
Sequence Alignment-Based Prediction of Myosin 7A: Structural Implications and Protein Interactions

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Conservation of Amino Acid Sequence in the Nucleotide-Binding Sites of Myosin 7A Motors
2.2. Prediction of the Actin Binding Sites in Actomyosin-7A Complex
2.3. Analysis of Myosin Motors to Define the Core Sites of the Actomyosin-7A Interface
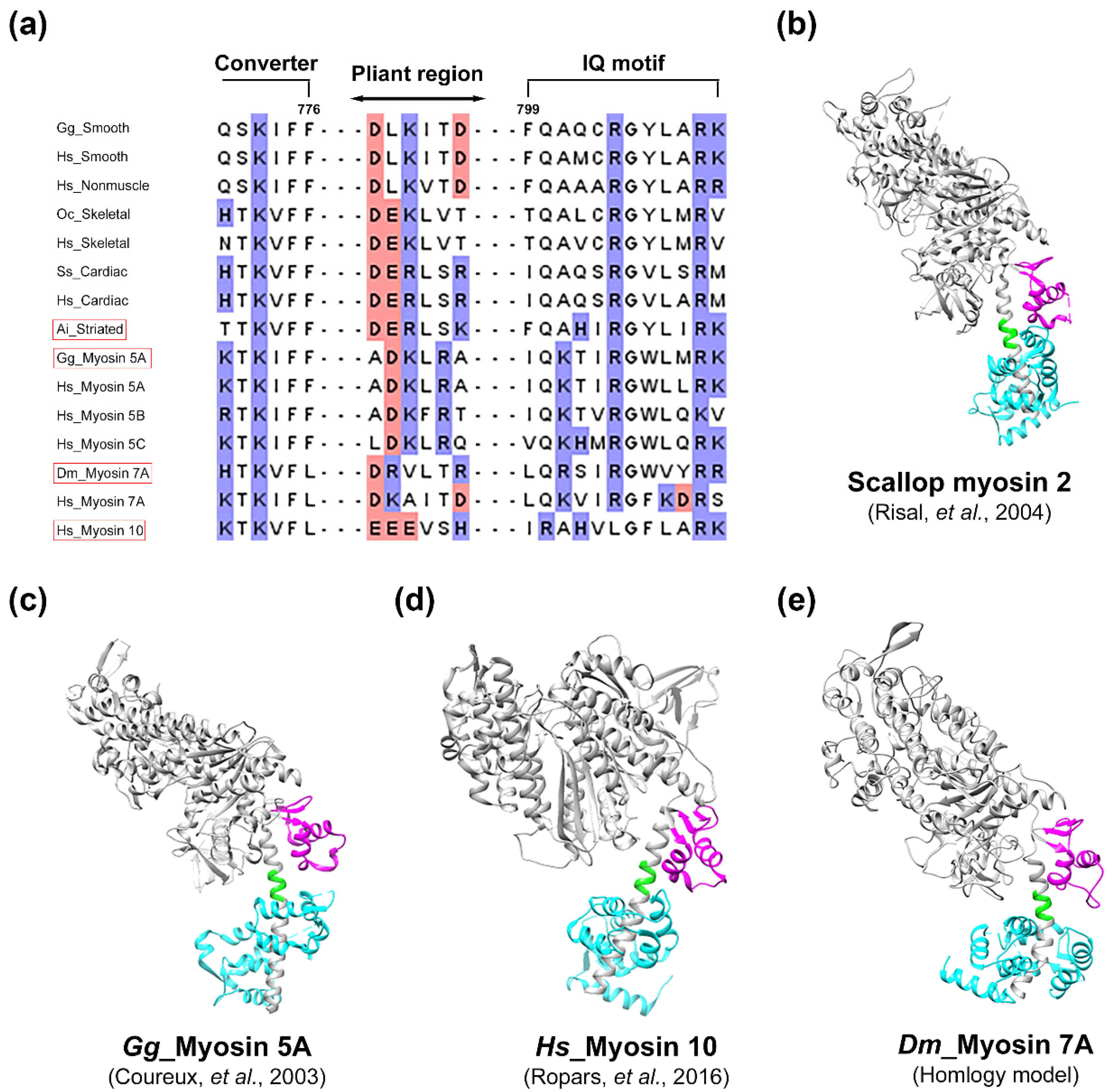
2.4. Suggestive Evidences for the Presence of a Pliant Region of Myosin 7A
3. Discussion
4. Materials and Methods
4.1. Multiple Sequence Alignment and Analysis
4.2. Model Building and Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Mermall, V.; Post, P.L.; Mooseker, M.S. Unconventional myosins in cell movement, membrane traffic, and signal transduction. Science 1998, 279, 527–533. [Google Scholar] [CrossRef]
- Sellers, J.R. Myosins: A diverse superfamily. Biochim. Biophys. Acta 2000, 1496, 3–22. [Google Scholar] [CrossRef]
- Cope, M.J.; Whisstock, J.; Rayment, I.; Kendrick-Jones, J. Conservation within the myosin motor domain: Implications for structure and function. Structure 1996, 4, 969–987. [Google Scholar] [CrossRef]
- Korn, E.D. Coevolution of head, neck, and tail domains of myosin heavy chains. Proc. Natl. Acad. Sci. USA 2000, 97, 12559–12564. [Google Scholar] [CrossRef]
- Lymn, R.W.; Taylor, E.W. Transient state phosphate production in the hydrolysis of nucleoside triphosphates by myosin. Biochemistry 1970, 9, 2975–2983. [Google Scholar] [CrossRef]
- Lymn, R.W.; Taylor, E.W. Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry 1971, 10, 4617–4624. [Google Scholar] [CrossRef]
- Okimoto, N.; Yamanaka, K.; Ueno, J.; Hata, M.; Hoshino, T.; Tsuda, M. Theoretical studies of the ATP hydrolysis mechanism of myosin. Biophys. J. 2001, 81, 2786–2794. [Google Scholar] [CrossRef] [PubMed]
- Houdusse, A.; Szent-Gyorgyi, A.G.; Cohen, C. Three conformational states of scallop myosin S1. Proc. Natl. Acad. Sci. USA 2000, 97, 11238–11243. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Walker, M.; Wang, F.; Sellers, J.R.; White, H.D.; Knight, P.J.; Trinick, J. The prepower stroke conformation of myosin V. J. Cell Biol. 2002, 159, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Gourinath, S.; Himmel, D.M.; Brown, J.H.; Reshetnikova, L.; Szent-Györgyi, A.G.; Cohen, C. Crystal structure of scallop Myosin s1 in the pre-power stroke state to 2.6 a resolution: Flexibility and function in the head. Structure 2003, 11, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Oke, O.A.; Burgess, S.A.; Forgacs, E.; Knight, P.J.; Sakamoto, T.; Sellers, J.R.; White, H.; Trinick, J. Influence of lever structure on myosin 5a walking. Proc. Natl. Acad. Sci. USA 2010, 107, 2509–2514. [Google Scholar] [CrossRef]
