
Impact of α-Globin Gene Expression and α-Globin Modifiers on the Phenotype of β-Thalassemia and Other Hemoglobinopathies: Implications for Patient Management

 , , , and
, , , and  on behalf of the International Hemoglobinopathy Research Network (INHERENT)
on behalf of the International Hemoglobinopathy Research Network (INHERENT) {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Genetics, Pathophysiology, and Clinical Picture of Beta-Thalassemia
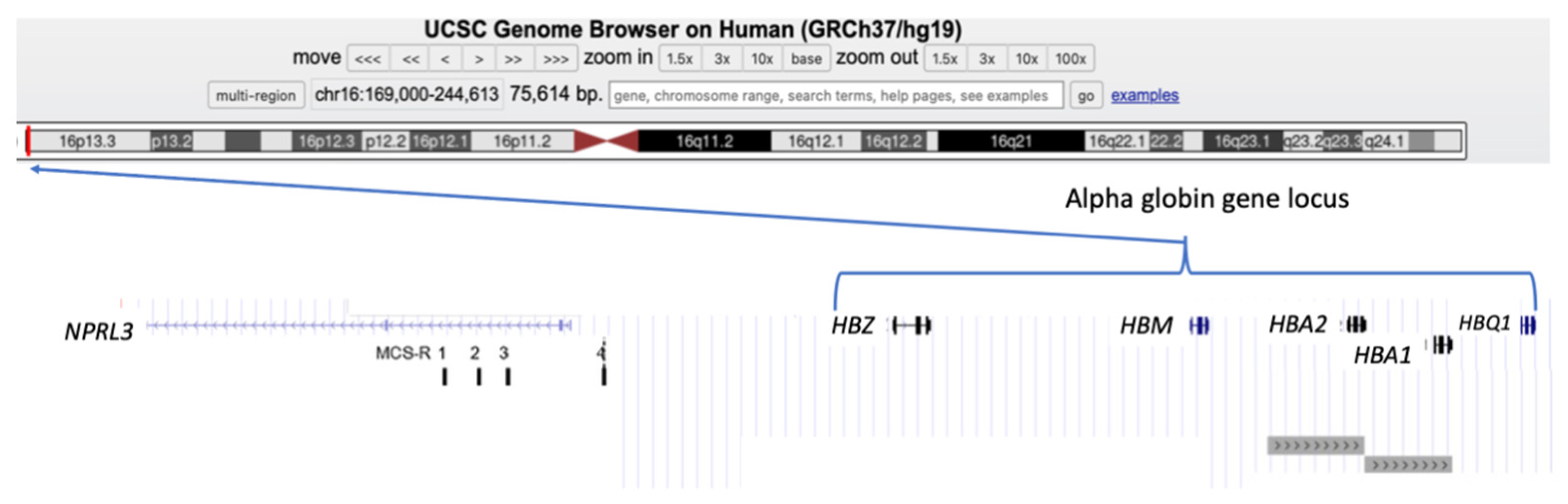
3. Alpha-Globin Gene Cluster and Alpha-Globin Biosynthesis
3.1. Normal Expression of Alpha-Globin Genes
3.2. Genomic Variants in the Alpha-Globin Gene Cluster
4. The Impact of Changes in the Expression of Alpha-Globin Genes on Hematopoietic Pathologies: Genetic Evidence
5. The Impact of Changes in the Expression of Alpha-Globin Genes on Hematopoietic Pathologies: Evidence in the “Gene Editing Era”
6. Alpha-Hemoglobin-Stabilizing Protein (AHSP): Expression, Function, and Molecular Genetics
7. Alpha-Hemoglobin-Stabilizing Protein as a Modifier of Beta-Thalassemia
8. Alpha-Hemoglobin-Stabilizing Protein as a Modifier of Sickle Cell Disease
8.1. Interaction of Alpha-Thalassemia and Homozygous Sickle Cell Disease
8.2. Levels of Alpha-Hemoglobin-Stabilizing Protein in Sickle Cell Disease
9. Role of Alpha-Hemoglobin-Stabilizing Protein in Normal and Pathological Erythropoiesis: Updates from Studies Based on Transgenic Mouse Model Systems
10. Inducers of Alpha-Hemoglobin-Stabilizing Protein
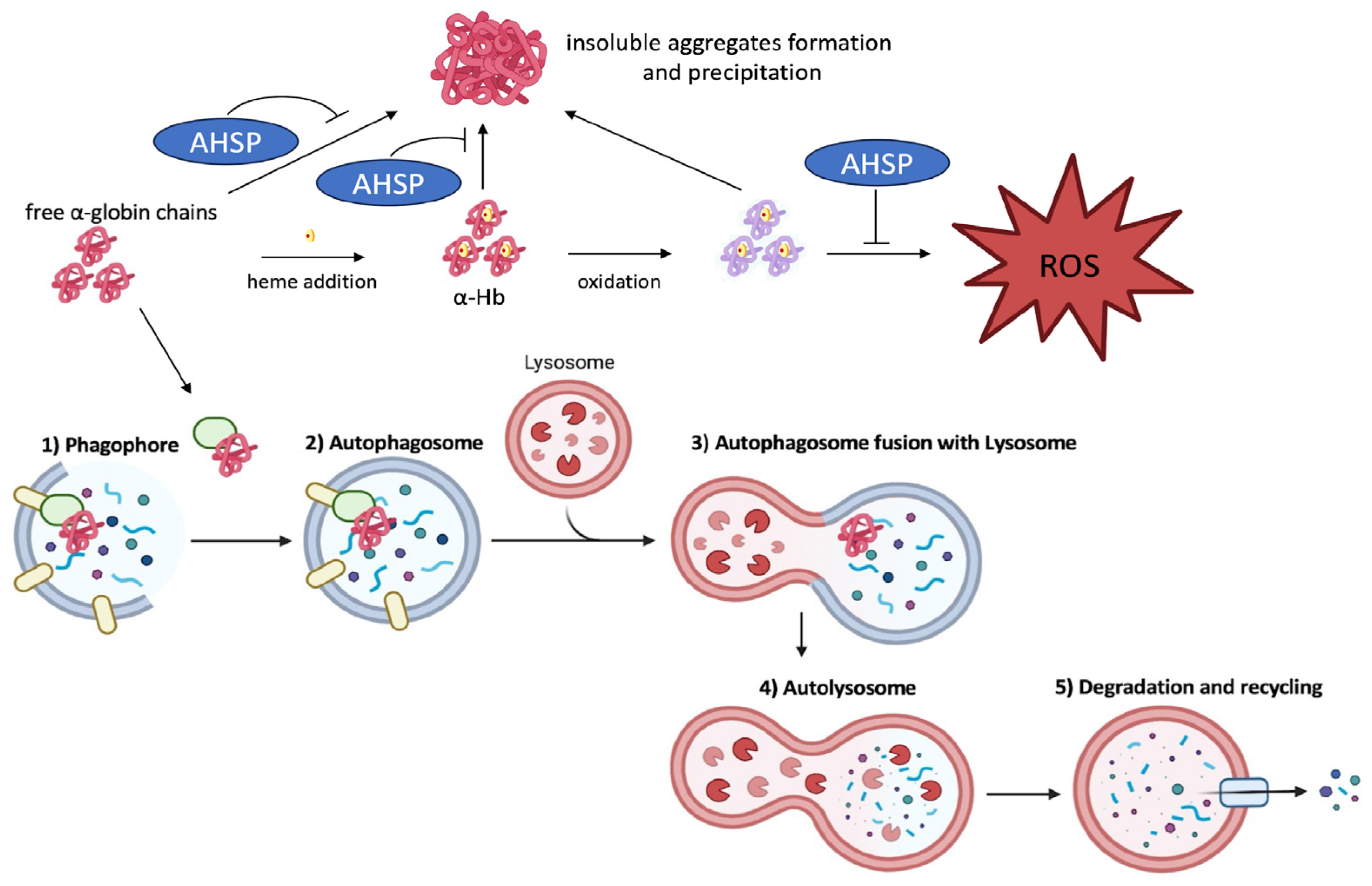
11. The Clearance of Free Alpha-Globin Is Activated by Autophagy in Erythroid Cells from Beta-Thalassemia Patients
12. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weatherall, D.J. Phenotype-genotype relationships in monogenic disease: Lessons from the thalassaemias. Nat. Rev. Genet. 2001, 2, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Higgs, D.R.; Engel, J.D.; Stamatoyannopoulos, G. Thalassaemia. Lancet 2012, 379, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L. Molecular basis of β thalassemia and potential therapeutic targets. Blood Cells Mol. Dis. 2018, 70, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sagar, C.; Sharma, D.; Kishor, P. β-globin genes: Mutation hot-spots in the global thalassemia belt. Hemoglobin 2015, 39, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rao, E.; Kumar Chandraker, S.; Misha Singh, M.; Kumar, R. Global distribution of β-thalassemia mutations: An update. Gene 2023, 896, 148022. [Google Scholar] [CrossRef] [PubMed]
- Daniels, R.J.; Peden, J.F.; Lloyd, C.; Horsley, S.W.; Clark, K.; Tufarelli, C.; Kearney, L.; Buckle, V.J.; Doggett, N.A.; Flint, J.; et al. Sequence, structure and pathology of the fully annotated terminal 2 Mb of the short arm of human chromosome 16. Hum. Mol. Genet. 2001, 10, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Xu, L.; Zhang, Y.; Liang, G.; Wei, X.; Li, L.; Jin, W.; Shang, X. Investigation of the mechanism of copy number variations involving the α-globin gene cluster on chromosome 16: Two case reports and literature review. Mol. Genet. Genom. 2023, 298, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Higgs, D.R.; Garrick, D.; Anguita, E.; De Gobbi, M.; Hughes, J.; Muers, M.; Vernimmen, D.; Lower, K.; Law, M.; Argentaro, A.; et al. Understanding alpha-globin gene regulation: Aiming to improve the management of thalassemia. Ann. N. Y. Acad. Sci. 2005, 1054, 92–102. [Google Scholar] [CrossRef]
- Harteveld, C.L.; Higgs, D.R. Alpha-thalassaemia. Orphanet J. Rare Dis. 2010, 5, 13. [Google Scholar] [CrossRef]
