneumonia. Genomics 2021, 113, 1219–1233. [Google Scholar] [CrossRef]
- Ogura, H.; Gohda, J.; Lu, X.; Yamamoto, M.; Takesue, Y.; Son, A.; Doi, S.; Matsushita, K.; Isobe, F.; Fukuda, Y.; et al. Dysfunctional Sars-CoV-2-M protein-specific cytotoxic T lymphocytes in patients recovering from severe COVID-19. Nat. Commun. 2022, 13, 7063. [Google Scholar] [CrossRef]
- Maucourant, C.; Filipovic, I.; Ponzetta, A.; Aleman, S.; Cornillet, M.; Hertwig, L.; Strunz, B.; Lentini, A.; Reinius, B.; Brownlie, D.; et al. Natural killer cell immunotypes related to COVID-19 disease severity. Sci. Immunol. 2020, 5, eabd6832. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, X.; Fu, Z.; Chen, J.; Chen, S.; Tan, Y. PathogenTrack and Yeskit: Tools for identifying intracellular pathogens from single-cell RNA-sequencing datasets as illustrated by application to COVID-19. Front. Med. 2022, 16, 251–262. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32 (Suppl. S1), D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Papatheodorou, I.; Moreno, P.; Manning, J.; Fuentes, A.M.; George, N.; Fexova, S.; Fonseca, N.A.; Fullgrabe, A.; Green, M.; Huang, N.; et al. Expression Atlas update: From tissues to single cells. Nucleic Acids Res. 2020, 48, D77–D83. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Forrest, A.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; de Hoon, M.J.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; Itoh, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [PubMed]
- Marschner, I.C. Estimating age-specific COVID-19 fatality risk and time to death by comparing population diagnosis and death patterns: Australian data. BMC Med. Res. Methodol. 2021, 21, 126. [Google Scholar] [CrossRef] [PubMed]
| Keyword | Species | Total | NGS | Microarray | ChIP-seq |
|---|---|---|---|---|---|
| SARS-CoV-2 | All | 808 | 642 | 22 | 52 |
| H. sapiens | 596 | 472 | 16 | 42 | |
| C. sabaeus | 14 | 10 | n/a | 1 | |
| M. musculus | 16 | 14 | n/a | 2 | |
| SARS-CoV-2 | 29 | 20 | n/a | n/a | |
| COVID-19 | All | 536 | 407 | 22 | 33 |
| H. sapiens | 536 | 407 | 22 | 33 | |
| C. sabaeus | 6 | 5 | n/a | 1 | |
| M. musculus | 10 | 9 | 1 | 1 | |
| SARS-CoV-2 | 19 | 14 | n/a | n/a |
| Cell or Tissue Type | Differentially Expressed Genes | Differentially Expressed Pathways or Gene Sets | Reference |
|---|---|---|---|
| PBMCs | Interferon-stimulated gene 15 (ISG15) ↑ Interferon-induced protein 44-like (IFI44L) ↑ MX dynamin-like GTPase 1 (MX1) ↑ XIAP-associated factor 1 (XAF1) ↑ | Pro-inflammatory cytokines ↑ Cytokine receptors ↑ Interferon-responsive TFs ↑ Response to type 1 interferon signaling ↑ Defense response to virus signaling ↑ Endoplasm and protein unfolding ↑ Regulation of chromosome organization ↑ DNA conformation change ↑ | [31]. |
| Thrombospondin 1 (THBS1) ↑ | Neutrophil degranulation ↑ Plate activation, signaling, and aggregation ↑ Semaphorin interactions ↑ Crosslinking of collagen fibrils ↑ Interleuking-1 family signaling ↑ Platelet degranulation ↑ | [27]. | |
| lncRNA LUCAT1 ↑ CXCL2 ↑ IL-6 ↑ lncRNA PIRAT ↓ | Hematopoietic cell lineage ↑ Cytokine–cytokine receptor interaction ↑ Chemokine signaling pathway ↑ | [39]. | |
| Immunoglobulin heavy constant alpha 1 (IGHA1) ↑ Immunoglobulin heavy constant gamma 1 (IGHG1) ↑ Lactotransferrin (LTF) ↑ Interferon regulatory factor 1 (IRF1) ↑ Signal transducer and activator of transcription 3 (STAT3) ↑ Hypoxia inducible factor 1 subunit alpha (HIF1A) ↑ RAR-related orphan receptor C (RORC) ↓ | IL-1β and vasodilatory signaling ↑ IFN-related transcripts ↑ Myeloid cell-mediated immunity ↑ Neutrophil degranulation ↑ Erythroid cell differentiation ↑ Cell differentiation pathway ↑ Hypoxic signaling ↑ Inflammation and IFN signaling ↑ Ribosomal structural proteins ↓ | [25]. | |
| HLA-DPB1 and HLA-DMA in monocytes ↓ | Type I interferon-driven inflammatory signature in monocytes ↑ | [26]. | |
| Interferon-induced transmembrane protein 2 (IFITM2) ↑ Interferon-induced transmembrane protein 3 (IFITM3) ↑ Interferon-induced protein 20 (ISG20) ↑ Interferon-induced protein 15 (ISG15) ↑ | Interferon gamma response ↑ | [41]. | |
| Interferon-ɑ response upregulation ↑ | HLA-class II downregulation in CD14+ monocytes ↓ | [53]. | |
