

Computational Characterization of Membrane Proteins as Anticancer Targets: Current Challenges and Opportunities

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Key Experimental and Computational Challenges in the Study of Membrane Proteins
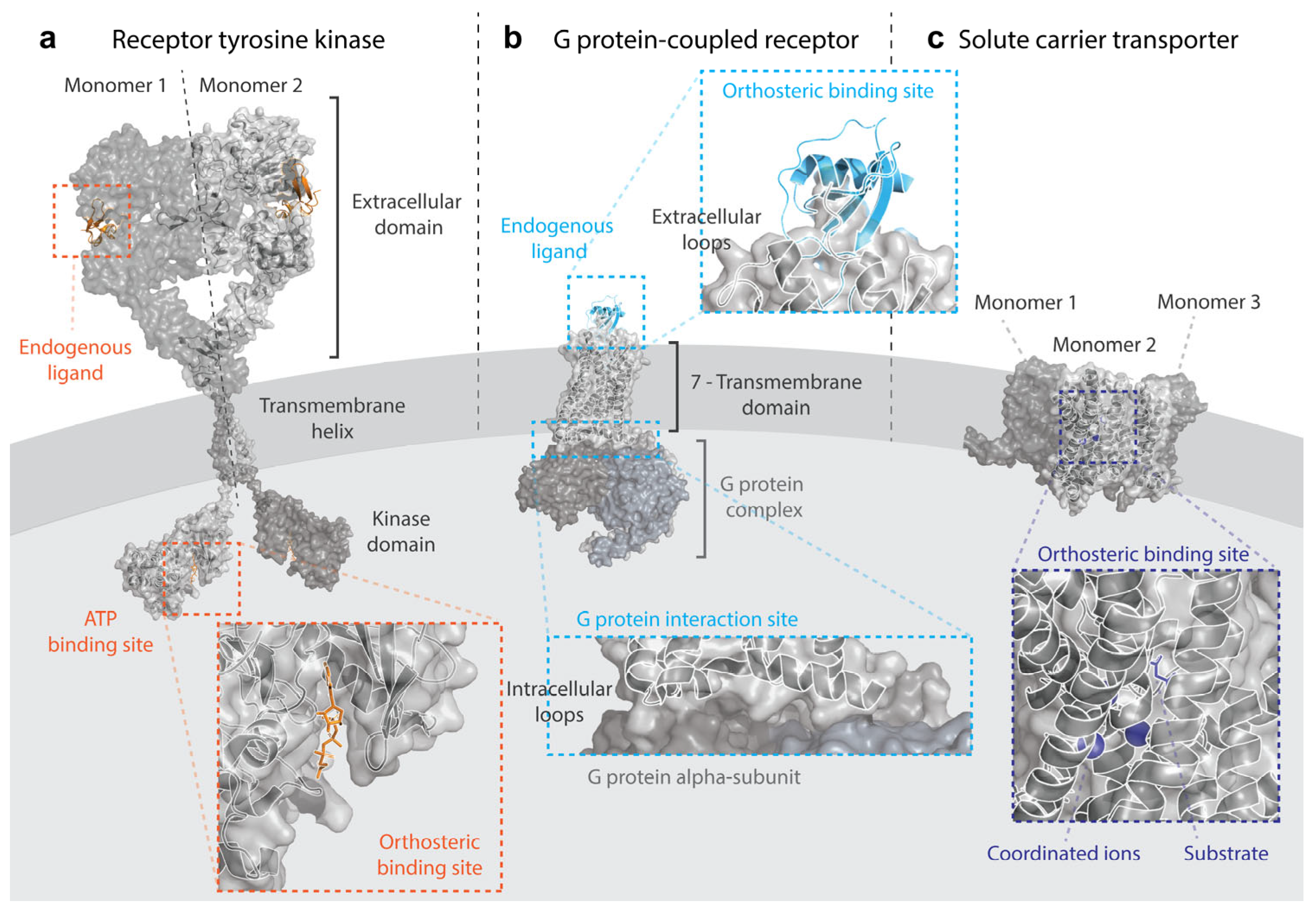
3. Receptor Tyrosine Kinases
4. G Protein-Coupled Receptors
5. Solute Carriers
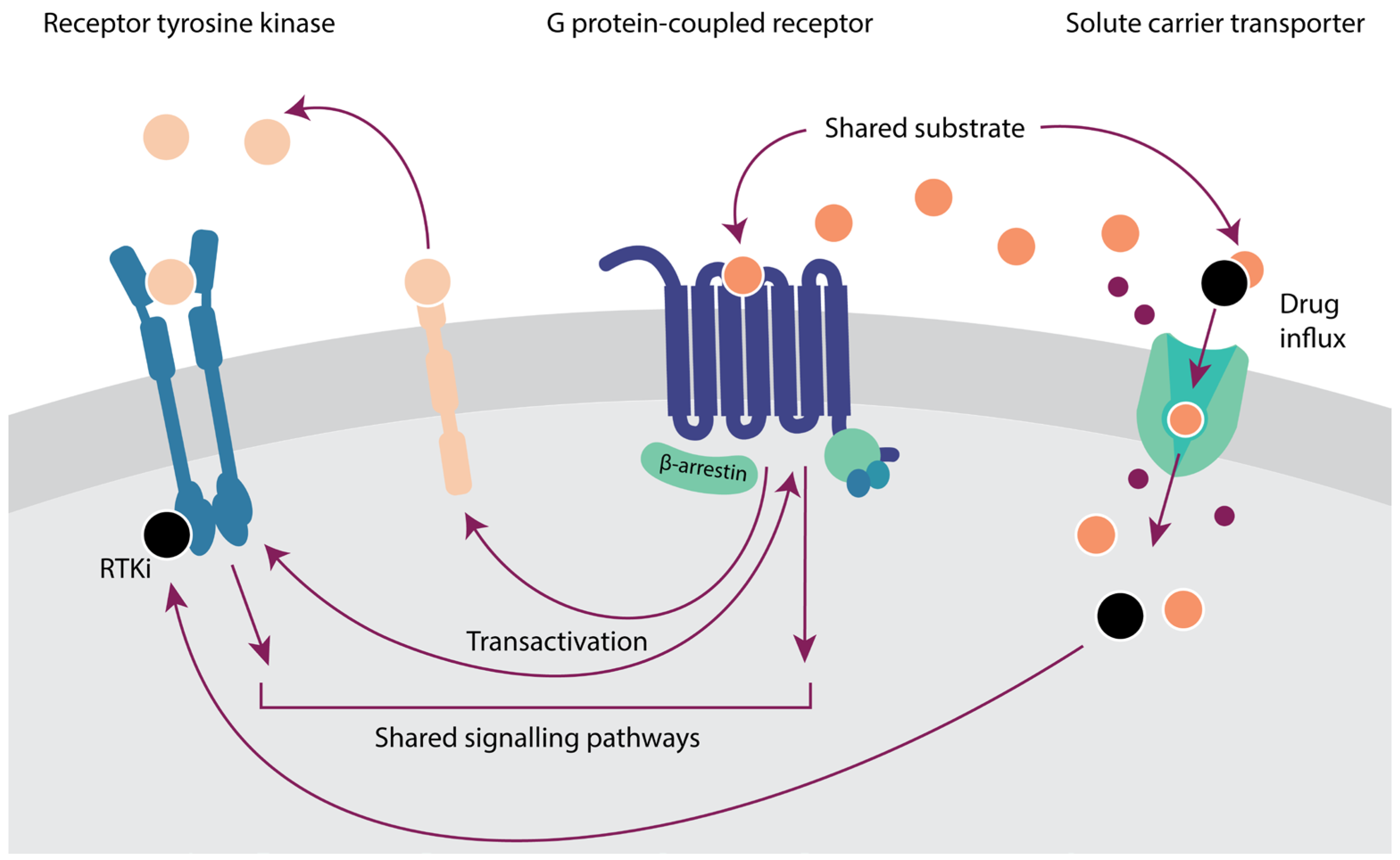
6. Crosstalk between Membrane Proteins
7. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Kampen, K.R. Membrane Proteins: The Key Players of a Cancer Cell. J. Membr. Biol. 2011, 242, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Lee, C.-H.; Chuang, Y.-H.; Lee, J.-Y.; Chiu, Y.-Y.; Wu Lee, Y.-H.; Jong, Y.-J.; Hwang, J.-K.; Huang, S.-H.; Chen, L.-C.; et al. Membrane Protein-Regulated Networks across Human Cancers. Nat. Commun. 2019, 10, 3131. [Google Scholar] [CrossRef] [PubMed]
- De Jong, E.; Kocer, A. Current Methods for Identifying Plasma Membrane Proteins as Cancer Biomarkers. Membranes 2023, 13, 409. [Google Scholar] [CrossRef] [PubMed]
- Sojo, V.; Dessimoz, C.; Pomiankowski, A.; Lane, N. Membrane Proteins Are Dramatically Less Conserved than Water-Soluble Proteins across the Tree of Life. Mol. Biol. Evol. 2016, 33, 2874–2884. [Google Scholar] [CrossRef]
- Hedin, L.E.; Illergård, K.; Elofsson, A. An Introduction to Membrane Proteins. J. Proteome Res. 2011, 10, 3324–3331. [Google Scholar] [CrossRef]
- Sowlati-Hashjin, S.; Gandhi, A.; Garton, M. Dawn of a New Era for Membrane Protein Design. BioDesign Res. 2022, 2022, 9791435. [Google Scholar] [CrossRef]
- Rahman, M.M.; Islam, M.R.; Rahman, F.; Rahaman, M.S.; Khan, M.S.; Abrar, S.; Ray, T.K.; Uddin, M.B.; Kali, M.S.K.; Dua, K.; et al. Emerging Promise of Computational Techniques in Anti-Cancer Research: At a Glance. Bioengineering 2022, 9, 335. [Google Scholar] [CrossRef]
- Sadybekov, A.V.; Katritch, V. Computational Approaches Streamlining Drug Discovery. Nature 2023, 616, 673–685. [Google Scholar] [CrossRef]
- Gorostiola González, M.; Janssen, A.P.A.; IJzerman, A.P.; Heitman, L.H.; van Westen, G.J.P. Oncological Drug Discovery: AI Meets Structure-Based Computational Research. Drug Discov. Today 2022, 27, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B.; RenéRen, I.; Rachou, R.; Oswaldo Cruz, F. DynaMut: Predicting the Impact of Mutations on Protein Conformation, Flexibility and Stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef] [PubMed]
- Gorostiola González, M.; Sijben, H.J.; Dall’ Acqua, L.; Liu, R.; IJzerman, A.P.; Heitman, L.H.; van Westen, G.J.P. Molecular Insights into Disease-Associated Glutamate Transporter (EAAT1/SLC1A3) Variants Using in Silico and in Vitro Approaches. Front. Mol. Biosci. 2023, 10, 3389. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Shi, S.; Sun, X.; Lu, M.; Liao, Y.; Zhu, S.; Zhang, H.; Pan, Z.; Fang, P.; Zeng, Z.; et al. MoDAFold: A Strategy for Predicting the Structure of Missense Mutant Protein Based on AlphaFold2 and Molecular Dynamics. Brief. Bioinform. 2024, 25, bbae006. [Google Scholar] [CrossRef] [PubMed]