- Higgs, D.R.; Gibbons, R.J. The molecular basis of α-thalassemia: A model for understanding human molecular genetics. Hematol. Oncol. Clin. N. Am. 2010, 24, 1033–1054. [Google Scholar] [CrossRef]
- Kanavakis, E.; Wainscoat, J.S.; Wood, W.G.; Weatherall, D.J.; Cao, A.; Furbetta, M.; Galanello, R.; Georgiou, D.; Sophocleous, T. The interaction of alpha thalassaemia with heterozygous beta thalassaemia. Br. J. Haematol. 1982, 52, 465–473. [Google Scholar] [CrossRef]
- Wainscoat, J.S.; Thein, S.L.; Weatherall, D.J. Thalassaemia Intermedia. Blood Rev. 1987, 1, 273–279. [Google Scholar] [CrossRef]
- Kanavakis, E.; Traeger-Synodinos, J.; Lafioniatis, S.; Lazaropoulou, C.; Liakopoulou, T.; Paleologos, G.; Metaxotou-Mavrommati, A.; Stamoulakatou, A.; Papassotiriou, I. A rare example that coinheritance of a severe form of beta-thalassemia and alpha-thalassemia interact in a ‘‘synergistic’’ manner to balance the phenotype of classic thalassemic syndromes. Blood Cells Mol. Dis. 2004, 32, 319–324. [Google Scholar] [CrossRef]
- Loukopoulos, D.; Loutradi, A.; Fessas, P. A unique thalassaemic syndrome: Homozygous alpha-thalassaemia + homozygous beta-thalassaemia. Br. J. Haematol. 1978, 39, 377–389. [Google Scholar] [CrossRef]
- Panyasai, S.; Jaiping, K.; Pornprasert, S. Elevated Hb A₂ Levels in a Patient with a Compound Heterozygosity for the (β⁺)-31 (A > G) and (β⁰) Codon 17 (A > T) Mutations Together with a Single α-Globin Gene. Hemoglobin 2015, 39, 292–295. [Google Scholar] [CrossRef]
- Traeger-Synodinos, J.; Kanavakis, E.; Vrettou, C.; Maragoudaki, E.; Michael, T.; Metaxotou-Mavromati, A.; Kattamis, C. The triplicated alpha-globin gene locus in beta-thalassaemia heterozygotes: Clinical, haematological, biosynthetic and molecular studies. Br. J. Haematol. 1996, 95, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Kattamis, A.C.; Petroni, D.; Roetto, A.; Sivera, P.; Sbaiz, L.; Cohen, A.; Ohene-Frempong, K.; Trifillis, P.; Surrey, S.; et al. Different hematological phenotypes caused by the interaction of triplicated alpha-globin genes and heterozygous beta-thalassemia. Am. J. Hematol. 1997, 55, 83–88. [Google Scholar] [CrossRef]
- Ma, S.K.; Au, W.Y.; Chan, A.Y.; Chan, L.C. Clinical phenotype of triplicated alpha-globin genes and heterozygosity for beta0-thalassemia in Chinese subjects. Int. J. Mol. Med. 2001, 8, 171–175. [Google Scholar] [PubMed]
- Theodoridou, S.; Balassopoulou, A.; Boutou, E.; Delaki, E.E.; Yfanti, E.; Vyzantiadis, T.A.; Vetsiou, E.; Voskaridou, E.; Vlachaki, E. Coinheritance of Triplicated Alpha-Globin Gene and Beta-Thalassemia Mutations in Adulthood: Ten Years of Referrals in Northern Greece. J. Pediatr. Hematol. Oncol. 2020, 42, e762–e764. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, D.D.; Hira, J.K.; Chhabra, S.; Trehan, A.; Khadwal, A.R.; Malhotra, P.; Sharma, P.; Das, R. Hematological and genetic profiles of persons with co-inherited heterozygous β-thalassemia and supernumerary α-globin genes. Eur. J. Haematol. 2023, 110, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Ropero, P.; González Fernández, F.A.; Nieto, J.M.; Torres-Jiménez, W.M.; Benavente, C. β-Thalassemia Intermedia: Interaction of α-Globin Gene Triplication With β-thalassemia Heterozygous in Spain. Front. Med. 2022, 9, 866396. [Google Scholar] [CrossRef]
- Gurunathan, A.; Tarango, C.; McGann, P.T.; Niss, O.; Quinn, C.T. Non-transfusion-dependent β-Thalassemia Because of a Single β-Thalassemia Mutation and Coinherited α-Globin Gene Triplication: Need for Increased Awareness to Prevent Incorrect and Delayed Diagnosis. J. Pediatr. Hematol. Oncol. 2020, 42, e494–e496. [Google Scholar] [CrossRef]