- Ménétrey, J.; Bahloul, A.; Wells, A.L.; Yengo, C.M.; Morris, C.A.; Sweeney, H.L.; Houdusse, A. The structure of the myosin VI motor reveals the mechanism of directionality reversal. Nature 2005, 435, 779–785. [Google Scholar] [CrossRef]
- Sun, Y.; Goldman, Y.E. Lever-arm mechanics of processive myosins. Biophys. J. 2011, 101, 1–11. [Google Scholar] [CrossRef]
- Ménétrey, J.; Isabet, T.; Ropars, V.; Mukherjea, M.; Pylypenko, O.; Liu, X.; Perez, J.; Vachette, P.; Sweeney, H.L.; Houdusse, A.M. Processive steps in the reverse direction require uncoupling of the lead head lever arm of myosin VI. Mol. Cell 2012, 48, 75–86. [Google Scholar] [CrossRef]
- Coluccio, L.M. Myosins and Disease. Adv. Exp. Med. Biol. 2020, 1239, 245–316. [Google Scholar] [PubMed]
- Warrick, H.M.; Spudich, J.A. Myosin structure and function in cell motility. Annu. Rev. Cell Biol. 1987, 3, 379–421. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.E.; Bridgman, P.C. Myosin function in nervous and sensory systems. J. Neurobiol. 2004, 58, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.; Kitamoto, J.; Williams, D.S. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin VIIa, the Usher syndrome 1B protein. Proc. Natl. Acad. Sci. USA 2003, 100, 6481–6486. [Google Scholar] [CrossRef]
- Weil, D.; Blanchard, S.; Kaplan, J.; Guilford, P.; Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Levilliers, J.; Weston, M.D.; et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 1995, 374, 60–61. [Google Scholar] [CrossRef]
- Hasson, T.; Heintzelman, M.B.; Santos-Sacchi, J.; Corey, D.P.; Mooseker, M.S. Expression in cochlea and retina of myosin VIIa, the gene product defective in Usher syndrome type 1B. Proc. Natl. Acad. Sci. USA 1995, 92, 9815–9819. [Google Scholar] [CrossRef]
- Hasson, T.; Gillespie, P.G.; Garcia, J.A.; MacDonald, R.B.; Zhao, Y.; Yee, A.G.; Mooseker, M.S.; Corey, D.P. Unconventional myosins in inner-ear sensory epithelia. J. Cell Biol. 1997, 137, 1287–1307. [Google Scholar] [CrossRef]
- Liu, X.; Ondek, B.; Williams, D.S. Mutant myosin VIIa causes defective melanosome distribution in the RPE of shaker-1 mice. Nat. Genet. 1998, 19, 117–118. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef]
- Edman, P. A method for the determination of amino acid sequence in peptides. Arch. Biochem. 1949, 22, 475. [Google Scholar]
- Hunt, D.F.; Yates, J.R., 3rd; Shabanowitz, J.; Winston, S.; Hauer, C.R. Protein sequencing by tandem mass spectrometry. Proc. Natl. Acad. Sci. USA 1986, 83, 6233–6237. [Google Scholar] [CrossRef]
- Whisstock, J.C.; Lesk, A.M. Prediction of protein function from protein sequence and structure. Q. Rev. Biophys. 2003, 36, 307–340. [Google Scholar] [CrossRef] [PubMed]