- Burggraaff, L.; Lenselink, E.B.; Jespers, W.; van Engelen, J.; Bongers, B.J.; Gorostiola González, M.; Liu, R.; Hoos, H.H.; van Vlijmen, H.W.T.; IJzerman, A.P.; et al. Successive Statistical and Structure-Based Modeling to Identify Chemically Novel Kinase Inhibitors. J. Chem. Inf. Model. 2020, 60, 4283–4295. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.-W.; Li, J.-H.; Tsai, J.-Y.; Lin, S.-H.; Chang, G.-C.; Liu, C.-C.; Chen, J.J. Pharmacophore-Based Virtual Screening for the Identification of the Novel Src Inhibitor SJG-136 against Lung Cancer Cell Growth and Motility. Am. J. Cancer Res. 2020, 10, 1668–1690. [Google Scholar] [PubMed]
- Mohanan, A.; Melge, A.R.; Mohan, C.G. Predicting the Molecular Mechanism of EGFR Domain II Dimer Binding Interface by Machine Learning to Identify Potent Small Molecule Inhibitor for Treatment of Cancer. J. Pharm. Sci. 2020, 110, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Sakellaropoulos, T.; Vougas, K.; Narang, S.; Koinis, F.; Kotsinas, A.; Polyzos, A.; Moss, T.J.; Piha-Paul, S.; Zhou, H.; Kardala, E.; et al. A Deep Learning Framework for Predicting Response to Therapy in Cancer. Cell Rep. 2019, 29, 3367–3373.e4. [Google Scholar] [CrossRef]
- Wu, V.; Yeerna, H.; Nohata, N.; Chiou, J.; Harismendy, O.; Raimondi, F.; Inoue, A.; Russell, R.B.; Tamayo, P.; Gutkind, J.S. Illuminating the Onco-GPCRome: Novel G Protein-Coupled Receptor-Driven Oncocrine Networks and Targets for Cancer Immunotherapy. J. Biol. Chem. 2019, 294, 11062–11086. [Google Scholar] [CrossRef]
- Lavoro, A.; Falzone, L.; Tomasello, B.; Conti, G.N.; Libra, M.; Candido, S. In Silico Analysis of the Solute Carrier (SLC) Family in Cancer Indicates a Link among DNA Methylation, Metabolic Adaptation, Drug Response, and Immune Reactivity. Front. Pharmacol. 2023, 14, 1191262. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of Receptor Tyrosine Kinase Activation in Cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Saraon, P.; Pathmanathan, S.; Snider, J.; Lyakisheva, A.; Wong, V.; Stagljar, I. Receptor Tyrosine Kinases and Cancer: Oncogenic Mechanisms and Therapeutic Approaches. Oncogene 2021, 40, 4079–4093. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gallo, L.; Jadhav, A.; Hawkins, R.; Parker, C.G. The Druggability of Solute Carriers. J. Med. Chem. 2020, 63, 3834–3867. [Google Scholar] [CrossRef]
- Born, J.; Manica, M. Trends in Deep Learning for Property-Driven Drug Design. Curr. Med. Chem. 2021, 28, 7862–7886. [Google Scholar] [CrossRef]
- Lee, A.G. How Lipids Affect the Activities of Integral Membrane Proteins. Biochim. Biophys. Acta 2004, 1666, 62–87. [Google Scholar] [CrossRef] [PubMed]
- Kermani, A.A. A Guide to Membrane Protein X-Ray Crystallography. FEBS J. 2021, 288, 5788–5804. [Google Scholar] [CrossRef] [PubMed]
- Errasti-Murugarren, E.; Bartoccioni, P.; Palacín, M. Membrane Protein Stabilization Strategies for Structural and Functional Studies. Membranes 2021, 11, 155. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, version 2.5.2; Schrödinger, LLC: New York, NY, USA, 2022.