- Giordano, P.C.; Bakker-Verwij, M.; Harteveld, C.L. Frequency of alpha-globin gene triplications and their interaction with betathalassemia mutations. Hemoglobin 2009, 33, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Hamid, M.; Keikhaei, B.; Galehdari, H.; Saberi, A.; Sedaghat, A.; Shariati, G.; Mohammadi-Anaei, M. Alpha-globin gene triplication and its effect in beta-thalassemia carrier, sickle cell trait, and healthy individual. EJHaem 2021, 2, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Beris, P.; Darbellay, R.; Hochmann, A.; Pradervand, E.; Pugin, P. Interaction of heterozygous beta (0)-thalassemia and triplicated alpha globin loci in a Swiss-Spanish family. Klin. Wochenschr. 1991, 69, 710–714. [Google Scholar] [CrossRef]
- Beris, P.; Solenthaler, M.; Deutsch, S.; Darbellay, R.; Tobler, A.; Bochaton-Pialat, M.L.; Gabbiani, G. Severe inclusion body beta-thalassaemia with haemolysis in a patient double heterozygous for beta(0)-thalassaemia and quadruplicated alpha-globin gene arrangement of the anti-4.2 type. Br. J. Haematol. 1999, 105, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Galanello, R.; Ruggeri, R.; Paglietti, E.; Addis, M.; Malis, M.A.; Cao, A. A Family with Segregating Triplicated Alpha Globin Loci and Beta Thalassemia. Blood 1983, 62, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Oron, V.; Filon, D.; Oppenheim, A.; Rund, D. Severe thalassaemia intermedia caused by interaction of homozygosity for alpha-globin gene triplication with heterozygosity for beta zero-thalassaemia. Br. J. Haematol. 1994, 86, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L.; Al-Hakim, I.; Hoffbrand, A.V. Thalassaemia intermedia: A new molecular basis. Br. J. Haematol. 1984, 56, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Harteveld, C.L.; Refaldi, C.; Cassinerio, E.; Cappellini, M.D.; Giordano, P.C. Segmental duplications involving the alpha-globin gene cluster are causing beta-thalassemia intermedia phenotypes in beta-thalassemia heterozygous patients. Blood Cells Mol. Dis. 2008, 40, 312–316. [Google Scholar] [CrossRef]
- Gu, Y.C.; Landman, H.; Huisman, T.H. Two different quadruplicated alpha globin gene arrangements. Br. J. Haematol. 1987, 66, 245–250. [Google Scholar] [CrossRef]
- Farashi, S.; Vakili, S.; Faramarzi Garous, N.; Ashki, M.; Imanian, H.; Azarkeivan, A.; Najmabadi, H. Copy number variations of six and seven α-globin genes in a family with intermedia and major thalassemia phenotypes. Expert Rev. Hematol. 2015, 8, 693–698. [Google Scholar] [CrossRef]
- Mettananda, S.; Gibbons, R.J.; Higgs, D.R. Understanding alpha-globin gene regulation and implications for the treatment of beta-thalassemia. Ann. N.Y. Acad. Sci. 2016, 1368, 16–24. [Google Scholar] [CrossRef]
- Khiabani, A.; Kohansal, M.H.; Keshavarzi, A.; Shahraki, H.; Kooshesh, M.; Karimzade, M.; Gholizadeh Navashenaq, J. CRISPR/Cas9, a promising approach for the treatment of β-thalassemia: A systematic review. Mol. Genet. Genom. 2023, 298, 1–11. [Google Scholar] [CrossRef]
- Zakaria, N.A.; Bahar, R.; Abdullah, W.Z.; Mohamed Yusoff, A.A.; Shamsuddin, S.; Abdul Wahab, R.; Johan, M.F. Genetic Manipulation Strategies for β-Thalassemia: A Review. Front. Pediatr. 2022, 10, 901605. [Google Scholar] [CrossRef]
- Rahimmanesh, I.; Boshtam, M.; Kouhpayeh, S.; Khanahmad, H.; Dabiri, A.; Ahangarzadeh, S.; Esmaeili, Y.; Bidram, E.; Vaseghi, G.; Haghjooy Javanmard, S.; et al. Gene Editing-Based Technologies for Beta-hemoglobinopathies Treatment. Biology 2022, 11, 862. [Google Scholar] [CrossRef]