| ECs | Heat shock protein 90 alpha family class A member 1 (HSP90AA1) ↑ Heat shock protein family A (Hsp70) member 1A (HSPA1A) ↑ TIMP metallopeptidase inhibitor 1 (TIMP1) ↑ Fibrillin 1 (FBN1) ↑ Matrix metallopeptidase 16 (MMP16) ↑ Collagen type XV alpha 1 chain (COL15A1) ↑ Indoleamine 2,3-dioxygenase 1 (IDO1) ↑ Intercellular adhesion molecule 1 (ICAM1) ↓ Interferon regulatory factor 1 (IRF1) ↓ Cadherin 5 (CDH5) ↓ Integrin subunit beta 1 (ITGB1) ↓ Member of RAS oncogene family (RAP1B) ↓ Cell division cycle 42 (CDC42) ↓ Occludin (OCLN) ↓ Vinculin (VCL) ↓ Sphingosine-1-phosphate receptor 1 (S1PR1) ↓ Protein C receptor (PROCR) ↓ Thrombomodulin (THBD) ↓ | Genes involved in cellular stress ↑ Heat shock proteins ↑ Genes involved in antigen presentation ↑ Hypoxia signaling ↑ Extracellular matrix (ECM) interactions ↑ ECM production/remodeling ↑ Immune system regulation ↓ Vessel maintenance/integrity ↓ Inflammation ↓ Angiogenesis ↓ Cell–cell adhesion ↓ Chemokines/cytokines ↓ TNF and JAK/STAT signaling ↓ | [45]. |
| Lungs | ACE2 ↑ | Interferon response ↑ | [46,47,48]. |
| ACE2 ↑ TMPRSS2 ↑ CTSL ↑ | Interferon response ↑ | [49]. | |
| Lung cancer cell lines | Interferon gamma receptor 1 (IFNGR1) ↑ Interferon gamma receptor 2 (IFNGR2) ↑ Colony stimulating factor 2 (CSF2) ↑ Colony stimulating factor 3 (CSF3) ↑ C-X-C motif chemokine ligand 1 (CXCL1) ↑ C-X-C motif chemokine ligand 2 (CXCL2) ↑ Interleukin 1 alpha (IL1A) ↑ Interleukin 1 beta (IL1B) ↑ Interleukin 6 (IL6) ↑ Tumor necrosis factor superfamily member 14 (TNFSF14) ↑ | Type II interferon signaling ↑ Immune response ↑ Response to stress ↑ Cytokine-mediated signaling ↑ Inflammatory response ↑ Cytokine activity ↑ Extracellular space ↑ Growth factor receptor binding ↑ Response to virus ↑ | [52]. |
| Kidney | ACE2 ↑ | Interferon response ↑ | [50]. |
| Differentially Expressed Gene Sets | References |
|---|---|
| Increased interferon response, interferon signaling, interferon-responsive TFs. | [25,26,31,46,47,48,49,50,52,63]. |
| Increased immune or inflammatory responses. | [26,31,52]. |
| Increased expression of cytokines, chemokines, or receptors. | [31,39,45,52]. |
| Increased interleukin-1 family signaling. | [27,52]. |
| Increased hypoxic signaling. | [25,45]. |
| Diminished immune system regulation, angiogenesis, and vessel integrity. | [45,53]. |
| Tissue Type | Reference | Cell Type Increasing in Frequency in Severe COVID-19 | Less Common Cell Type |
|---|---|---|---|
| PBMCs | [53]. | Plasma cells. Increased B-cell clonal expansion. | Regulatory T-cells. |
| [31]. | Plasmablasts. | Lymphocytes. | |
| [63]. | Plasmablasts. | n/a | |
| [27]. | Myeloid cells. | n/a | |
| [26]. | Developing neutrophils. CD14+ monocytes. Plasmablasts. | CD16+ monocytes. Plasmacytoid dendritic cells, conventional dendritic cells, and NK cells. | |
| [35]. | Cytotoxic follicular helper cells, and cytotoxic T helper cells. | Regulatory T-cells. | |
| [65]. | Highly cytotoxic NK cells containing high levels of cytotoxic proteins, such as perforin. | Unarmed NK cells. | |
| [41]. | Activated macrophages. | n/a | |
| BALF | [40]. | Recruited macrophages, monocytes, or neutrophils. | Alveolar macrophages. |
| [41]. | Neutrophils. | Basal epithelial cells. | |
| CSF | [29]. | Dedifferentiated monocytes, and CD4+ T-cells. | n/a |
| Lungs | [55]. | Fibroblasts, myeloid, and neuronal cells. | Antigen presenting cells, epithelia. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huminiecki, Ł. Bulk and Single-Cell RNA Sequencing Elucidate the Etiology of Severe COVID-19. Int. J. Mol. Sci. 2024, 25, 3280. https://doi.org/10.3390/ijms25063280
Huminiecki Ł. Bulk and Single-Cell RNA Sequencing Elucidate the Etiology of Severe COVID-19. International Journal of Molecular Sciences. 2024; 25(6):3280. https://doi.org/10.3390/ijms25063280
Chicago/Turabian StyleHuminiecki, Łukasz. 2024. "Bulk and Single-Cell RNA Sequencing Elucidate the Etiology of Severe COVID-19" International Journal of Molecular Sciences 25, no. 6: 3280. https://doi.org/10.3390/ijms25063280
APA StyleHuminiecki, Ł. (2024). Bulk and Single-Cell RNA Sequencing Elucidate the Etiology of Severe COVID-19. International Journal of Molecular Sciences, 25(6), 3280. https://doi.org/10.3390/ijms25063280