- Piper, S.J.; Johnson, R.M.; Wootten, D.; Sexton, P.M. Membranes under the Magnetic Lens: A Dive into the Diverse World of Membrane Protein Structures Using Cryo-EM. Chem. Rev. 2022, 122, 13989–14017. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Kanev, G.K.; Van Linden, O.P.J.; Leurs, R.; De Esch, I.J.P.; De Graaf, C. KLIFS: A Structural Kinase-Ligand Interaction Database. Nucleic Acids Res. 2015, 44, 365–371. [Google Scholar] [CrossRef]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR Structure Models and Ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef]
- Schlessinger, A.; Zatorski, N.; Hutchinson, K.; Colas, C. Targeting SLC Transporters: Small Molecules as Modulators and Therapeutic Opportunities. Trends Biochem. Sci. 2023, 48, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Jambrich, M.A.; Tusnady, G.E.; Dobson, L. How AlphaFold2 Shaped the Structural Coverage of the Human Transmembrane Proteome. Sci. Rep. 2023, 13, 20283. [Google Scholar] [CrossRef] [PubMed]
- Goossens, K.; De Winter, H. Molecular Dynamics Simulations of Membrane Proteins: An Overview. J. Chem. Inf. Model. 2018, 58, 2193–2202. [Google Scholar] [CrossRef]
- Škerle, J.; Humpolíčková, J.; Johnson, N.; Rampírová, P.; Poláchová, E.; Fliegl, M.; Dohnálek, J.; Suchánková, A.; Jakubec, D.; Strisovsky, K. Membrane Protein Dimerization in Cell-Derived Lipid Membranes Measured by FRET with MC Simulations. Biophys. J. 2020, 118, 1861–1875. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor-Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Regad, T. Targeting RTK Signaling Pathways in Cancer. Cancers 2015, 7, 1758–1784. [Google Scholar] [CrossRef] [PubMed]
- Hoyos, D.; Greenbaum, B.; Levine, A.J. The Genotypes and Phenotypes of Missense Mutations in the Proline Domain of the P53 Protein. Cell Death Differ. 2022, 29, 938–945. [Google Scholar] [CrossRef]
- Browne, B.C.; O’Brien, N.; Duffy, M.J.; Crown, J.; O’Donovan, N. HER-2 Signaling and Inhibition in Breast Cancer. Curr. Cancer Drug Targets 2009, 9, 419–438. [Google Scholar] [CrossRef]
- Wang, Z.; Longo, P.A.; Tarrant, M.K.; Kim, K.; Head, S.; Leahy, D.J.; Cole, P.A. Mechanistic Insights into the Activation of Oncogenic Forms of EGF Receptor. Nat. Struct. Mol. Biol. 2011, 18, 1388–1393. [Google Scholar] [CrossRef]
- Lopez-Gines, C.; Gil-Benso, R.; Ferrer-Luna, R.; Benito, R.; Serna, E.; Gonzalez-Darder, J.; Quilis, V.; Monleon, D.; Celda, B.; Cerdá-Nicolas, M. New Pattern of EGFR Amplification in Glioblastoma and the Relationship of Gene Copy Number with Gene Expression Profile. Mod. Pathol. 2010, 23, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2024 Update. Pharmacol. Res. 2024, 200, 107059. [Google Scholar] [CrossRef] [PubMed]
- Kreutzfeldt, J.; Rozeboom, B.; Dey, N.; De, P. The Trastuzumab Era: Current and Upcoming Targeted HER2+ Breast Cancer Therapies. Am. J. Cancer Res. 2020, 10, 1045–1067. [Google Scholar] [PubMed]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal Growth Factor Receptor Mutations in Lung Cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Akula, S.; Mullaguri, S.C.; Melton, N.M.; Katta, A.; Naga, V.S.G.R.; Kandula, S.; Pedada, R.K.; Subramanian, J.; Kancha, R.K. Large-Scale Pathogenicity Prediction Analysis of Cancer-Associated Kinase Mutations Reveals Variability in Sensitivity and Specificity of Computational Methods. Cancer Med. 2023, 12, 17468–17474. [Google Scholar] [CrossRef] [PubMed]
- Raghav, P.K.; Singh, A.K.; Gangenahalli, G. A Change in Structural Integrity of C-Kit Mutant D816V Causes Constitutive Signaling. Mutat. Res. 2018, 808, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Trummell, H.Q.; Rajbhandari, R.; Thudi, N.K.; Nozell, S.E.; Warram, J.M.; Willey, C.D.; Yang, E.S.; Placzek, W.J.; Bonner, J.A.; et al. Novel EGFR Ectodomain Mutations Associated with Ligand-Independent Activation and Cetuximab Resistance in Head and Neck Cancer. PLoS ONE 2020, 15, e0229077. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.M.; Raghavan, S.; Wu, Q.; Li, Y.Y.; Spurr, L.F.; Gupta, H.V.; Rubinson, D.A.; Fetter, I.J.; Hornick, J.L.; Nowak, J.A.; et al. FGFR2 Extracellular Domain In-Frame Deletions Are Therapeutically Targetable Genomic Alterations That Function as Oncogenic Drivers in Cholangiocarcinoma. Cancer Discov. 2021, 11, 2488–2505. [Google Scholar] [CrossRef]
- Ishiyama, N.; O’Connor, M.; Salomatov, A.; Romashko, D.; Thakur, S.; Mentes, A.; Hopkins, J.F.; Frampton, G.M.; Albacker, L.A.; Kohlmann, A.; et al. Computational and Functional Analyses of HER2 Mutations Reveal Allosteric Activation Mechanisms and Altered Pharmacologic Effects. Cancer Res. 2023, 83, 1531–1542. [Google Scholar] [CrossRef]
- Wagner, A.; Galicia-Andrés, E.; Teufl, M.; Gold, L.; Obinger, C.; Sykacek, P.; Oostenbrink, C.; Traxlmayr, M.W. Identification of Activating Mutations in the Transmembrane and Extracellular Domains of EGFR. Biochemistry 2022, 61, 2049–2062. [Google Scholar] [CrossRef]
- Latysheva, N.S.; Babu, M.M. Discovering and Understanding Oncogenic Gene Fusions through Data Intensive Computational Approaches. Nucleic Acids Res. 2016, 44, 4487–4503. [Google Scholar] [CrossRef]