- Mussolino, C.; Strouboulis, J. Recent Approaches for Manipulating Globin Gene Expression in Treating Hemoglobinopathies. Front. Genome Ed. 2021, 3, 618111. [Google Scholar] [CrossRef]
- Koniali, L.; Lederer, C.W.; Kleanthous, M. Therapy Development by Genome Editing of Hematopoietic Stem Cells. Cells 2021, 10, 1492. [Google Scholar] [CrossRef] [PubMed]
- Karamperis, K.; Tsoumpeli, M.T.; Kounelis, F.; Koromina, M.; Mitropoulou, C.; Moutinho, C.; Patrinos, G.P. Genome-based therapeutic interventions for β-type hemoglobinopathies. Hum. Genom. 2021, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Finotti, A.; Gambari, R. Combined approaches for increasing fetal hemoglobin (HbF) and de novo production of adult hemoglobin (HbA) in erythroid cells from β-thalassemia patients: Treatment with HbF inducers and CRISPR-Cas9 based genome editing. Front. Genome Ed. 2023, 5, 1204536. [Google Scholar] [CrossRef] [PubMed]
- Christakopoulos, G.E.; Telange, R.; Yen, J.; Weiss, M.J. Gene Therapy and Gene Editing for β-Thalassemia. Hematol. Oncol. Clin. N. Am. 2023, 37, 433–447. [Google Scholar] [CrossRef]
- Paschoudi, K.; Yannaki, E.; Psatha, N. Precision Editing as a Therapeutic Approach for β-Hemoglobinopathies. Int. J. Mol. Sci. 2023, 24, 9527. [Google Scholar] [CrossRef]
- Gabr, H.; El Ghamrawy, M.K.; Almaeen, A.H.; Abdelhafiz, A.S.; Hassan, A.O.S.; El Sissy, M.H. CRISPR-mediated gene modification of hematopoietic stem cells with beta-thalassemia IVS-1-110 mutation. Stem Cell Res. Ther. 2020, 11, 390. [Google Scholar] [CrossRef]
- Lu, D.; Gong, X.; Fang, Y.; Guo, X.; Chen, Y.; Yang, F.; Zhao, G.; Ma, Q.; Zeng, Y.; Zeng, F. Correction of RNA splicing defect in β654-thalassemia mice using CRISPR/Cas9 gene-editing technology. Haematologica 2022, 107, 1427–1437. [Google Scholar] [CrossRef]
- Cosenza, L.C.; Gasparello, J.; Romanini, N.; Zurlo, M.; Zuccato, C.; Gambari, R.; Finotti, A. Efficient CRISPR-Cas9-based genome editing of β-globin gene on erythroid cells from homozygous β039-thalassemia patients. Mol. Ther. Methods Clin. Dev. 2021, 21, 507–523. [Google Scholar] [CrossRef]
- Mettananda, S. Genetic and Epigenetic Therapies for β-Thalassaemia by Altering the Expression of α-Globin Gene. Front. Genome Ed. 2021, 3, 752278. [Google Scholar] [CrossRef]
- Cromer, M.K.; Camarena, J.; Martin, R.M.; Lesch, B.J.; Vakulskas, C.A.; Bode, N.M.; Kurgan, G.; Collingwood, M.A.; Rettig, G.R.; Behlke, M.A.; et al. Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells. Nat. Med. 2021, 27, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Pavani, G.; Fabiano, A.; Laurent, M.; Amor, F.; Cantelli, E.; Chalumeau, A.; Maule, G.; Tachtsidi, A.; Concordet, J.P.; Cereseto, A.; et al. Correction of β-thalassemia by CRISPR/Cas9 editing of the α-globin locus in human hematopoietic stem cells. Blood Adv. 2021, 5, 1137–1153. [Google Scholar] [CrossRef]
- Mettananda, S.; Fisher, C.A.; Hay, D.; Badat, M.; Quek, L.; Clark, K.; Hublitz, P.; Downes, D.; Kerry, J.; Gosden, M.; et al. Editing an α-globin enhancer in primary human hematopoietic stem cells as a treatment for β-thalassemia. Nat. Commun. 2017, 8, 424. [Google Scholar] [CrossRef] [PubMed]
