
Chaperones—A New Class of Potential Therapeutic Targets in Alzheimer’s Disease
 , , , ,
, , , , 
Abstract
1. Introduction
2. Alzheimer’s Disease
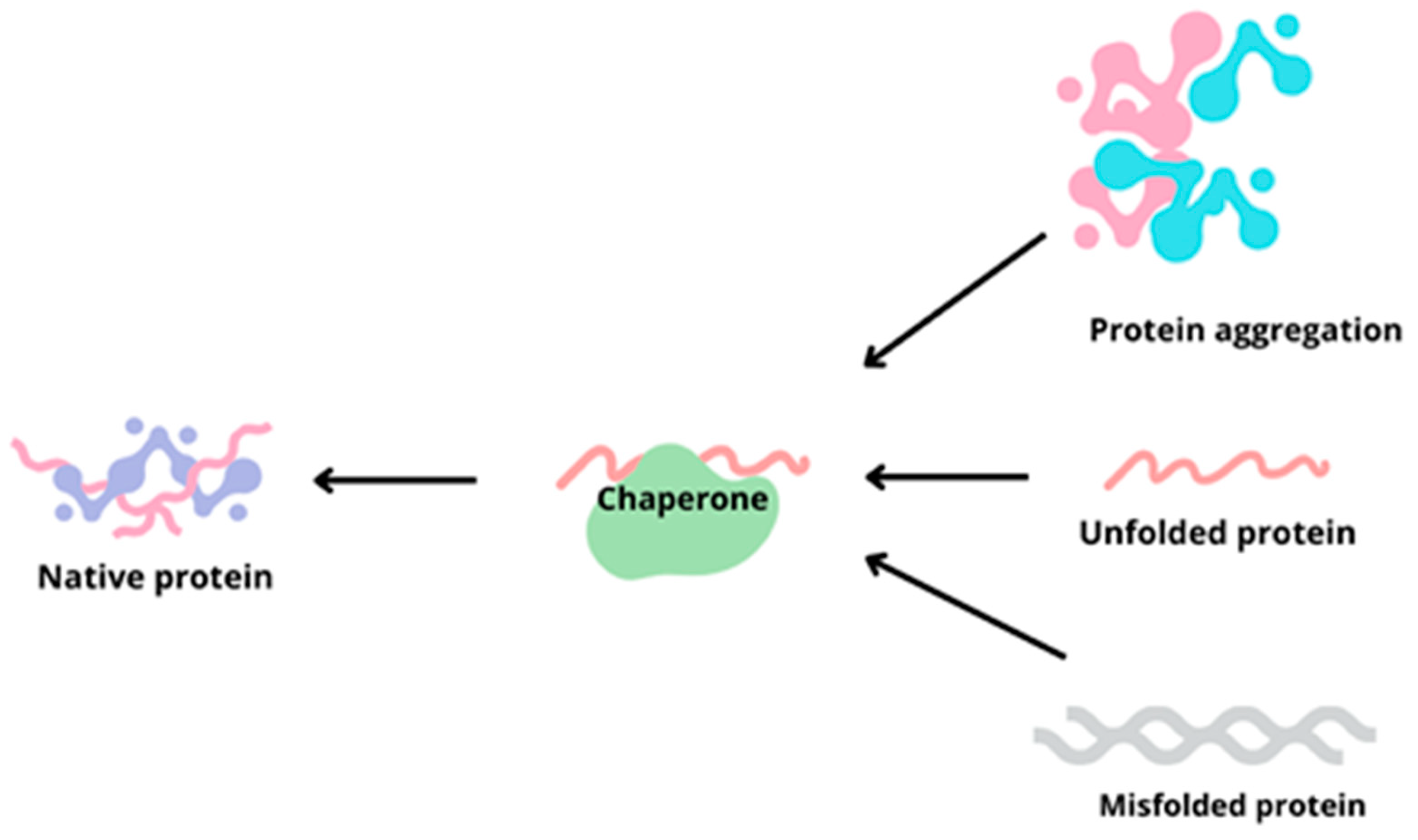
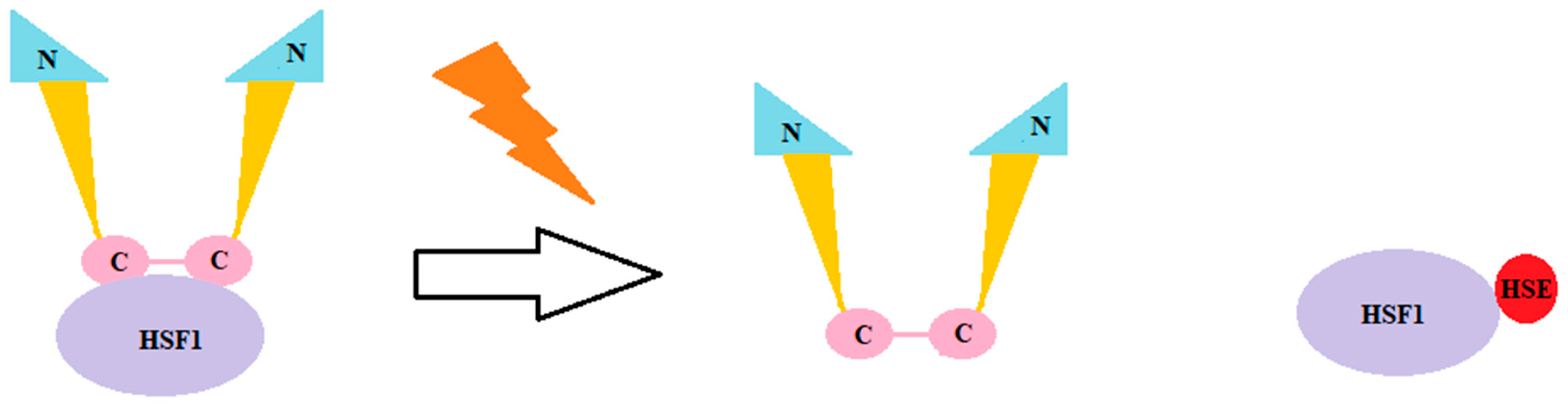
3. Chaperones
4. Chaperonopathies
5. Clusterin
6. Hsp90, Hsp70, Co-Chaperones
7. Chaperones as Diagnostic Markers
8. Targeting Chaperones in the Treatment of Alzheimer’s Disease—Possibilities
9. Targeting Chaperones in the Treatment of Alzheimer’s Disease—Limitations
10. Final Remarks and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2021, 17, 327–406. [CrossRef]
- Alzheimer’s Association’s Website. Available online: https://www.alz.org/alzheimers-dementia/facts-figures (accessed on 3 January 2024).