- Hernandez, A.; Muñoz-Mármol, A.M.; Esteve-Codina, A.; Alameda, F.; Carrato, C.; Pineda, E.; Arpí-Lluciá, O.; Martinez-García, M.; Mallo, M.; Gut, M.; et al. In Silico Validation of RNA-Seq Results Can Identify Gene Fusions with Oncogenic Potential in Glioblastoma. Sci. Rep. 2022, 12, 14439. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lu, H.; Ng, P.K.-S.; Pantazi, A.; Ip, C.K.M.; Jeong, K.J.; Amador, B.; Tran, R.; Tsang, Y.H.; Yang, L.; et al. A Functional Genomic Approach to Actionable Gene Fusions for Precision Oncology. Sci. Adv. 2022, 8, eabm2382. [Google Scholar] [CrossRef] [PubMed]
- Hafstað, V.; Häkkinen, J.; Larsson, M.; Staaf, J.; Vallon-Christersson, J.; Persson, H. Improved Detection of Clinically Relevant Fusion Transcripts in Cancer by Machine Learning Classification. BMC Genom. 2023, 24, 783. [Google Scholar] [CrossRef] [PubMed]
- Lovino, M.; Urgese, G.; Macii, E.; Di Cataldo, S.; Ficarra, E. A Deep Learning Approach to the Screening of Oncogenic Gene Fusions in Humans. Int. J. Mol. Sci. 2019, 20, 1645. [Google Scholar] [CrossRef] [PubMed]
- Diwanji, D.; Thaker, T.; Jura, N. More than the Sum of the Parts: Towards Full-Length Receptor Tyrosine Kinase Structures. IUBMB Life 2019, 71, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Cruz, V.L.; Souza-Egipsy, V.; Gion, M.; Pérez-García, J.; Cortes, J.; Ramos, J.; Vega, J.F. Binding Affinity of Trastuzumab and Pertuzumab Monoclonal Antibodies to Extracellular HER2 Domain. Int. J. Mol. Sci. 2023, 24, 12031. [Google Scholar] [CrossRef]
- Hao, Y.; Yu, X.; Bai, Y.; McBride, H.J.; Huang, X. Cryo-EM Structure of HER2-Trastuzumab-Pertuzumab Complex. PLoS ONE 2019, 14, e0216095. [Google Scholar] [CrossRef] [PubMed]
- Shanehsazzadeh, A.; Bachas, S.; Kasun, G.; Sutton, J.M.; Steiger, A.K.; Shuai, R.; Kohnert, C.; Morehead, A.; Brown, A.; Chung, C.; et al. Unlocking de Novo Antibody Design with Generative Artificial Intelligence. bioRxiv 2023. [Google Scholar] [CrossRef]
- Balakrishnan, N.; Baskar, G.; Balaji, S.; Kullappan, M.; Krishna Mohan, S. Machine Learning Modeling to Identify Affinity Improved Biobetter Anticancer Drug Trastuzumab and the Insight of Molecular Recognition of Trastuzumab towards Its Antigen HER2. J. Biomol. Struct. Dyn. 2022, 40, 11638–11652. [Google Scholar] [CrossRef]
- Majumdar, S.; Di Palma, F.; Spyrakis, F.; Decherchi, S.; Cavalli, A. Molecular Dynamics and Machine Learning Give Insights on the Flexibility–Activity Relationships in Tyrosine Kinome. J. Chem. Inf. Model. 2023, 63, 4814–4826. [Google Scholar] [CrossRef]
- Reid, T.-E.; Fortunak, J.M.; Wutoh, A.; Wang, X.S. Cheminfomatic-Based Drug Discovery of Human Tyrosine Kinase Inhibitors. Curr. Top. Med. Chem. 2016, 16, 1452–1462. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.-W.; Wei, C.-H.; Tsai, J.-Y.; Lai, Y.-H.; Chang, G.-C.; Chen, J.J.W. Hybrid Pharmacophore- and Structure-Based Virtual Screening Pipeline to Identify Novel EGFR Inhibitors That Suppress Non-Small Cell Lung Cancer Cell Growth. Int. J. Mol. Sci. 2022, 23, 3487. [Google Scholar] [CrossRef] [PubMed]
- Ferrato, M.H.; Marsh, A.G.; Franke, K.R.; Huang, B.J.; Kolb, E.A.; DeRyckere, D.; Grahm, D.K.; Chandrasekaran, S.; Crowgey, E.L. Machine Learning Classifier Approaches for Predicting Response to RTK-Type-III Inhibitors Demonstrate High Accuracy Using Transcriptomic Signatures and Ex Vivo Data. Bioinform. Adv. 2023, 3, vbad034. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep Learning Enables Rapid Identification of Potent DDR1 Kinase Inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.H.; Wang, R.; Tan, C.Y.; Jiang, Y.Y.; Lu, T.; Rao, H.B.; Li, X.Y.; Go, M.L.; Low, B.C.; Chen, Y.Z. Virtual Screening of Selective Multitarget Kinase Inhibitors by Combinatorial Support Vector Machines. Mol. Pharm. 2010, 7, 1545–1560. [Google Scholar] [CrossRef] [PubMed]
- Robichaux, J.P.; Le, X.; Vijayan, R.S.K.; Hicks, J.K.; Heeke, S.; Elamin, Y.Y.; Lin, H.Y.; Udagawa, H.; Skoulidis, F.; Tran, H.; et al. Structure-Based Classification Predicts Drug Response in EGFR-Mutant NSCLC. Nature 2021, 597, 732–737. [Google Scholar] [CrossRef]
- Light, T.P.; Gomez-Soler, M.; Wang, Z.; Karl, K.; Zapata-Mercado, E.; Gehring, M.P.; Lechtenberg, B.C.; Pogorelov, T.V.; Hristova, K.; Pasquale, E.B. A Cancer Mutation Promotes EphA4 Oligomerization and Signaling by Altering the Conformation of the SAM Domain. J. Biol. Chem. 2021, 297, 100876. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Wang, D.D.; Yan, H. Genotype-Determined EGFR-RTK Heterodimerization and Its Effects on Drug Resistance in Lung Cancer Treatment Revealed by Molecular Dynamics Simulations. BMC Mol. Cell Biol. 2021, 22, 34. [Google Scholar] [CrossRef]
- Lam, I.; Pickering, C.M.; Mac Gabhann, F. Context-Dependent Regulation of Receptor Tyrosine Kinases: Insights from Systems Biology Approaches. Wiley Interdiscip. Rev. Syst. Biol. Med. 2019, 11, e1437. [Google Scholar] [CrossRef]
- Schertler, G.F.X.; Villa, C.; Henderson, R. Projection Structure of Rhodopsin. Nature 1993, 362, 770–772. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The Structure and Function of G-Protein-Coupled Receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef]