- Kihm, A.J.; Kong, Y.; Hong, W.; Russell, J.E.; Rouda, S.; Adachi, K.; Simon, M.C.; Blobel, G.A.; Weiss, M.J. An abundant erythroid protein that stabilizes free alpha-haemoglobin. Nature 2002, 417, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Favero, M.E.; Costa, F.F. Alpha-hemoglobin-stabilizing protein: An erythroid molecular chaperone. Biochem. Res. Int. 2011, 2011, 373859. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.G.; Liem, R.I.; Wong, E.; Weiss, M.J. GATA-1 and Oct-1 are required for expression of the human alpha-hemoglobin-stabilizing protein gene. J. Biol. Chem. 2005, 280, 39016–39023. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, C.O.; Duarte, A.S.; Saad, S.T.; Costa, F.F. Expression of alpha-hemoglobin stabilizing protein gene during human erythropoiesis. Exp. Hematol. 2004, 32, 157–162. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, C.O.; Costa, F.F. AHSP and beta-thalassemia: A possible genetic modifier. Hematology 2005, 10, 157–161. [Google Scholar] [CrossRef]
- Varricchio, L.; Fabucci, M.E.; Alfani, E.; Godbold, J.; Migliaccio, A.R. Compensated variability in the expression of globin-related genes in erythroblasts generated ex vivo from different donors. Transfusion 2010, 50, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Cao, C.; Yang, X.; Zhao, G.W.; Hu, X.J.; Yu, D.L.; Yang, R.F.; Yang, K.; Zhang, Y.Y.; Wang, W.-T.; et al. Nrf2 expands the intracellular pool of the chaperone AHSP in a cellular model of β-thalassemia. Redox Biol. 2022, 50, 102239. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, C.O.; Dore, L.C.; Valentine, E.; Shelat, S.G.; Hardison, R.C.; Ghosh, M.; Wang, W.; Eisenstein, R.S.; Costa, F.F.; Weiss, M.J. An iron responsive element-like stem-loop regulates alpha-hemoglobin-stabilizing protein mRNA. J. Biol. Chem. 2008, 283, 26956–26964. [Google Scholar] [CrossRef] [PubMed]
- Viprakasit, V.; Tanphaichitr, V.S.; Chinchang, W.; Sangkla, P.; Weiss, M.J.; Higgs, D.R. Evaluation of alpha hemoglobin stabilizing protein (AHSP) as a genetic modifier in patients with beta thalassemia. Blood 2004, 103, 3296–3299. [Google Scholar] [CrossRef]
- dos Santos, C.O.; Zhou, S.; Secolin, R.; Wang, X.; Cunha, A.F.; Higgs, D.R.; Kwiatkowski, J.L.; Thein, S.L.; Gallagher, P.G.; Costa, F.F.; et al. Population analysis of the alpha hemoglobin stabilizing protein (AHSP) gene identifies sequence variants that alter expression and function. Am. J. Hematol. 2008, 83, 103–108. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, W.; Li, Y.; Shang, X.; Zhang, X.; Xiong, F.; Xu, X. Analysis of alpha-hemoglobin-stabilizing protein (AHSP) gene as a genetic modifier to the phenotype of beta-thalassemia in Southern China. Blood Cells Mol. Dis. 2010, 45, 128–132. [Google Scholar] [CrossRef]
- Brillet, T.; Baudin-Creuza, V.; Vasseur, C.; Domingues-Hamdi, E.; Kiger, L.; Wajcman, H.; Pissard, S.; Marden, M.C. α-Hemoglobin stabilizing protein (AHSP), a kinetic scheme of the action of a human mutant, AHSPV56G. J. Biol. Chem. 2010, 285, 17986–17992. [Google Scholar] [CrossRef]
- Ray, R.; Kalantri, S.A.; Bhattacharjee, S.; Biswas, A.; Shahab, A.; Biswas, S.; Bhattacharyya, M. Association of alpha hemoglobin-stabilizing protein (AHSP) gene mutation and disease severity among HbE-beta thalassemia patients. Ann. Hematol. 2019, 98, 1827–1834. [Google Scholar] [CrossRef]
- Lai, M.I.; Jiang, J.; Silver, N.; Best, S.; Menzel, S.; Mijovic, A.; Colella, S.; Ragoussis, J.; Garner, C.; Weiss, M.J.; et al. Alpha-haemoglobin stabilising protein is a quantitative trait gene that modifies the phenotype of beta-thalassaemia. Br. J. Haematol. 2006, 133, 675–682. [Google Scholar] [CrossRef]