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Quan, S.; Bardwell, J.C.A. Chaperone Discovery. BioEssays 2012, 34, 973–981. [Google Scholar] [CrossRef] [PubMed]
- 2023 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [CrossRef] [PubMed]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef] [PubMed]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Med. Clin. N. Am. 2019, 103, 263–293. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s Disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Kravats, A.N.; Wickner, S.; Camberg, J.L. Molecular Chaperones. Ref. Modul. Life Sci. 2022. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Ulrich Hartl, F. Molecular Chaperone Functions in Protein Folding and Proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Scalia, F.; Vitale, A.M.; Santonocito, R.; de Macario, E.C.; Macario, A.J.L.; Cappello, F. The Neurochaperonopathies: Anomalies of the Chaperone System with Pathogenic Effects in Neurodegenerative and Neuromuscular Disorders. Appl. Sci. 2021, 11, 898. [Google Scholar] [CrossRef]
- Camberg, J.L.; Doyle, S.M.; Johnston, D.M.; Wickner, S. Molecular Chaperones. In Brenner’s Encyclopedia of Genetics; Elsevier: Amsterdam, The Netherlands, 2013; pp. 456–460. [Google Scholar] [CrossRef]
- Muronetz, V.I.; Kudryavtseva, S.S.; Leisi, E.V.; Kurochkina, L.P.; Barinova, K.V.; Schmalhausen, E.V. Regulation by Different Types of Chaperones of Amyloid Transformation of Proteins Involved in the Development of Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 2747. [Google Scholar] [CrossRef]
- Gupta, A.; Bansal, A.; Hashimoto-Torii, K. HSP70 and HSP90 in Neurodegenerative Diseases. Neurosci. Lett. 2020, 716, 134678. [Google Scholar] [CrossRef] [PubMed]
- Bascos, N.A.D.; Landry, S.J. A History of Molecular Chaperone Structures in the Protein Data Bank. Int. J. Mol. Sci. 2019, 20, 6195. [Google Scholar] [CrossRef] [PubMed]
- Keyzor, I.; Shohet, S.; Castelli, J.; Sitaraman, S.; Veleva-Rotse, B.; Weimer, J.M.; Fox, B.; Willer, T.; Tuske, S.; Crathorne, L.; et al. Therapeutic Role of Pharmacological Chaperones in Lysosomal Storage Disorders: A Review of the Evidence and Informed Approach to Reclassification. Biomolecules 2023, 13, 1227. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef]
- Truttmann, M.C.; Pincus, D.; Ploegh, H.L. Chaperone AMPylation Modulates Aggregation and Toxicity of Neurodegenerative Disease-Associated Polypeptides. Proc. Natl. Acad. Sci. USA 2018, 115, E5008–E5017. [Google Scholar] [CrossRef] [PubMed]
- Verba, K.A.; Agard, D.A. How Hsp90 and Cdc37 Lubricate Kinase Molecular Switches. Trends Biochem. Sci. 2017, 42, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Shim, Y.-J.; Tae, Y.-K.; Kang, B.-H.; Park, J.-S.; Jeon, S.-Y.; Min, B.-H. Toll-like Receptor 4 Signaling Is Required for Clusterin-Induced Tumor Necrosis Factor-α Secretion in Macrophage. Biochem. Biophys. Res. Commun. 2017, 482, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Che, R.; Liang, W.; Zhang, Y.; Wu, L.; Han, C.; Lu, H.; Song, W.; Wu, Y.; Wang, Z. Clusterin Transduces Alzheimer-Risk Signals to Amyloidogenesis. Signal Transduct. Target. Ther. 2022, 7, 325. [Google Scholar] [CrossRef]
- Yang, C.; Wang, H.; Li, C.; Niu, H.; Luo, S.; Guo, X. Association between Clusterin Concentration and Dementia: A Systematic Review and Meta-Analysis. Metab. Brain Dis. 2019, 34, 129–140. [Google Scholar] [CrossRef]
- Sampedro, F.; Marín-Lahoz, J.; Martínez-Horta, S.; Pérez-González, R.; Pagonabarraga, J.; Kulisevsky, J. CLU Rs11136000 Promotes Early Cognitive Decline in Parkinson’s Disease. Mov. Disord. 2020, 35, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Lee, W.J.; Liao, Y.C.; Wang, S.J.; Fuh, J.L. The Clinical Significance of Plasma Clusterin and Aβ in the Longitudinal Follow-up of Patients with Alzheimer’s Disease. Alzheimers Res. Ther. 2017, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Yuste-Checa, P.; Trinkaus, V.A.; Riera-Tur, I.; Imamoglu, R.; Schaller, T.F.; Wang, H.; Dudanova, I.; Hipp, M.S.; Bracher, A.; Hartl, F.U. The Extracellular Chaperone Clusterin Enhances Tau Aggregate Seeding in a Cellular Model. Nat. Commun. 2021, 12, 4863. [Google Scholar] [CrossRef] [PubMed]
- Praharaj, P.P.; Patra, S.; Panigrahi, D.P.; Patra, S.K.; Bhutia, S.K. Clusterin as Modulator of Carcinogenesis: A Potential Avenue for Targeted Cancer Therapy. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188500. [Google Scholar] [CrossRef] [PubMed]