- Guzzo, A.V. The influence of amino-acid sequence on protein structure. Biophys. J. 1965, 5, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Zhou, B.; Lai, L.; Pei, J. Sequence-based prediction of protein protein interaction using a deep-learning algorithm. BMC Bioinform. 2017, 18, 277. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Thornton, J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef]
- Nooren, I.M.; Thornton, J.M. Diversity of protein-protein interactions. EMBO J. 2003, 22, 3486–3492. [Google Scholar] [CrossRef]
- Obenauer, J.C.; Yaffe, M.B. Computational prediction of protein-protein interactions. Methods Mol. Biol. 2004, 261, 445–468. [Google Scholar]
- Smith, G.R.; Sternberg, M.J. Prediction of protein-protein interactions by docking methods. Curr. Opin. Struct. Biol. 2002, 12, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Gschwend, D.A.; Good, A.C.; Kuntz, I.D. Molecular docking towards drug discovery. J. Mol. Recognit. 1996, 9, 175–186. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, T. Nucleotide-induced closure of the ATP-binding pocket in myosin subfragment-1. J. Biol. Chem. 1994, 269, 27251–27257. [Google Scholar] [CrossRef] [PubMed]
- Goodson, H.V.; Spudich, J.A. Molecular evolution of the myosin family: Relationships derived from comparisons of amino acid sequences. Proc. Natl. Acad. Sci. USA 1993, 90, 659–663. [Google Scholar] [CrossRef]
- Espreafico, E.M.; Cheney, R.E.; Matteoli, M.; Nascimento, A.A.; De Camilli, P.V.; Larson, R.E.; Mooseker, M.S. Primary structure and cellular localization of chicken brain myosin-V (p190), an unconventional myosin with calmodulin light chains. J. Cell Biol. 1992, 119, 1541–1557. [Google Scholar] [CrossRef] [PubMed]
- Mueller, H.; Perry, S.V. The degradation of heavy meromyosin by trypsin. Biochem. J. 1962, 85, 431–439. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Fujii, T.; Namba, K. Structure of actomyosin rigour complex at 5.2 Å resolution and insights into the ATPase cycle mechanism. Nat. Commun. 2017, 8, 13969. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Behrmann, E.; Müller, M.; Penczek, P.A.; Mannherz, H.G.; Manstein, D.J.; Raunser, S. Structure of the rigor actin-tropomyosin-myosin complex. Cell 2012, 150, 327–338. [Google Scholar] [CrossRef]
- Lorenz, M.; Holmes, K.C. The actin-myosin interface. Proc. Natl. Acad. Sci. USA 2010, 107, 12529–12534. [Google Scholar] [CrossRef]
- Pospich, S.; Sweeney, H.L.; Houdusse, A.; Raunser, S. High-resolution structures of the actomyosin-V complex in three nucleotide states provide insights into the force generation mechanism. Elife 2021, 10, e73724. [Google Scholar] [CrossRef]