- Lai, M.I.; Garner, C.; Jiang, J.; Silver, N.; Best, S.; Menzel, S.; Thein, S.L. A twins heritability study on alpha hemoglobin stabilizing protein (AHSP) expression variability. Twin Res. Hum. Genet. 2010, 13, 567–572. [Google Scholar] [CrossRef]
- Kong, Y.; Zhou, S.; Kihm, A.J.; Katein, A.M.; Yu, X.; Gell, D.A.; Mackay, J.P.; Adachi, K.; Foster-Brown, L.; Louden, C.S.; et al. Loss of alpha-hemoglobin-stabilizing protein impairs erythropoiesis and exacerbates beta-thalassemia. J. Clin. Investig. 2004, 114, 1457–1466. [Google Scholar] [CrossRef]
- Galanello, R.; Perseu, L.; Giagu, N.; Sole, G. AHSP expression in beta-thalassemia carriers with thalassemia intermedia phenotype. Blood 2003, 102, 1881. [Google Scholar]
- Mahmoud, H.M.; Shoeib, A.A.; Abd El Ghany, S.M.; Reda, M.M.; Ragab, I.A. Study of alpha hemoglobin stabilizing protein expression in patients with β thalassemia and sickle cell anemia and its impact on clinical severity. Blood Cells Mol. Dis. 2015, 55, 358–362. [Google Scholar] [CrossRef]
- Che Yaacob, N.S.; Islam, M.A.; Alsaleh, H.; Ibrahim, I.K.; Hassan, R. Alpha-hemoglobin-stabilizing protein (AHSP): A modulatory factor in β-thalassemia. Int. J. Hematol. 2020, 111, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.; Johnston, J.M.; Hein, M.; Kipp, B.R.; Coon, L.; Savedra, M.E.; Hoyer, J.D.; He, R.; Rangan, A.; Shi, M.; et al. Further Characterization of Hb Bronovo [α103(G10)His→Leu; HBA2: C.311A>T] and First Report of the Homozygous State. Hemoglobin 2020, 44, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Cardiero, G.; Musollino, G.; Friscia, M.G.; Testa, R.; Virruso, L.; Di Girgenti, C.; Caldora, M.; Colella Bisogno, R.; Gaudiano, C.; Manco, G.; et al. Effect of Mutations on mRNA and Globin Stability: The Cases of Hb Bernalda/Groene Hart and Hb Southern Italy. Genes 2020, 11, 870. [Google Scholar] [CrossRef] [PubMed]
- Cardiero, G.; Musollino, G.; Prezioso, R.; Lacerra, G. mRNA Analysis of Frameshift Mutations with Stop Codon in the Last Exon: The Case of Hemoglobins Campania [α1 cod95 (-C)] and Sciacca [α1 cod109 (-C)]. Biomedicines 2021, 9, 1390. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, C.; Domingues-Hamdi, E.; Pakdaman, S.; Galactéros, F.; Baudin-Creuza, V. Alpha haemoglobin-stabilising protein concentration in the red blood cells of patients with sickle cell anaemia with and without hydroxycarbamide treatment. Br. J. Haematol. 2022, 196, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Higgs, D.R.; Aldridge, B.E.; Lamb, J.; Clegg, J.B.; Weatherall, D.J.; Hayes, R.J.; Grandison, Y.; Lowrie, Y.; Mason, K.P.; Serjeant, B.E.; et al. The interaction of alpha-thalassemia and homozygous sickle-cell disease. N. Engl. J. Med. 1982, 306, 1441–1446. [Google Scholar] [CrossRef]
- Embury, S.H.; Dozy, A.M.; Miller, J.; Davis, J.R., Jr.; Kleman, K.M.; Preisler, H.; Vichinsky, E.; Lande, W.N.; Lubin, B.H.; Kan, Y.W.; et al. Concurrent sickle-cell anemia and alpha-thalassemia: Effect on severity of anemia. N. Engl. J. Med. 1982, 306, 270–274. [Google Scholar] [CrossRef]
- de Ceulaer, K.; Higgs, D.R.; Weatherall, D.J.; Hayes, R.J.; Serjeant, B.E.; Serjeant, G.R. alpha-Thalassemia reduces the hemolytic rate in homozygous sickle-cell disease. N. Engl. J. Med. 1983, 309, 189–190. [Google Scholar]
- Kirkham, J.K.; Estepp, J.H.; Weiss, M.J.; Rashkin, S.R. Genetic Variation and Sickle Cell Disease Severity: A Systematic Review and Meta-Analysis. JAMA Netw. Open 2023, 6, e2337484. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E. Overview of Compound Sickle Cell Syndromes. Available online: https://medilib.ir/uptodate/show/7115 (accessed on 24 January 2024).