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons from Other Pathologies. Front. Neurosci. 2019, 13, 164. [Google Scholar] [CrossRef] [PubMed]
- Nilselid, A.M.; Davidsson, P.; Nägga, K.; Andreasen, N.; Fredman, P.; Blennow, K. Clusterin in Cerebrospinal Fluid: Analysis of Carbohydrates and Quantification of Native and Glycosylated Forms. Neurochem. Int. 2006, 48, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Zandl-Lang, M.; Fanaee-Danesh, E.; Sun, Y.; Albrecher, N.M.; Gali, C.C.; Čančar, I.; Kober, A.; Tam-Amersdorfer, C.; Stracke, A.; Storck, S.M.; et al. Regulatory Effects of Simvastatin and ApoJ on APP Processing and Amyloid-β Clearance in Blood-Brain Barrier Endothelial Cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 40–60. [Google Scholar] [CrossRef]
- Haight, T.; Bryan, R.N.; Meirelles, O.; Tracy, R.; Fornage, M.; Richard, M.; Nasrallah, I.; Yaffe, K.; Jacobs, D.R.; Lewis, C.; et al. Associations of Plasma Clusterin and Alzheimer’s Disease-Related MRI Markers in Adults at Mid-Life: The CARDIA Brain MRI Sub-Study. PLoS ONE 2018, 13, e0190478. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, X.; Zhu, B.; Fu, P. Association of Clusterin Levels in Cerebrospinal Fluid with Synaptic Degeneration across the Alzheimer’s Disease Continuum. Neuropsychiatr. Dis. Treat. 2020, 16, 183–190. [Google Scholar] [CrossRef]
- Zhou, Y.; Hayashi, I.; Wong, J.; Tugusheva, K.; Renger, J.J.; Zerbinatti, C. Intracellular Clusterin Interacts with Brain Isoforms of the Bridging Integrator 1 and with the Microtubule-Associated Protein Tau in Alzheimer’s Disease. PLoS ONE 2014, 9, e103187. [Google Scholar] [CrossRef]
- Jackson, R.J.; Rose, J.; Tulloch, J.; Henstridge, C.; Smith, C.; Spires-Jones, T.L. Clusterin Accumulates in Synapses in Alzheimer’s Disease and Is Increased in Apolipoprotein E4 Carriers. Brain Commun. 2019, 1, fcz003. [Google Scholar] [CrossRef]
- Borchardt, T.; Camakaris, J.; Cappai, R.; Masters, C.L.; Beyreuther, K.; Multhaup, G. Copper Inhibits Beta-Amyloid Production and Stimulates the Non-Amyloidogenic Pathway of Amyloid-Precursor-Protein Secretion. Biochem. J. 1999, 344 Pt 2, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Greenough, M.A.; Ramírez Munoz, A.; Bush, A.I.; Opazo, C.M. Metallo-Pathways to Alzheimer’s Disease: Lessons from Genetic Disorders of Copper Trafficking. Metallomics 2016, 8, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Telianidis, J.; Hung, Y.H.; Materia, S.; Fontaine, S. La Role of the P-Type ATPases, ATP7A and ATP7B in Brain Copper Homeostasis. Front. Aging Neurosci. 2013, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Materia, S.; Cater, M.A.; Klomp, L.W.J.; Mercer, J.F.B.; La Fontaine, S. Clusterin (Apolipoprotein J), a Molecular Chaperone That Facilitates Degradation of the Copper-ATPases ATP7A and ATP7B. J. Biol. Chem. 2011, 286, 10073–10083. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Kumano, M.; Beraldi, E.; Fazli, L.; Du, C.; Moore, S.; Sorensen, P.; Zoubeidi, A.; Gleave, M.E. Clusterin Facilitates Stress-Induced Lipidation of LC3 and Autophagosome Biogenesis to Enhance Cancer Cell Survival. Nat. Commun. 2014, 5, 5775. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E. Hsp90: Structure and Function. In Molecular Chaperones; Springer: Berlin/Heidelberg, Germany, 2012; pp. 155–240. [Google Scholar]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular Chaperones in Protein Folding and Proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Gorantla, N.V.; Chinnathambi, S. Tau Protein Squired by Molecular Chaperones During Alzheimer’s Disease. J. Mol. Neurosci. 2018, 66, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Bohush, A.; Bieganowski, P.; Filipek, A. Hsp90 and Its Co-Chaperones in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4976. [Google Scholar] [CrossRef]
- Castro, J.P.; Wardelmann, K.; Grune, T.; Kleinridders, A. Mitochondrial Chaperones in the Brain: Safeguarding Brain Health and Metabolism? Front. Endocrinol. 2018, 9, 196. [Google Scholar] [CrossRef]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the Hub of Protein Homeostasis: Emerging Mechanistic Insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 Chaperone Machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Sima, S.; Richter, K. Regulation of the Hsp90 System. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2018, 1865, 889–897. [Google Scholar] [CrossRef]