- Risi, C.; Schäfer, L.U.; Belknap, B.; Pepper, I.; White, H.D.; Schröder, G.F.; Galkin, V.E. High-Resolution Cryo-EM Structure of the Cardiac Actomyosin Complex. Structure 2021, 29, 50–60.e4. [Google Scholar] [CrossRef]
- von der Ecken, J.; Heissler, S.M.; Pathan-Chhatbar, S.; Manstein, D.J.; Raunser, S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature 2016, 534, 724–728. [Google Scholar] [CrossRef]
- Gurel, P.S.; Kim, L.Y.; Ruijgrok, P.V.; Omabegho, T.; Bryant, Z.; Alushin, G.M. Cryo-EM structures reveal specialization at the myosin VI-actin interface and a mechanism of force sensitivity. Elife 2017, 6, e31125. [Google Scholar] [CrossRef] [PubMed]
- Furch, M.; Remmel, B.; Geeves, M.A.; Manstein, D.J. Stabilization of the actomyosin complex by negative charges on myosin. Biochemistry 2000, 39, 11602–11608. [Google Scholar] [CrossRef] [PubMed]
- Joel, P.B.; Trybus, K.M.; Sweeney, H.L. Two conserved lysines at the 50/20-kDa junction of myosin are necessary for triggering actin activation. J. Biol. Chem. 2001, 276, 2998–3003. [Google Scholar] [CrossRef] [PubMed]
- Onishi, H.; Mikhailenko, S.V.; Morales, M.F. Toward understanding actin activation of myosin ATPase: The role of myosin surface loops. Proc. Natl. Acad. Sci. USA 2006, 103, 6136–6141. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Risal, D.; Gourinath, S.; Himmel, D.M.; Szent-Györgyi, A.G.; Cohen, C. Myosin subfragment 1 structures reveal a partially bound nucleotide and a complex salt bridge that helps couple nucleotide and actin binding. Proc. Natl. Acad. Sci. USA 2004, 101, 8930–8935. [Google Scholar] [CrossRef] [PubMed]
- Coureux, P.D.; Wells, A.L.; Ménétrey, J.; Yengo, C.M.; Morris, C.A.; Sweeney, H.L.; Houdusse, A. A structural state of the myosin V motor without bound nucleotide. Nature 2003, 425, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Ropars, V.; Yang, Z.; Isabet, T.; Blanc, F.; Zhou, K.; Lin, T.; Liu, X.; Hissier, P.; Samazan, F.; Amigues, B.; et al. The myosin X motor is optimized for movement on actin bundles. Nat. Commun. 2016, 7, 12456. [Google Scholar] [CrossRef]
- Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Brown, K.A.; Antonio, M.; Beisel, K.W.; Steel, K.P.; Brown, S.D. A type VII myosin encoded by the mouse deafness gene shaker-1. Nature 1995, 374, 62–64. [Google Scholar] [CrossRef]
- Self, T.; Mahony, M.; Fleming, J.; Walsh, J.; Brown, S.D.; Steel, K.P. Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development 1998, 125, 557–566. [Google Scholar] [CrossRef]
- Liu, X.; Udovichenko, I.P.; Brown, S.D.; Steel, K.P.; Williams, D.S. Myosin VIIa participates in opsin transport through the photoreceptor cilium. J. Neurosci. 1999, 19, 6267–6274. [Google Scholar] [CrossRef] [PubMed]