- Vasseur, C.; Domingues-Hamdi, E.; Pakdaman, S.; Barau, C.; Pissard, S.; Le Corvoisier, P.; Pirenne, F.; Galactéros, F.; Baudin-Creuza, V. Elevated soluble α-hemoglobin pool in sickle cell anemia. Am. J. Hematol. 2017, 92, E593–E595. [Google Scholar] [CrossRef]
- Domingues-Hamdi, E.; Vasseur, C.; Pakdaman, S.; Moutereau, S.; Habibi, A.; Bartolucci, P.; Galactéros, F.; Baudin-Creuza, V. Hydroxycarbamide decreases the free alpha-hemoglobin pool in red blood cells of adult patients with sickle cell anemia. Am. J. Hematol. 2020, 95, E302–E305. [Google Scholar] [CrossRef]
- Wang, B.; Fang, Y.; Guo, X.; Ren, Z.; Zhang, J. Transgenic human alpha-hemoglobin stabilizing protein could partially relieve betaIVS-2-654-thalassemia syndrome in model mice. Hum. Gene Ther. 2010, 21, 149–156. [Google Scholar] [CrossRef]
- Nasimuzzaman, M.; Khandros, E.; Wang, X.; Kong, Y.; Zhao, H.; Weiss, D.; Rivella, S.; Weiss, M.J.; Persons, D.A. Analysis of alpha hemoglobin stabilizing protein overexpression in murine β-thalassemia. Am. J. Hematol. 2010, 85, 820–822. [Google Scholar] [CrossRef]
- Weiss, M.J.; Zhou, S.; Feng, L.; Gell, D.A.; Mackay, J.P.; Shi, Y.; Gow, A.J. Role of alpha-hemoglobin-stabilizing protein in normal erythropoiesis and beta-thalassemia. Ann. N. Y. Acad. Sci. 2005, 1054, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Li, P.; Zang, S.; Liu, Q.; Ma, D.; Sun, X.; Ji, C. Novel agent nitidine chloride induces erythroid differentiation and apoptosis in CML cells through c-Myc-miRNAs axis. PLoS ONE 2015, 10, e0116880. [Google Scholar] [CrossRef] [PubMed]
- Treasure, K.; Harris, J.; Williamson, G. Exploring the anti-inflammatory activity of sulforaphane. Immunol. Cell Biol. 2023, 101, 805–828. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, W.; Zennadi, R. Keap1-Nrf2 Heterodimer: A Therapeutic Target to Ameliorate Sickle Cell Disease. Antioxidants 2023, 12, 740. [Google Scholar] [CrossRef]
- Maciel, T.T.; Carvalho, C.; Rignault, R.; Andemariam, B. IMR-261, a Novel Oral Nrf2 Activator, Induces Fetal Hemoglobin in Human Erythroblasts, Reduces VOCs, and Ameliorates Ineffective Erythropoiesis in Experimental Mouse Models of Sickle Cell Disease and Beta-Thalassemia. Blood 2021, 138 (Suppl. S1), 853. [Google Scholar] [CrossRef]
- Hodge, D.; Coghill, E.; Keys, J.; Maguire, T. A global role for EKLF in definitive and primitive erythropoiesis. Blood 2006, 107, 3359–3370. [Google Scholar] [CrossRef]
- Pilon, A.M.; Nilson, D.G.; Zhou, D.; Sangerman, J. Alterations in expression and chromatin configuration of the alpha hemoglobin-stabilizing protein gene in erythroid Kruppel-like factor-deficient mice. Mol. Cell Biol. 2006, 26, 4368–4377. [Google Scholar] [CrossRef]
- Cao, C.; Zhao, G.; Yu, W.; Xie, X.; Wang, W.; Yang, R.; Lv, X.; Liu, D. Activation of STAT3 stimulates AHSP expression in K562 cells. Sci. China Life. Sci. 2014, 57, 488–494. [Google Scholar] [CrossRef]
- Pinho, F.O.; de Albuquerque, D.M.; Olalla Saad, S.T.; Costa, F.F. Reduction of AHSP synthesis in hemin-induced K562 cells and EPO-induced CD34(+) cells leads to alpha-globin precipitation, impairment of normal hemoglobin production, and increased cell death. Exp. Hematol. 2008, 36, 265–272. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar]
- Grosso, R.; Fader, C.M.; Colombo, M.I. Autophagy: A necessary event during erythropoiesis. Blood Rev. 2017, 31, 300–305. [Google Scholar] [CrossRef]
- Li, J.; Quan, C.; He, Y.L.; Cao, Y.; Chen, Y.; Wang, Y.F.; Wu, L.Y. Autophagy regulated by the HIF/REDD1/mTORC1 signaling is progressively increased during erythroid differentiation under hypoxia. Front. Cell Dev. Biol. 2022, 10, 896893. [Google Scholar] [CrossRef]
- Stolla, M.C.; Reilly, A.; Bergantinos, R.; Stewart, S.; Thom, N.; Clough, C.A.; Wellington, R.C.; Stolitenko, R.; Abkowitz, J.L.; Doulatov, S. ATG4A regulates human erythroid maturation and mitochondrial clearance. Blood Adv. 2022, 6, 3579–3589. [Google Scholar] [CrossRef]
- Castillo-Castellanos, F.; Ramírez, L.; Lomelí, H. zmiz1a zebrafish mutants have defective erythropoiesis, altered expression of autophagy genes, and a deficient response to vitamin D. Life Sci. 2021, 284, 119900. [Google Scholar] [CrossRef] [PubMed]
- Matte, A.; De Franceschi, L. Oxidation and erythropoiesis. Curr. Opin. Hematol. 2019, 26, 145–151. [Google Scholar] [CrossRef]
- Grosso, R.A.; Caldarone, P.V.S.; Sánchez, M.C.; Chiabrando, G.A.; Colombo, M.I.; Fader, C.M. Hemin induces autophagy in a leukemic erythroblast cell line through the LRP1 receptor. Biosci. Rep. 2019, 39, BSR20181156. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, K.; Xiao, X.; Liao, J.; Hu, Q.; Chen, H.; Liu, J.; An, X. Autophagy as a regulatory component of erythropoiesis. Int. J. Mol. Sci. 2015, 16, 4083–4094. [Google Scholar] [CrossRef] [PubMed]
- Lechauve, C.; Keith, J.; Khandros, E.; Fowler, S.; Mayberry, K.; Freiwan, A.; Thom, C.S.; Delbini, P.; Romero, E.B.; Zhang, J.; et al. The autophagy-activating kinase ULK1 mediates clearance of free α-globin in β-thalassemia. Sci. Transl. Med. 2019, 11, eaav4881. [Google Scholar] [CrossRef] [PubMed]