- Prodromou, C. Regulatory Mechanisms of Hsp90. Biochem. Mol. Biol. J. 2017, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Mollapour, M.; Neckers, L. Post-Translational Modifications of Hsp90 and Their Contributions to Chaperone Regulation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 648–655. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Schenk, D. Alzheimer’s Disease: Molecular Understanding Predicts Amyloid-Based Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 545–584. [Google Scholar] [CrossRef]
- Esch, F.S.; Keim, P.S.; Beattie, E.C.; Blacher, R.W.; Culwell, A.R.; Oltersdorf, T.; McClure, D.; Ward, P.J. Cleavage of Amyloid β Peptide During Constitutive Processing of Its Precursor. Science 1990, 248, 1122–1124. [Google Scholar] [CrossRef]
- Tanemura, K.; Murayama, M.; Akagi, T.; Hashikawa, T.; Tominaga, T.; Ichikawa, M.; Yamaguchi, H.; Takashima, A. Neurodegeneration with Tau Accumulation in a Transgenic Mouse Expressing V337M Human Tau. J. Neurosci. 2002, 22, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Callejas, J.D.; Fuchs, E.; Perez-Cruz, C. Evidence of Tau Hyperphosphorylation and Dystrophic Microglia in the Common Marmoset. Front. Aging Neurosci. 2016, 8, 315. [Google Scholar] [CrossRef]
- Alonso, A.D.; Cohen, L.S.; Corbo, C.; Morozova, V.; ElIdrissi, A.; Phillips, G.; Kleiman, F.E. Hyperphosphorylation of Tau Associates with Changes in Its Function Beyond Microtubule Stability. Front. Cell Neurosci. 2018, 12, 338. [Google Scholar] [CrossRef]
- Basso, A.D.; Solit, D.B.; Chiosis, G.; Giri, B.; Tsichlis, P.; Rosen, N. Akt Forms an Intracellular Complex with Heat Shock Protein 90 (Hsp90) and Cdc37 and Is Destabilized by Inhibitors of Hsp90 Function. J. Biol. Chem. 2002, 277, 39858–39866. [Google Scholar] [CrossRef]
- Wasik, U.; Schneider, G.; Mietelska-Porowska, A.; Mazurkiewicz, M.; Fabczak, H.; Weis, S.; Zabke, C.; Harrington, C.R.; Filipek, A.; Niewiadomska, G. Calcyclin Binding Protein and Siah-1 Interacting Protein in Alzheimer’s Disease Pathology: Neuronal Localization and Possible Function. Neurobiol. Aging 2013, 34, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Jinwal, U.K.; Trotter, J.H.; Abisambra, J.F.; Koren, J.; Lawson, L.Y.; Vestal, G.D.; O’Leary, J.C.; Johnson, A.G.; Jin, Y.; Jones, J.R.; et al. The Hsp90 Kinase Co-Chaperone Cdc37 Regulates Tau Stability and Phosphorylation Dynamics. J. Biol. Chem. 2011, 286, 16976–16983. [Google Scholar] [CrossRef] [PubMed]
- Dickey, C.A.; Koren, J.; Zhang, Y.-J.; Xu, Y.; Jinwal, U.K.; Birnbaum, M.J.; Monks, B.; Sun, M.; Cheng, J.Q.; Patterson, C.; et al. Akt and CHIP Coregulate Tau Degradation through Coordinated Interactions. Proc. Natl. Acad. Sci. USA 2008, 105, 3622–3627. [Google Scholar] [CrossRef] [PubMed]
- Ambegaokar, S.S.; Jackson, G.R. Functional Genomic Screen and Network Analysis Reveal Novel Modifiers of Tauopathy Dissociated from Tau Phosphorylation. Hum. Mol. Genet. 2011, 20, 4947–4977. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.; Guillemeau, K.; Dounane, O.; Sazdovitch, V.; Duyckaerts, C.; Chambraud, B.; Baulieu, E.E.; Giustiniani, J. Caspase-Cleaved Tau-D421 Is Colocalized with the Immunophilin FKBP52 in the Autophagy-Endolysosomal System of Alzheimer’s Disease Neurons. Neurobiol. Aging 2016, 46, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Taglialatela, G.; Rastellini, C.; Cicalese, L. Reduced Incidence of Dementia in Solid Organ Transplant Patients Treated with Calcineurin Inhibitors. J. Alzheimer’s Dis. 2015, 47, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Oroz, J.; Chang, B.J.; Wysoczanski, P.; Lee, C.-T.; Pérez-Lara, Á.; Chakraborty, P.; Hofele, R.V.; Baker, J.D.; Blair, L.J.; Biernat, J.; et al. Structure and Pro-Toxic Mechanism of the Human Hsp90/PPIase/Tau Complex. Nat. Commun. 2018, 9, 4532. [Google Scholar] [CrossRef] [PubMed]
- Lackie, R.E.; Maciejewski, A.; Ostapchenko, V.G.; Marques-Lopes, J.; Choy, W.Y.; Duennwald, M.L.; Prado, V.F.; Prado, M.A.M. The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci. 2017, 11, 254. [Google Scholar] [CrossRef]