- Reubold, T.F.; Eschenburg, S.; Becker, A.; Kull, F.J.; Manstein, D.J. A structural model for actin-induced nucleotide release in myosin. Nat. Struct. Biol. 2003, 10, 826–830. [Google Scholar] [CrossRef]
- Onishi, H.; Morales, M.F.; Kojima, S.; Katoh, K.; Fujiwara, K. Functional transitions in myosin: Role of highly conserved Gly and Glu residues in the active site. Biochemistry 1997, 36, 3767–3772. [Google Scholar] [CrossRef] [PubMed]
- Guzik-Lendrum, S.; Heissler, S.M.; Billington, N.; Takagi, Y.; Yang, Y.; Knight, P.J.; Homsher, E.; Sellers, J.R. Mammalian myosin-18A, a highly divergent myosin. J. Biol. Chem. 2013, 288, 9532–9548. [Google Scholar] [CrossRef] [PubMed]
- Llinas, P.; Isabet, T.; Song, L.; Ropars, V.; Zong, B.; Benisty, H.; Sirigu, S.; Morris, C.; Kikuti, C.; Safer, D.; et al. How actin initiates the motor activity of Myosin. Dev. Cell 2015, 33, 401–412. [Google Scholar] [CrossRef]
- Sasaki, N.; Ohkura, R.; Sutoh, K. Dictyostelium myosin II as a model to study the actin-myosin interactions during force generation. J. Muscle Res. Cell Motil. 2002, 23, 697–702. [Google Scholar] [CrossRef]
- Gong, R.; Jiang, F.; Moreland, Z.G.; Reynolds, M.J.; de Los Reyes, S.E.; Gurel, P.; Shams, A.; Heidings, J.B.; Bowl, M.R.; Bird, J.E.; et al. Structural basis for tunable control of actin dynamics by myosin-15 in mechanosensory stereocilia. Sci. Adv. 2022, 8, eabl4733. [Google Scholar] [CrossRef]
- Morck, M.M.; Bhowmik, D.; Pathak, D.; Dawood, A.; Spudich, J.; Ruppel, K.M. Hypertrophic cardiomyopathy mutations in the pliant and light chain-binding regions of the lever arm of human β-cardiac myosin have divergent effects on myosin function. Elife 2022, 11, e76805. [Google Scholar] [CrossRef]
- Yang, Y.; Kovács, M.; Sakamoto, T.; Zhang, F.; Kiehart, D.P.; Sellers, J.R. Dimerized Drosophila myosin VIIa: A processive motor. Proc. Natl. Acad. Sci. USA 2006, 103, 5746–5751. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, C.J.; Park, Y.H.; Ryu, B.; Jung, H.S. Sequence Alignment-Based Prediction of Myosin 7A: Structural Implications and Protein Interactions. Int. J. Mol. Sci. 2024, 25, 3365. https://doi.org/10.3390/ijms25063365
Yu CJ, Park YH, Ryu B, Jung HS. Sequence Alignment-Based Prediction of Myosin 7A: Structural Implications and Protein Interactions. International Journal of Molecular Sciences. 2024; 25(6):3365. https://doi.org/10.3390/ijms25063365
Chicago/Turabian StyleYu, Chan Jong, Yoon Ho Park, Bumhan Ryu, and Hyun Suk Jung. 2024. "Sequence Alignment-Based Prediction of Myosin 7A: Structural Implications and Protein Interactions" International Journal of Molecular Sciences 25, no. 6: 3365. https://doi.org/10.3390/ijms25063365