- Wheatley, M.; Wootten, D.; Conner, M.; Simms, J.; Kendrick, R.; Logan, R.; Poyner, D.; Barwell, J. Lifting the Lid on GPCRs: The Role of Extracellular Loops. Br. J. Pharmacol. 2012, 165, 1688–1703. [Google Scholar] [CrossRef] [PubMed]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Hedderich, J.B.; Persechino, M.; Becker, K.; Heydenreich, F.M.; Gutermuth, T.; Bouvier, M.; Bünemann, M.; Kolb, P. The Pocketome of G-Protein-Coupled Receptors Reveals Previously Untargeted Allosteric Sites. Nat. Commun. 2022, 13, 2567. [Google Scholar] [CrossRef]
- Schwartz, T.W.; Frimurer, T.M.; Holst, B.; Rosenkilde, M.M.; Elling, C.E. Molecular Mechanism of 7tm Receptor Activation—A Global Toggle Switch Model. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 481–519. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef]
- Parma, J.; Duprez, L.; Sande, J.V.; Cochaux, P.; Gervy, C.; Mockel, J.; Dumont, J.; Vassart, G. Somatic Mutations in the Thyrotropin Receptor Gene Cause Hyperfunctioning Thyroid Adenomas. Nature 1993, 365, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, C.; Liao, H.; Wang, Q. Activation of GPER by E2 Promotes Proliferation, Invasion and Migration of Breast Cancer Cells by Regulating the miR-124/CD151 Pathway. Oncol. Lett. 2021, 21, 432. [Google Scholar] [CrossRef]
- Steiman, J.; Peralta, E.A.; Louis, S.; Kamel, O. Biology of the Estrogen Receptor, GPR30, in Triple Negative Breast Cancer. Am. J. Surg. 2013, 206, 698–703. [Google Scholar] [CrossRef]
- Cook, T.; Sheridan, W.P. Development of GnRH Antagonists for Prostate Cancer: New Approaches to Treatment. Oncologist 2000, 5, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Usman, S.; Khawer, M.; Rafique, S.; Naz, Z.; Saleem, K. The Current Status of Anti-GPCR Drugs against Different Cancers. J. Pharm. Anal. 2020, 10, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Komachi, M.; Sato, K.; Tobo, M.; Mogi, C.; Yamada, T.; Ohta, H.; Tomura, H.; Kimura, T.; Im, D.-S.; Yanagida, K.; et al. Orally Active Lysophosphatidic Acid Receptor Antagonist Attenuates Pancreatic Cancer Invasion and Metastasis in Vivo. Cancer Sci. 2012, 103, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.K.; Kim, S. An Insight into GPCR and G-Proteins as Cancer Drivers. Cells 2021, 10, 3288. [Google Scholar] [CrossRef] [PubMed]
- Arang, N.; Gutkind, J.S. G Protein-Coupled Receptors and Heterotrimeric G Proteins as Cancer Drivers. FEBS Lett. 2020, 594, 4201–4232. [Google Scholar] [CrossRef] [PubMed]
- Qualliotine, J.R.; Nakagawa, T.; Rosenthal, S.B.; Sadat, S.; Ballesteros-Merino, C.; Xu, G.; Mark, A.; Nasamran, A.; Gutkind, J.S.; Fisch, K.M.; et al. A Network Landscape of HPVOPC Reveals Methylation Alterations as Significant Drivers of Gene Expression via an Immune-Mediated GPCR Signal. Cancers 2023, 15, 4379. [Google Scholar] [CrossRef]
- Huang, C.; Zhu, F.; Zhang, H.; Wang, N.; Huang, Q. Identification of S1PR4 as an Immune Modulator for Favorable Prognosis in HNSCC through Machine Learning. iScience 2023, 26, 107693. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Wang, Q.; Wang, L.; Yang, Y.; Ren, M.; Li, Y.; Gao, Z.; Zheng, S.; Ding, Y.; Ji, J.; et al. Prediction of Survival and Immunotherapy Response by the Combined Classifier of G Protein-Coupled Receptors and Tumor Microenvironment in Melanoma. Eur. J. Med. Res. 2023, 28, 352. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, E.A.; Tan, C.Y.R.; Gregory, K.J.; Christopoulos, A.; White, P.J.; May, L.T. Ligand-Independent Adenosine A2B Receptor Constitutive Activity as a Promoter of Prostate Cancer Cell Proliferation. J. Pharmacol. Exp. Ther. 2016, 357, 36–44. [Google Scholar] [CrossRef]
- Lappano, R.; Maggiolini, M. GPCRs and Cancer. Acta Pharmacol. Sin. 2012, 33, 351–362. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/Beta-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.A.; Yaari, A.U.; Priebe, O.; Dietlein, F.; Loh, P.-R.; Berger, B. Genome-Wide Mapping of Somatic Mutation Rates Uncovers Drivers of Cancer. Nat. Biotechnol. 2022, 40, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, X.; Chen, J.; Wu, B.; Liu, J.; Guo, Y.; Li, M.; Pu, X. Multi-Omics Integration Analysis of GPCRs in Pan-Cancer to Uncover Inter-Omics Relationships and Potential Driver Genes. Comput. Biol. Med. 2023, 161, 106988. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, F.; Inoue, A.; Kadji, F.M.N.; Shuai, N.; Gonzalez, J.C.; Singh, G.; de la Vega, A.A.; Sotillo, R.; Fischer, B.; Aoki, J.; et al. Rare, Functional, Somatic Variants in Gene Families Linked to Cancer Genes: GPCR Signaling as a Paradigm. Oncogene 2019, 38, 6491–6506. [Google Scholar] [CrossRef]
- Suteau, V.; Munier, M.; Ben Boubaker, R.; Wery, M.; Henrion, D.; Rodien, P.; Briet, C. Identification of Dysregulated Expression of G Protein Coupled Receptors in Endocrine Tumors by Bioinformatics Analysis: Potential Drug Targets? Cells 2022, 11, 703. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.K.; Newman, D.; Libonate, G.; Borns-Stern, M.; Bevan, D.R.; Brown, A.M.; Anandakrishnan, R. Biophysical Insights into OR2T7: Investigation of a Potential Prognostic Marker for Glioblastoma. Biophys. J. 2022, 121, 3706–3718. [Google Scholar] [CrossRef]
- Rebolledo-Bustillo, M.; Garcia-Gomez, D.; Dávila, E.M.; Castro, M.E.; Caballero, N.A.; Melendez, F.J.; Baizabal-Aguirre, V.M.; Sanchez-Gaytan, B.L.; Perez-Aguilar, J.M. Structural Basis of the Binding Mode of the Antineoplastic Compound Motixafortide (BL-8040) in the CXCR4 Chemokine Receptor. Int. J. Mol. Sci. 2023, 24, 4393. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Mitra, A.; Bulusu, G. Structural Dynamics of Smoothened (SMO) in the Ciliary Membrane and Its Interaction with Membrane Lipids. Biochim. Biophys. Acta Biomembr. 2022, 1864, 183946. [Google Scholar] [CrossRef]