- Ostapchenko, V.G.; Beraldo, F.H.; Mohammad, A.H.; Xie, Y.-F.; Hirata, P.H.F.; Magalhaes, A.C.; Lamour, G.; Li, H.; Maciejewski, A.; Belrose, J.C.; et al. The Prion Protein-Ligand, Stress-Inducible Phosphoprotein 1, Regulates Amyloid-β Oligomer Toxicity. J. Neurosci. 2013, 33, 16552–16564. [Google Scholar] [CrossRef]
- Kang, S.; Kang, B.H. Structure, Function, and Inhibitors of the Mitochondrial Chaperone TRAP1. J. Med. Chem. 2022, 65, 16155–16172. [Google Scholar] [CrossRef]
- Joshi, A.; Dai, L.; Liu, Y.; Lee, J.; Ghahhari, N.M.; Segala, G.; Beebe, K.; Jenkins, L.M.; Lyons, G.C.; Bernasconi, L.; et al. The Mitochondrial HSP90 Paralog TRAP1 Forms an OXPHOS-Regulated Tetramer and Is Involved in Mitochondrial Metabolic Homeostasis. BMC Biol. 2020, 18, 10. [Google Scholar] [CrossRef]
- Dekker, F.A.; Rüdiger, S.G.D. The Mitochondrial Hsp90 TRAP1 and Alzheimer’s Disease. Front. Mol. Biosci. 2021, 8, 697913. [Google Scholar] [CrossRef]
- Yoshida, S.; Tsutsumi, S.; Muhlebach, G.; Sourbier, C.; Lee, M.-J.; Lee, S.; Vartholomaiou, E.; Tatokoro, M.; Beebe, K.; Miyajima, N.; et al. Molecular Chaperone TRAP1 Regulates a Metabolic Switch between Mitochondrial Respiration and Aerobic Glycolysis. Proc. Natl. Acad. Sci. USA 2013, 110, E1604–E1612. [Google Scholar] [CrossRef]
- Rasola, A.; Neckers, L.; Picard, D. Mitochondrial Oxidative Phosphorylation TRAP(1)Ped in Tumor Cells. Trends Cell Biol. 2014, 24, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Avolio, R.; Matassa, D.S.; Criscuolo, D.; Landriscina, M.; Esposito, F. Modulation of Mitochondrial Metabolic Reprogramming and Oxidative Stress to Overcome Chemoresistance in Cancer. Biomolecules 2020, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative Toxicity in Diabetes and Alzheimer’s Disease: Mechanisms behind ROS/ RNS Generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef]
- Altieri, D.C.; Stein, G.S.; Lian, J.B.; Languino, L.R. TRAP-1, the Mitochondrial Hsp90. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.-H. TRAP1 Regulation of Mitochondrial Life or Death Decision in Cancer Cells and Mitochondria-Targeted TRAP1 Inhibitors. BMB Rep. 2012, 45, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D Deficiency Attenuates Mitochondrial and Neuronal Perturbation and Ameliorates Learning and Memory in Alzheimer’s Disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Pridgeon, J.W.; Olzmann, J.A.; Chin, L.-S.; Li, L. PINK1 Protects against Oxidative Stress by Phosphorylating Mitochondrial Chaperone TRAP1. PLoS Biol. 2007, 5, e172. [Google Scholar] [CrossRef]
- Grimm, S. The ER–Mitochondria Interface: The Social Network of Cell Death. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 327–334. [Google Scholar] [CrossRef]
- LaFerla, F.M. Calcium Dyshomeostasis and Intracellular Signalling in Alzheimer’s Disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Park, H.-K.; Yoon, N.G.; Lee, J.-E.; Hu, S.; Yoon, S.; Kim, S.Y.; Hong, J.-H.; Nam, D.; Chae, Y.C.; Park, J.B.; et al. Unleashing the Full Potential of Hsp90 Inhibitors as Cancer Therapeutics through Simultaneous Inactivation of Hsp90, Grp94, and TRAP1. Exp. Mol. Med. 2020, 52, 79–91. [Google Scholar] [CrossRef]
- Siegelin, M.D. Inhibition of the Mitochondrial Hsp90 Chaperone Network: A Novel, Efficient Treatment Strategy for Cancer? Cancer Lett. 2013, 333, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Saharan, S.; Mandal, P.K. The Emerging Role of Glutathione in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 40, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of Pro-Inflammatory Cytokines Released from Microglia in Alzheimer’s Disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Boche, D.; Perry, V.H.; Nicoll, J.A.R. Review: Activation Patterns of Microglia and Their Identification in the Human Brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Czeh, M.; Gressens, P.; Kaindl, A.M. The Yin and Yang of Microglia. Dev. Neurosci. 2011, 33, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Bian, J.-S. Hydrogen Sulfide Protects Amyloid-β Induced Cell Toxicity in Microglia. J. Alzheimer’s Dis. 2011, 22, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Degradation of Tau Protein by Autophagy and Proteasomal Pathways. Biochem. Soc. Trans. 2012, 40, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Marino Gammazza, A.; Restivo, V.; Baschi, R.; Caruso Bavisotto, C.; Cefalù, A.B.; Accardi, G.; Conway de Macario, E.; Macario, A.J.L.; Cappello, F.; Monastero, R. Circulating Molecular Chaperones in Subjects with Amnestic Mild Cognitive Impairment and Alzheimer’s Disease: Data from the Zabùt Aging Project. J. Alzheimer’s Dis. 2022, 87, 161–172. [Google Scholar] [CrossRef]