- Chaichompoo, P.; Nithipongvanitch, R.; Kheansaard, W.; Tubsuwan, A.; Srinoun, K.; Vadolas, J.; Fucharoen, S.; Smith, D.R.; Winichagoon, P.; Svasti, S. Increased autophagy leads to decreased apoptosis during β-thalassaemic mouse and patient erythropoiesis. Sci. Rep. 2022, 12, 18628. [Google Scholar] [CrossRef]
- Chaichompoo, P.; Svasti, S.; Smith, D.R. The Roles of Mitophagy and Autophagy in Ineffective Erythropoiesis in β-Thalassemia. Int. J. Mol. Sci. 2022, 23, 10811. [Google Scholar] [CrossRef]
- Wu, L.; Xu, W.; Xu, L.; Kong, Q.; Fang, J. Mitophagy is increased during erythroid differentiation in β-thalassemia. Int. J. Hematol. 2017, 105, 162–173. [Google Scholar] [CrossRef]
- Lithanatudom, P.; Wannatung, T.; Leecharoenkiat, A.; Svasti, S.; Fucharoen, S.; Smith, D.R. Enhanced activation of autophagy in β-thalassemia/Hb E erythroblasts during erythropoiesis. Ann. Hematol. 2011, 90, 747–758. [Google Scholar] [CrossRef]
- Khandros, E.; Thom, C.S.; D’Souza, J.; Weiss, M.J. Integrated protein quality-control pathways regulate free α-globin in murine β-thalassemia. Blood 2012, 119, 5265–5275. [Google Scholar] [CrossRef]
- Martínez-Borra, J.; López-Larrea, C. Autophagy and self-defense. Adv. Exp. Med. Biol. 2012, 738, 169–184. [Google Scholar] [PubMed]
- Zurlo, M.; Gasparello, J.; Cosenza, L.C.; Breveglieri, G.; Papi, C.; Zuccato, C.; Gambari, R.; Finotti, A. Production and Characterization of K562 Cellular Clones Hyper-Expressing the Gene Encoding α-Globin: Preliminary Analysis of Biomarkers Associated with Autophagy. Genes 2023, 14, 556. [Google Scholar] [CrossRef] [PubMed]
- Fibach, E.; Bianchi, N.; Borgatti, M.; Zuccato, C.; Finotti, A.; Lampronti, I.; Prus, E.; Mischiati, C.; Gambari, R. Effects of rapamycin on accumulation of alpha-, beta- and gamma-globin mRNAs in erythroid precursor cells from beta-thalassaemia patients. Eur. J. Haematol. 2006, 77, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cosenza, L.C.; Zurlo, M.; Gasparello, J.; Papi, C.; D’Aversa, E.; Breveglieri, G.; Lampronti, I.; Finotti, A.; Borgatti, M.; et al. Expression of γ-globin genes in β-thalassemia patients treated with sirolimus: Results from a pilot clinical trial (Sirthalaclin). Ther. Adv. Hematol. 2022, 13, 20406207221100648. [Google Scholar] [CrossRef] [PubMed]
- Zurlo, M.; Zuccato, C.; Cosenza, L.C.; Gasparello, J.; Gamberini, M.R.; Stievano, A.; Fortini, M.; Prosdocimi, M.; Finotti, A.; Gambari, R. Decrease of α-globin and increase of the autophagy-activating kinase ULK1 mRNA in erythroid precursors from β-thalassemia patients treated with sirolimus. Int. J. Mol. Sci. 2023, 24, 15049. [Google Scholar] [CrossRef] [PubMed]
- Keith, J.; Christakopoulos, G.E.; Fernandez, A.G.; Yao, Y.; Zhang, J.; Mayberry, K.; Telange, R.; Sweileh, R.B.A.; Dudley, M.; Westbrook, C.; et al. Loss of miR-144/451 alleviates β-thalassemia by stimulating ULK1-mediated autophagy of free α-globin. Blood 2023, 142, 918–932. [Google Scholar] [CrossRef] [PubMed]
- Kountouris, P.; Stephanou, C.; Archer, N.; Bonifazi, F.; Giannuzzi, V.; Kuo, K.H.M.; Maggio, A.; Makani, J.; Mañú-Pereira, M.D.M.; Michailidou, K.; et al. The International Hemoglobinopathy Research Network (INHERENT): An international initiative to study the role of genetic modifiers in hemoglobinopathies. Am. J. Hematol. 2021, 96, E416–E420. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Traeger-Synodinos, J.; Vrettou, C.; Sofocleous, C.; Zurlo, M.; Finotti, A.; Gambari, R., on behalf of the International Hemoglobinopathy Research Network (INHERENT). Impact of α-Globin Gene Expression and α-Globin Modifiers on the Phenotype of β-Thalassemia and Other Hemoglobinopathies: Implications for Patient Management. Int. J. Mol. Sci. 2024, 25, 3400. https://doi.org/10.3390/ijms25063400
Traeger-Synodinos J, Vrettou C, Sofocleous C, Zurlo M, Finotti A, Gambari R on behalf of the International Hemoglobinopathy Research Network (INHERENT). Impact of α-Globin Gene Expression and α-Globin Modifiers on the Phenotype of β-Thalassemia and Other Hemoglobinopathies: Implications for Patient Management. International Journal of Molecular Sciences. 2024; 25(6):3400. https://doi.org/10.3390/ijms25063400
Chicago/Turabian StyleTraeger-Synodinos, Joanne, Christina Vrettou, Christalena Sofocleous, Matteo Zurlo, Alessia Finotti, and Roberto Gambari on behalf of the International Hemoglobinopathy Research Network (INHERENT). 2024. "Impact of α-Globin Gene Expression and α-Globin Modifiers on the Phenotype of β-Thalassemia and Other Hemoglobinopathies: Implications for Patient Management" International Journal of Molecular Sciences 25, no. 6: 3400. https://doi.org/10.3390/ijms25063400
APA StyleTraeger-Synodinos, J., Vrettou, C., Sofocleous, C., Zurlo, M., Finotti, A., & Gambari, R., on behalf of the International Hemoglobinopathy Research Network (INHERENT). (2024). Impact of α-Globin Gene Expression and α-Globin Modifiers on the Phenotype of β-Thalassemia and Other Hemoglobinopathies: Implications for Patient Management. International Journal of Molecular Sciences, 25(6), 3400. https://doi.org/10.3390/ijms25063400