- Matic, M.; Singh, G.; Carli, F.; De Oliveira Rosa, N.; Miglionico, P.; Magni, L.; Gutkind, J.S.; Russell, R.B.; Inoue, A.; Raimondi, F. PRECOGx: Exploring GPCR Signaling Mechanisms with Deep Protein Representations. Nucleic Acids Res. 2022, 50, W598–W610. [Google Scholar] [CrossRef]
- Seyedabadi, M.; Gharghabi, M.; Gurevich, E.V.; Gurevich, V.V. Structural Basis of GPCR Coupling to Distinct Signal Transducers: Implications for Biased Signaling. Trends Biochem. Sci. 2022, 47, 570–581. [Google Scholar] [CrossRef]
- Coke, C.J.; Scarlett, K.A.; Chetram, M.A.; Jones, K.J.; Sandifer, B.J.; Davis, A.S.; Marcus, A.I.; Hinton, C.V. Simultaneous Activation of Induced Heterodimerization between CXCR4 Chemokine Receptor and Cannabinoid Receptor 2 (CB2) Reveals a Mechanism for Regulation of Tumor Progression. J. Biol. Chem. 2016, 291, 9991–10005. [Google Scholar] [CrossRef] [PubMed]
- Gahbauer, S.; Pluhackova, K.; Böckmann, R.A. Closely Related, yet Unique: Distinct Homo- and Heterodimerization Patterns of G Protein Coupled Chemokine Receptors and Their Fine-Tuning by Cholesterol. PLoS Comput. Biol. 2018, 14, e1006062. [Google Scholar] [CrossRef] [PubMed]
- Di Marino, D.; Conflitti, P.; Motta, S.; Limongelli, V. Structural Basis of Dimerization of Chemokine Receptors CCR5 and CXCR4. Nat. Commun. 2023, 14, 6439. [Google Scholar] [CrossRef] [PubMed]
- Paradis, J.S.; Feng, X.; Murat, B.; Jefferson, R.E.; Sokrat, B.; Szpakowska, M.; Hogue, M.; Bergkamp, N.D.; Heydenreich, F.M.; Smit, M.J.; et al. Computationally Designed GPCR Quaternary Structures Bias Signaling Pathway Activation. Nat. Commun. 2022, 13, 6826. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Curtin, J.F.; Kinsella, G.K. In Silico and in Vitro Screening for Potential Anticancer Candidates Targeting GPR120. Bioorg. Med. Chem. Lett. 2021, 31, 127672. [Google Scholar] [CrossRef] [PubMed]
- Muthiah, I.; Rajendran, K.; Dhanaraj, P. In Silico Molecular Docking and Physicochemical Property Studies on Effective Phytochemicals Targeting GPR116 for Breast Cancer Treatment. Mol. Cell Biochem. 2021, 476, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Muthiah, I.; Rajendran, K.; Dhanaraj, P.; Vallinayagam, S. In Silico Structure Prediction, Molecular Docking and Dynamic Simulation Studies on G Protein-Coupled Receptor 116: A Novel Insight into Breast Cancer Therapy. J. Biomol. Struct. Dyn. 2021, 39, 4807–4815. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulos, A.A.; Konstantinou, E.; Pirintsos, S.A.; Castanas, E.; Kampa, M. Mining the ZINC Database of Natural Products for Specific, Testosterone-like, OXER1 Antagonists. Steroids 2023, 199, 109309. [Google Scholar] [CrossRef]
- Tan, M.; Gao, S.; Ru, X.; He, M.; Zhao, J.; Zheng, L. Prediction and Identification of GPCRs Targeting for Drug Repurposing in Osteosarcoma. Front. Oncol. 2022, 12, 828849. [Google Scholar] [CrossRef]
- Cornwell, A.C.; Feigin, M.E. Unintended Effects of GPCR-Targeted Drugs on the Cancer Phenotype. Trends Pharmacol. Sci. 2020, 41, 1006–1022. [Google Scholar] [CrossRef]
- Schlessinger, A.; Matsson, P.; Shima, J.E.; Pieper, U.; Yee, S.W.; Kelly, L.; Apeltsin, L.; Stroud, R.M.; Ferrin, T.E.; Giacomini, K.M.; et al. Comparison of Human Solute Carriers. Protein Sci. 2010, 19, 412–428. [Google Scholar] [CrossRef] [PubMed]
- Meixner, E.; Goldmann, U.; Sedlyarov, V.; Scorzoni, S.; Rebsamen, M.; Girardi, E.; Superti-Furga, G. A Substrate-Based Ontology for Human Solute Carriers. Mol. Syst. Biol. 2020, 16, e9652. [Google Scholar] [CrossRef] [PubMed]
- Höglund, P.J.; Nordström, K.J.V.; Schiöth, H.B.; Fredriksson, R. The Solute Carrier Families Have a Remarkably Long Evolutionary History with the Majority of the Human Families Present before Divergence of Bilaterian Species. Mol. Biol. Evol. 2011, 28, 1531–1541. [Google Scholar] [CrossRef]
- Colas, C.; Ung, P.M.U.; Schlessinger, A. SLC Transporters: Structure, Function, and Drug Discovery. MedChemComm 2016, 7, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- El-Gebali, S.; Bentz, S.; Hediger, M.A.; Anderle, P. Solute Carriers (SLCs) in Cancer. Mol. Asp. Med. 2013, 34, 719–734. [Google Scholar] [CrossRef]
- Scafoglio, C.; Hirayama, B.A.; Kepe, V.; Liu, J.; Ghezzi, C.; Satyamurthy, N.; Moatamed, N.A.; Huang, J.; Koepsell, H.; Barrio, J.R.; et al. Functional Expression of Sodium-Glucose Transporters in Cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4111–E4119. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A Small-Molecule Inhibitor of Glucose Transporter 1 Downregulates Glycolysis, Induces Cell-Cycle Arrest, and Inhibits Cancer Cell Growth in Vitro and in Vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC Transporters as Therapeutic Targets: Emerging Opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef]
- Xie, M.; Wang, F.; Chen, B.; Wu, Z.; Chen, C.; Xu, J. Systematic Pan-Cancer Analysis Identifies SLC35C1 as an Immunological and Prognostic Biomarker. Sci. Rep. 2023, 13, 5331. [Google Scholar] [CrossRef]