- Zimbone, S.; Di Rosa, M.C.; Chiechio, S.; Giuffrida, M.L. Exploring the Role of Hsp60 in Alzheimer’s Disease and Type 2 Diabetes: Suggestion for Common Drug Targeting. Int. J. Mol. Sci. 2023, 24, 12456. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.-C.; Tan, M.-S.; Wang, H.; Xie, A.-M.; Yu, J.-T.; Tan, L. Heat Shock Protein 70 in Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 435203. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Kodam, A.; Gong, P.; Chen, Y.; Parent, A.T.; Kar, S.; Thinakaran, G. Localization and Regional Distribution of P23/TMP21 in the Brain. Neurobiol. Dis. 2008, 32, 37–49. [Google Scholar] [CrossRef]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Rossie, S.; Gong, C.-X. Dephosphorylation of Tau by Protein Phosphatase 5. J. Biol. Chem. 2005, 280, 1790–1796. [Google Scholar] [CrossRef]
- Lackie, R.E.; Marques-Lopes, J.; Ostapchenko, V.G.; Good, S.; Choy, W.-Y.; van Oosten-Hawle, P.; Pasternak, S.H.; Prado, V.F.; Prado, M.A.M. Increased Levels of Stress-Inducible Phosphoprotein-1 Accelerates Amyloid-β Deposition in a Mouse Model of Alzheimer’s Disease. Acta Neuropathol. Commun. 2020, 8, 143. [Google Scholar] [CrossRef]
- Chang, H.C.; Nathan, D.F.; Lindquist, S. In Vivo Analysis of the Hsp90 Cochaperone Sti1 (P60). Mol. Cell Biol. 1997, 17, 318–325. [Google Scholar] [CrossRef]
- Blair, L.J.; Nordhues, B.A.; Hill, S.E.; Scaglione, K.M.; O’Leary, J.C.; Fontaine, S.N.; Breydo, L.; Zhang, B.; Li, P.; Wang, L.; et al. Accelerated Neurodegeneration through Chaperone-Mediated Oligomerization of Tau. J. Clin. Investig. 2013, 123, 4158–4169. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Tseng, B.; Cheng, D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking Abeta42 Accumulation Delays the Onset and Progression of Tau Pathology via the C Terminus of Heat Shock Protein70-Interacting Protein: A Mechanistic Link between Abeta and Tau Pathology. J. Neurosci. 2008, 28, 12163–12175. [Google Scholar] [CrossRef] [PubMed]
- Basset, C.A.; Cappello, F.; Rappa, F.; Lentini, V.L.; Jurjus, A.R.; Conway de Macario, E.; Macario, A.J.L.; Leone, A. Molecular Chaperones in Tumors of Salivary Glands. J. Mol. Histol. 2020, 51, 109–115. [Google Scholar] [CrossRef]
- Alberti, G.; Vergilio, G.; Paladino, L.; Barone, R.; Cappello, F.; Conway de Macario, E.; Macario, A.J.L.; Bucchieri, F.; Rappa, F. The Chaperone System in Breast Cancer: Roles and Therapeutic Prospects of the Molecular Chaperones Hsp27, Hsp60, Hsp70, and Hsp90. Int. J. Mol. Sci. 2022, 23, 7792. [Google Scholar] [CrossRef]
- Holbrook, N.J.; Carlson, S.G.; Choi, A.M.; Fargnoli, J. Induction of HSP70 Gene Expression by the Antiproliferative Prostaglandin PGA2: A Growth-Dependent Response Mediated by Activation of Heat Shock Transcription Factor. Mol. Cell Biol. 1992, 12, 1528–1534. [Google Scholar] [CrossRef]
- Wei, J.C.; Leong, P.; Liu, G. Chaperone/Scaffolding/Adaptor Protein 14-3-3η (Eta): A Diagnostic Marker of Rheumatoid Arthritis. Int. J. Rheum. Dis. 2020, 23, 1439–1442. [Google Scholar] [CrossRef]
- Macario, A.J.L. Heat-Shock Proteins and Molecular Chaperones: Implications for Pathogenesis, Diagnostics, and Therapeutics. Int. J. Clin. Lab. Res. 1995, 25, 59–70. [Google Scholar] [CrossRef]
- Gezen-Ak, D.; Dursun, E.; Hanağası, H.; Bilgiç, B.; Lohman, E.; Araz, Ö.S.; Atasoy, İ.L.; Alaylıoğlu, M.; Önal, B.; Gürvit, H.; et al. BDNF, TNFα, HSP90, CFH, and IL-10 Serum Levels in Patients with Early or Late-Onset Alzheimer’s Disease or Mild Cognitive Impairment. J. Alzheimer’s Dis. 2013, 37, 185–195. [Google Scholar] [CrossRef]
- Cappello, F. Hsp60 and Human Aging: Les Liaisons Dangereuses. Front. Biosci. 2013, 18, 626. [Google Scholar] [CrossRef]
- Wojsiat, J.; Prandelli, C.; Laskowska-Kaszub, K.; Martín-Requero, A.; Wojda, U. Oxidative Stress and Aberrant Cell Cycle in Alzheimer’s Disease Lymphocytes: Diagnostic Prospects. J. Alzheimer’s Dis. 2015, 46, 329–350. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.-R.; Tan, M.-S.; Xie, A.-M.; Yu, J.-T.; Tan, L. Heat Shock Protein 90 in Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 796869. [Google Scholar] [CrossRef] [PubMed]