- Li, J.; Xie, J.; Wu, D.; Chen, L.; Gong, Z.; Wu, R.; Hu, Y.; Zhao, J.; Xu, Y. A Pan-Cancer Analysis Revealed the Role of the SLC16 Family in Cancer. Channels 2021, 15, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Mou, Y.; Ye, S.; Hu, H.; Wang, R.; Yang, Q.; Hu, Y. Identification of a Six-Gene SLC Family Signature with Prognostic Value in Patients With Lung Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9, 803198. [Google Scholar] [CrossRef]
- Zhao, X.; Jin, L.; Liu, Y.; Liu, Z.; Liu, Q. Bioinformatic Analysis of the Role of Solute Carrier-Glutamine Transporters in Breast Cancer. Ann. Transl. Med. 2022, 10, 777. [Google Scholar] [CrossRef]
- Sun, T.; Bi, F.; Liu, Z.; Yang, Q. SLC7A2 Serves as a Potential Biomarker and Therapeutic Target for Ovarian Cancer. Aging 2020, 12, 13281–13296. [Google Scholar] [CrossRef] [PubMed]
- Samaržija, I.; Trošelj, K.G.; Konjevoda, P. Prognostic Significance of Amino Acid Metabolism-Related Genes in Prostate Cancer Retrieved by Machine Learning. Cancers 2023, 15, 1309. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Z.; Li, L.; Zeng, D.; Sun, H.; Wu, J.; Zhou, R.; Liao, W. Co-Expression Pattern of SLC Transporter Genes Associated with the Immune Landscape and Clinical Outcomes in Gastric Cancer. J. Cell. Mol. Med. 2023, 27, 4181–4194. [Google Scholar] [CrossRef]
- Zhou, R.; Li, L.; Zhang, Y.; Liu, Z.; Wu, J.; Zeng, D.; Sun, H.; Liao, W. Integrative Analysis of Co-Expression Pattern of Solute Carrier Transporters Reveals Molecular Subtypes Associated with Tumor Microenvironment Hallmarks and Clinical Outcomes in Colon Cancer. Heliyon 2024, 10, e22775. [Google Scholar] [CrossRef]
- Zhang, P.; Yang, H.; Zhu, K.; Chang, C.; Lv, W.; Li, R.; Li, X.; Ye, T.; Cao, D. SLC31A1 Identifying a Novel Biomarker with Potential Prognostic and Immunotherapeutic Potential in Pan-Cancer. Biomedicines 2023, 11, 2884. [Google Scholar] [CrossRef] [PubMed]
- Danzi, F.; Pacchiana, R.; Mafficini, A.; Scupoli, M.T.; Scarpa, A.; Donadelli, M.; Fiore, A. To Metabolomics and beyond: A Technological Portfolio to Investigate Cancer Metabolism. Sig. Transduct. Target. Ther. 2023, 8, 137. [Google Scholar] [CrossRef]
- Poplawski, P.; Alseekh, S.; Jankowska, U.; Skupien-Rabian, B.; Iwanicka-Nowicka, R.; Kossowska, H.; Fogtman, A.; Rybicka, B.; Bogusławska, J.; Adamiok-Ostrowska, A.; et al. Coordinated Reprogramming of Renal Cancer Transcriptome, Metabolome and Secretome Associates with Immune Tumor Infiltration. Cancer Cell Int. 2023, 23, 2. [Google Scholar] [CrossRef]
- Wang, W.; Rong, Z.; Wang, G.; Hou, Y.; Yang, F.; Qiu, M. Cancer Metabolites: Promising Biomarkers for Cancer Liquid Biopsy. Biomark. Res. 2023, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- Schaller, L.; Lauschke, V.M. The Genetic Landscape of the Human Solute Carrier (SLC) Transporter Superfamily. Hum. Genet. 2019, 138, 1359–1377. [Google Scholar] [CrossRef]
- Koleske, M.L.; McInnes, G.; Brown, J.E.H.; Thomas, N.; Hutchinson, K.; Chin, M.Y.; Koehl, A.; Arkin, M.R.; Schlessinger, A.; Gallagher, R.C.; et al. Functional Genomics of OCTN2 Variants Informs Protein-Specific Variant Effect Predictor for Carnitine Transporter Deficiency. Proc. Natl. Acad. Sci. USA 2022, 119, e2210247119. [Google Scholar] [CrossRef] [PubMed]
- Pasquadibisceglie, A.; Quadrotta, V.; Polticelli, F. In Silico Analysis of the Structural Dynamics and Substrate Recognition Determinants of the Human Mitochondrial Carnitine/Acylcarnitine SLC25A20 Transporter. Int. J. Mol. Sci. 2023, 24, 3946. [Google Scholar] [CrossRef]
- Wu, Q.; Akhter, A.; Pant, S.; Cho, E.; Zhu, J.X.; Garner, A.R.; Ohyama, T.; Tajkhorshid, E.; van Meyel, D.J.; Ryan, R.M. Ataxia-Linked SLC1A3 Mutations Alter EAAT1 Chloride Channel Activity and Glial Regulation of CNS Function. J. Clin. Investig. 2022, 132, e154891. [Google Scholar] [CrossRef] [PubMed]
- Tuerkova, A.; Bongers, B.J.; Norinder, U.; Ungvári, O.; Székely, V.; Tarnovskiy, A.; Szakács, G.; Özvegy-Laczka, C.; van Westen, G.J.P.; Zdrazil, B. Identifying Novel Inhibitors for Hepatic Organic Anion Transporting Polypeptides by Machine Learning-Based Virtual Screening. J. Chem. Inf. Model. 2022, 62, 6323–6335. [Google Scholar] [CrossRef]
- Burggraaff, L.; Oranje, P.; Gouka, R.; Van Der Pijl, P.; Geldof, M.; Van Vlijmen, H.W.T.; Ijzerman, A.P.; Van Westen, G.J.P. Identification of Novel Small Molecule Inhibitors for Solute Carrier SGLT1 Using Proteochemometric Modeling. J. Cheminform. 2019, 11, 15. [Google Scholar] [CrossRef]
- Xu, A.M.; Huang, P.H. Receptor Tyrosine Kinase Coactivation Networks in Cancer. Cancer Res. 2010, 70, 3857–3860. [Google Scholar] [CrossRef] [PubMed]
- Kisfalvi, K.; Rey, O.; Young, S.H.; Sinnett-Smith, J.; Rozengurt, E. Insulin Potentiates Ca2+ Signaling and Phosphatidylinositol 4,5-Bisphosphate Hydrolysis Induced by Gq Protein-Coupled Receptor Agonists through an mTOR-Dependent Pathway. Endocrinology 2007, 148, 3246–3257. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Maggiolini, M. Mechanisms of Estrogen Signaling and Gene Expression via GPR30. Mol. Cell Endocrinol. 2009, 308, 32–38. [Google Scholar] [CrossRef]
- Arora, P.; Cuevas, B.D.; Russo, A.; Johnson, G.L.; Trejo, J. Persistent Transactivation of EGFR and ErbB2/HER2 by Protease-Activated Receptor-1 Promotes Breast Carcinoma Cell Invasion. Oncogene 2008, 27, 4434–4445. [Google Scholar] [CrossRef]