- Campanella, C.; Pace, A.; Bavisotto, C.C.; Marzullo, P.; Gammazza, A.M.; Buscemi, S.; Piccionello, A.P. Heat Shock Proteins in Alzheimer’s Disease: Role and Targeting. Int. J. Mol. Sci. 2018, 19, 2603. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zheng, J.; Xu, Y.; Zhang, X. Paraquat-Induced Inflammatory Response of Microglia through HSP60/TLR4 Signaling. Hum. Exp. Toxicol. 2018, 37, 1161–1168. [Google Scholar] [CrossRef]
- Tanabe, M.; Ishida, R.; Izuhara, F.; Komatsuda, A.; Wakui, H.; Sawada, K.; Otaka, M.; Nakamura, N.; Itoh, H. The ATPase Activity of Molecular Chaperone HSP60 Is Inhibited by Immunosuppressant Mizoribine. Am. J. Mol. Biol. 2012, 02, 93–102. [Google Scholar] [CrossRef]
- Itoh, H.; Komatsuda, A.; Wakui, H.; Miura, A.B.; Tashima, Y. Mammalian HSP60 Is a Major Target for an Immunosuppressant Mizoribine. J. Biol. Chem. 1999, 274, 35147–35151. [Google Scholar] [CrossRef]
- Nagumo, Y.; Kakeya, H.; Shoji, M.; Hayashi, Y.; Dohmae, N.; Osada, H. Epolactaene Binds Human Hsp60 Cys442 Resulting in the Inhibition of Chaperone Activity. Biochem. J. 2005, 387, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Abisambra, J.; Jinwal, U.K.; Miyata, Y.; Rogers, J.; Blair, L.; Li, X.; Seguin, S.P.; Wang, L.; Jin, Y.; Bacon, J.; et al. Allosteric Heat Shock Protein 70 Inhibitors Rapidly Rescue Synaptic Plasticity Deficits by Reducing Aberrant Tau. Biol. Psychiatry 2013, 74, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Jindal, U.K.; Koren, J.; O’Leary, J.C.; Jones, J.R.; Abisambra, J.F.; Dickey, C.A. Hsp70 ATPase Modulators as Therapeutics for Alzheimer’s and Other Neurodegenerative Diseases. Mol. Cell Pharmacol. 2010, 2, 43–46. [Google Scholar]
- Lo Cascio, F.; Kayed, R. Azure C Targets and Modulates Toxic Tau Oligomers. ACS Chem. Neurosci. 2018, 9, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M.; Arai, T.; Masuda-Suzukake, M.; Nonaka, T.; Yamashita, M.; Akiyama, H.; Hasegawa, M. Methylene Blue Reduced Abnormal Tau Accumulation in P301L Tau Transgenic Mice. PLoS ONE 2012, 7, e52389. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Friedman, T.; Pitstick, R.; Polydoro, M.; Roe, A.; Carlson, G.A.; Hyman, B.T. Methylene Blue Does Not Reverse Existing Neurofibrillary Tangle Pathology in the RTg4510 Mouse Model of Tauopathy. Neurosci. Lett. 2014, 562, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Wang, Y.; Du, Y.; Zeng, C.; Liu, Y.; Pan, H.; Ke, C. Current Evidence for J147 as a Potential Therapeutic Agent in Nervous System Disease: A Narrative Review. BMC Neurol. 2023, 23, 317. [Google Scholar] [CrossRef]
- Blair, L.J.; Sabbagh, J.J.; Dickey, C.A. Targeting Hsp90 and Its Co-Chaperones to Treat Alzheimer’s Disease. Expert. Opin. Ther. Targets 2014, 18, 1219–1232. [Google Scholar] [CrossRef]
- Ansar, S.; Burlison, J.A.; Hadden, M.K.; Yu, X.M.; Desino, K.E.; Bean, J.; Neckers, L.; Audus, K.L.; Michaelis, M.L.; Blagg, B.S.J. A Non-Toxic Hsp90 Inhibitor Protects Neurons from Aβ-Induced Toxicity. Bioorg. Med. Chem. Lett. 2007, 17, 1984–1990. [Google Scholar] [CrossRef]
- Palihati, N.; Tang, Y.; Yin, Y.; Yu, D.; Liu, G.; Quan, Z.; Ni, J.; Yan, Y.; Qing, H. Clusterin Is a Potential Therapeutic Target in Alzheimer’s Disease. Mol. Neurobiol. 2023. [Google Scholar] [CrossRef]
- Repalli, J.; Meruelo, D. Screening Strategies to Identify HSP70 Modulators to Treat Alzheimer’s Disease. Drug Des. Devel Ther. 2015, 9, 321–331. [Google Scholar] [CrossRef]
- Bose, S.; Cho, J. Targeting Chaperones, Heat Shock Factor-1, and Unfolded Protein Response: Promising Therapeutic Approaches for Neurodegenerative Disorders. Ageing Res. Rev. 2017, 35, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Alam, Q.; Alam, M.Z.; Wali Sait, K.H.; Anfinan, N.; Noorwali, A.W.; Kamal, M.A.; Ahmad Khan, M.S.; Haque, A. Translational Shift of HSP90 as a Novel Therapeutic Target from Cancer to Neurodegenerative Disorders: An Emerging Trend in the Cure of Alzheimer’s and Parkinson’s Diseases. Curr. Drug Metab. 2017, 18, 868–876. [Google Scholar] [CrossRef]
- Wei, H.; Zhang, Y.; Jia, Y.; Chen, X.; Niu, T.; Chatterjee, A.; He, P.; Hou, G. Heat Shock Protein 90: Biological Functions, Diseases, and Therapeutic Targets. MedComm 2024, 5, e470. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Beretta, G.; Shala, A.L. Impact of Heat Shock Proteins in Neurodegeneration: Possible Therapeutical Targets. Ann. Neurosci. 2022, 29, 71–82. [Google Scholar] [CrossRef]
- Evans, C.G.; Wisén, S.; Gestwicki, J.E. Heat Shock Proteins 70 and 90 Inhibit Early Stages of Amyloid β-(1–42) Aggregation in Vitro. J. Biol. Chem. 2006, 281, 33182–33191. [Google Scholar] [CrossRef]
- Zhang, M.; Qian, C.; Zheng, Z.-G.; Qian, F.; Wang, Y.; Thu, P.M.; Zhang, X.; Zhou, Y.; Tu, L.; Liu, Q.; et al. Jujuboside A Promotes Aβ Clearance and Ameliorates Cognitive Deficiency in Alzheimer’s Disease through Activating Axl/HSP90/PPARγ Pathway. Theranostics 2018, 8, 4262–4278. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Chaperone: | Hsp60 | Hsp70 | Hsp90 | Hsp100 | Shop |
|---|---|---|---|---|---|
| Function: | Segregation of unfolded polypeptides; promotion of unfolding for misfolded polypeptides (by active and passive mechanisms) | The unfolding of misfolded polypeptides, translocation of unfolded polyproteins, dissociation of protein complexes | Modification of specific proteins and transcription factors. | Dissociation, refolding, un-aggregation. | Protection of proteins from irreversible aggregation |
| Localization: | Cytoplasm Mitochondria | Cytoplasm Endoplasmic Reticulum Mitochondria Nucleus | Cytoplasm Endoplasmic Reticulum Mitochondria Nucleus | Cytoplasm Nucleus | Cytoplasm Mitochondria |
| ATP-binding activity | + | + | + | + | - |
| Chaperone and Co-Chaperones | Level in AD | Function in Cell |
|---|---|---|
| Hsp60 | increased [85] | Inhibiting Aβ amyloid aggregation by closing molecular pathways leading to peptide fibrillogenesis [86] |
| Hsp70 | increased [85] | Protecting neurons from intracellular accumulation of Aβ through promoting the clearance of Aβ [87] |
| Hsp90 | increased [42] | Regulating tau phosphorylation [42] |
| p23 | decreased [88] | Facilitating the adenosine triphosphate-driven cycle of Hsp90 binding to client proteins [88] |
| PP5 | decreased [89] | Dephosphorylation of tau [89] |
| STI1/Hop | increased [90] | Clearance of tau [91] |
| Stg1 | decreased [42] | Clearance of tau [42] |
| CHIP | decreased [92] | Tau ubiquitination [42] |
| FKBP51 | increased [93] | Promoting, in coordination with Hsp90, the accumulation of non-ubiquitinated tau in the presence of a proteasome inhibitor [93] |
| Substance | Mechanism | Effect |
|---|---|---|
| Mizorbine | - Inhibiting the detachment of Hsp10 - decreasing the ATPase activity of Hsp60/Hsp10 [105,106] | Inhibition of protein folding function [105,106] |
| Parazolopyrimidine EC3016 | - blocking ATP binding and its hydrolysis [103,107] | Hsp60 inactivation, inhibition of protein folding function [103,107] |
| Epolactaene | - binding to Cys442 [103,107] | Hsp60 inactivation, inhibition of protein folding function [103,107] |
| Group of Molecules | Representatives | Mechanism | Effect |
|---|---|---|---|
| Compounds with rhodacyanine skelton | - MKT-077 - YM-01 - YM-08 | Inhibiting ATPase activity of Hsp70/Hsp40 complex [103,108] | Rapid reduction of tau levels [103,108] |
| Phenothiazines | - Methylene Blue - Azure C | - Inhibiting ATPase function of Hsp70 - Interacting with toxic tau oligomers [108,109,110,111] | - Reduction of tau levels - Inhibiting tau accumulation and aggregation [108,109,110,111] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batko, J.; Antosz, K.; Miśków, W.; Pszczołowska, M.; Walczak, K.; Leszek, J. Chaperones—A New Class of Potential Therapeutic Targets in Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 3401. https://doi.org/10.3390/ijms25063401
Batko J, Antosz K, Miśków W, Pszczołowska M, Walczak K, Leszek J. Chaperones—A New Class of Potential Therapeutic Targets in Alzheimer’s Disease. International Journal of Molecular Sciences. 2024; 25(6):3401. https://doi.org/10.3390/ijms25063401
Chicago/Turabian StyleBatko, Joanna, Katarzyna Antosz, Weronika Miśków, Magdalena Pszczołowska, Kamil Walczak, and Jerzy Leszek. 2024. "Chaperones—A New Class of Potential Therapeutic Targets in Alzheimer’s Disease" International Journal of Molecular Sciences 25, no. 6: 3401. https://doi.org/10.3390/ijms25063401
APA StyleBatko, J., Antosz, K., Miśków, W., Pszczołowska, M., Walczak, K., & Leszek, J. (2024). Chaperones—A New Class of Potential Therapeutic Targets in Alzheimer’s Disease. International Journal of Molecular Sciences, 25(6), 3401. https://doi.org/10.3390/ijms25063401