- Kalogriopoulos, N.A.; Lopez-Sanchez, I.; Lin, C.; Ngo, T.; Midde, K.K.; Roy, S.; Aznar, N.; Murray, F.; Garcia-Marcos, M.; Kufareva, I.; et al. Receptor Tyrosine Kinases Activate Heterotrimeric G Proteins via Phosphorylation within the Interdomain Cleft of Gαi. Proc. Natl. Acad. Sci. USA 2020, 117, 28763–28774. [Google Scholar] [CrossRef]
- Weihua, Z.; Tsan, R.; Huang, W.-C.; Wu, Q.; Chiu, C.-H.; Fidler, I.J.; Hung, M.-C. Survival of Cancer Cells Is Maintained by EGFR Independent of Its Kinase Activity. Cancer Cell 2008, 13, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Sijben, H.J.; Superti-Furga, G.; IJzerman, A.P.; Heitman, L.H. Targeting Solute Carriers to Modulate Receptor-Ligand Interactions. Trends Pharmacol. Sci. 2022, 43, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Ceong, H.-T.; Na, D.; Park, C. A Machine Learning Model for Classifying G-Protein-Coupled Receptors as Agonists or Antagonists. BMC Bioinform. 2022, 23, 346. [Google Scholar] [CrossRef]
- Van de Geer, W.S.; Mathijssen, R.H.J.; van Riet, J.; Steeghs, N.; Labots, M.; van Herpen, C.; Devriese, L.A.; Tjan-Heijnen, V.C.G.; Voest, E.E.; Sleijfer, S.; et al. Identifying Somatic Changes in Drug Transporters Using Whole Genome and Transcriptome Sequencing Data of Advanced Tumors. Biomed. Pharmacother. 2023, 159, 114210. [Google Scholar] [CrossRef]
- Qu, Y.-Y.; Guo, R.-Y.; Luo, M.-L.; Zhou, Q. Pan-Cancer Analysis of the Solute Carrier Family 39 Genes in Relation to Oncogenic, Immune Infiltrating, and Therapeutic Targets. Front. Genet. 2021, 12, 757582. [Google Scholar] [CrossRef] [PubMed]
- Buttarelli, M.; Ciucci, A.; Palluzzi, F.; Raspaglio, G.; Marchetti, C.; Perrone, E.; Minucci, A.; Giacò, L.; Fagotti, A.; Scambia, G.; et al. Identification of a Novel Gene Signature Predicting Response to First-Line Chemotherapy in BRCA Wild-Type High-Grade Serous Ovarian Cancer Patients. J. Exp. Clin. Cancer Res. 2022, 41, 50. [Google Scholar] [CrossRef]
- Alves, R.; Gonçalves, A.C.; Jorge, J.; Marques, G.; Ribeiro, A.B.; Tenreiro, R.; Coucelo, M.; Diamond, J.; Oliveiros, B.; Pereira, A.; et al. Genetic Variants of ABC and SLC Transporter Genes and Chronic Myeloid Leukaemia: Impact on Susceptibility and Prognosis. Int. J. Mol. Sci. 2022, 23, 9815. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Rodrigues, C.H.M.; Ascher, D.B. mCSM-Membrane: Predicting the Effects of Mutations on Transmembrane Proteins. Nucleic Acids Res. 2020, 48, W147–W153. [Google Scholar] [CrossRef]
- Ge, F.; Zhu, Y.-H.; Xu, J.; Muhammad, A.; Song, J.; Yu, D.-J. MutTMPredictor: Robust and Accurate Cascade XGBoost Classifier for Prediction of Mutations in Transmembrane Proteins. Comput. Struct. Biotechnol. J. 2021, 19, 6400–6416. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, S.; Liang, Q.; Huang, W.; Wang, H.; Pan, E.; Xu, P.; Zhang, S.; Tao, F.; Tang, J.; et al. CrMP-Sol Database: Classification, Bioinformatic Analyses and Comparison of Cancer-Related Membrane Proteins and Their Water-Soluble Variant Designs. BMC Bioinform. 2023, 24, 360. [Google Scholar] [CrossRef] [PubMed]
- Avsec, Ž.; Agarwal, V.; Visentin, D.; Ledsam, J.R.; Grabska-Barwinska, A.; Taylor, K.R.; Assael, Y.; Jumper, J.; Kohli, P.; Kelley, D.R. Effective Gene Expression Prediction from Sequence by Integrating Long-Range Interactions. Nat. Methods 2021, 18, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, B.R.; Tsai, C.-J.; Nussinov, R.; Tuncbag, N. Pan-Cancer Clinical Impact of Latent Drivers from Double Mutations. Commun. Biol. 2023, 6, 202. [Google Scholar] [CrossRef] [PubMed]
- Mateo, L.; Duran-Frigola, M.; Gris-Oliver, A.; Palafox, M.; Scaltriti, M.; Razavi, P.; Chandarlapaty, S.; Arribas, J.; Bellet, M.; Serra, V.; et al. Personalized Cancer Therapy Prioritization Based on Driver Alteration Co-Occurrence Patterns. Genome Med. 2020, 12, 78. [Google Scholar] [CrossRef]
- Qi, X.; Zhao, Y.; Qi, Z.; Hou, S.; Chen, J. Machine Learning Empowering Drug Discovery: Applications, Opportunities and Challenges. Molecules 2024, 29, 903. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorostiola González, M.; Rakers, P.R.J.; Jespers, W.; IJzerman, A.P.; Heitman, L.H.; van Westen, G.J.P. Computational Characterization of Membrane Proteins as Anticancer Targets: Current Challenges and Opportunities. Int. J. Mol. Sci. 2024, 25, 3698. https://doi.org/10.3390/ijms25073698
Gorostiola González M, Rakers PRJ, Jespers W, IJzerman AP, Heitman LH, van Westen GJP. Computational Characterization of Membrane Proteins as Anticancer Targets: Current Challenges and Opportunities. International Journal of Molecular Sciences. 2024; 25(7):3698. https://doi.org/10.3390/ijms25073698
Chicago/Turabian StyleGorostiola González, Marina, Pepijn R. J. Rakers, Willem Jespers, Adriaan P. IJzerman, Laura H. Heitman, and Gerard J. P. van Westen. 2024. "Computational Characterization of Membrane Proteins as Anticancer Targets: Current Challenges and Opportunities" International Journal of Molecular Sciences 25, no. 7: 3698. https://doi.org/10.3390/ijms25073698
APA StyleGorostiola González, M., Rakers, P. R. J., Jespers, W., IJzerman, A. P., Heitman, L. H., & van Westen, G. J. P. (2024). Computational Characterization of Membrane Proteins as Anticancer Targets: Current Challenges and Opportunities. International Journal of Molecular Sciences, 25(7), 3698. https://doi.org/10.3390/ijms25073698