
An Escherichia coli-Based Phosphorylation System for Efficient Screening of Kinase Substrates


 and
and
Abstract
:1. Introduction
2. Results
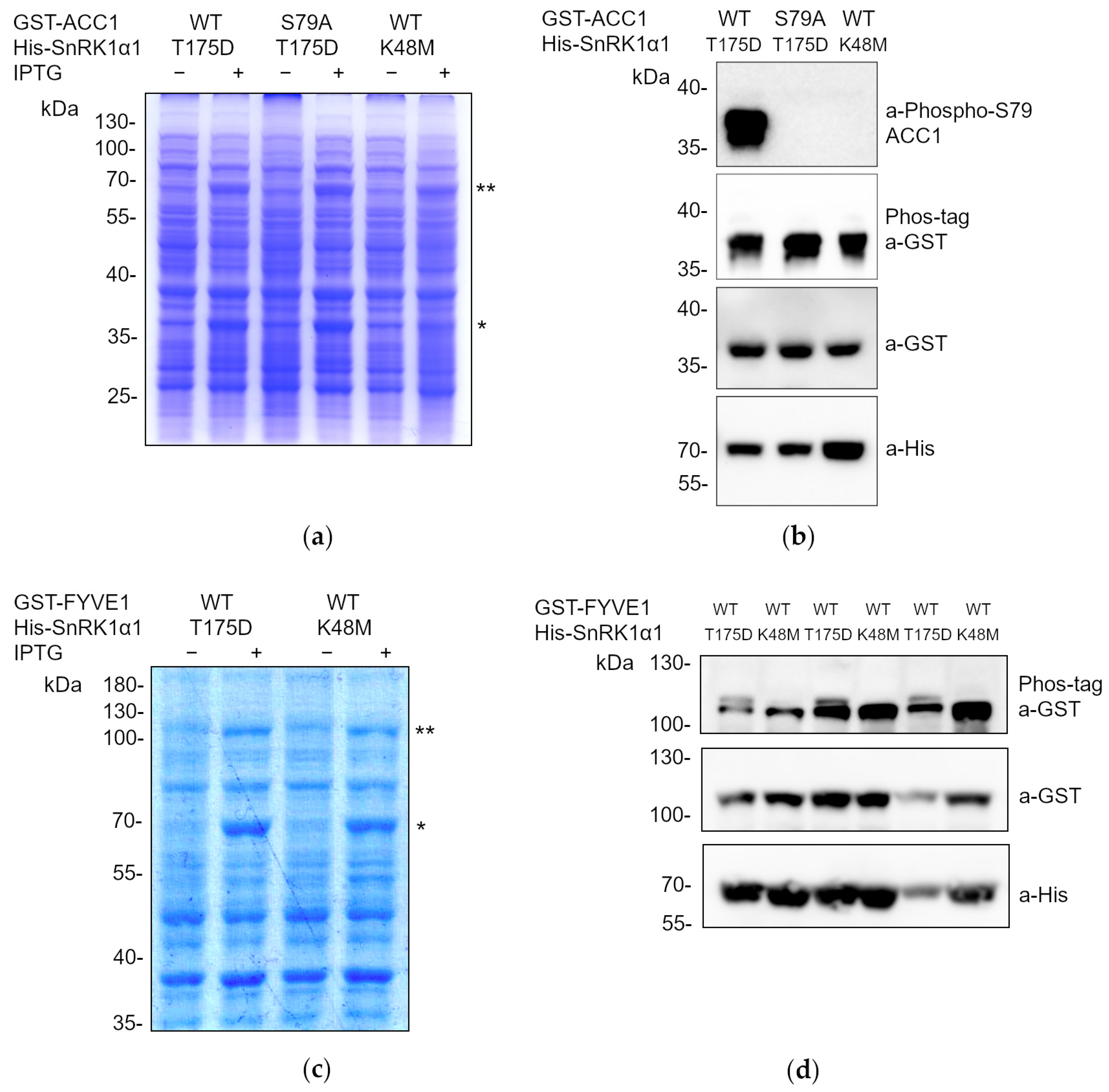
2.1. Development of a Methodology for Analyzing the Phosphorylation of Potential SnRK1 Substrates in Escherichia coli Cells Co-Expressing Both the Kinase and the Substrate in the Same Vector
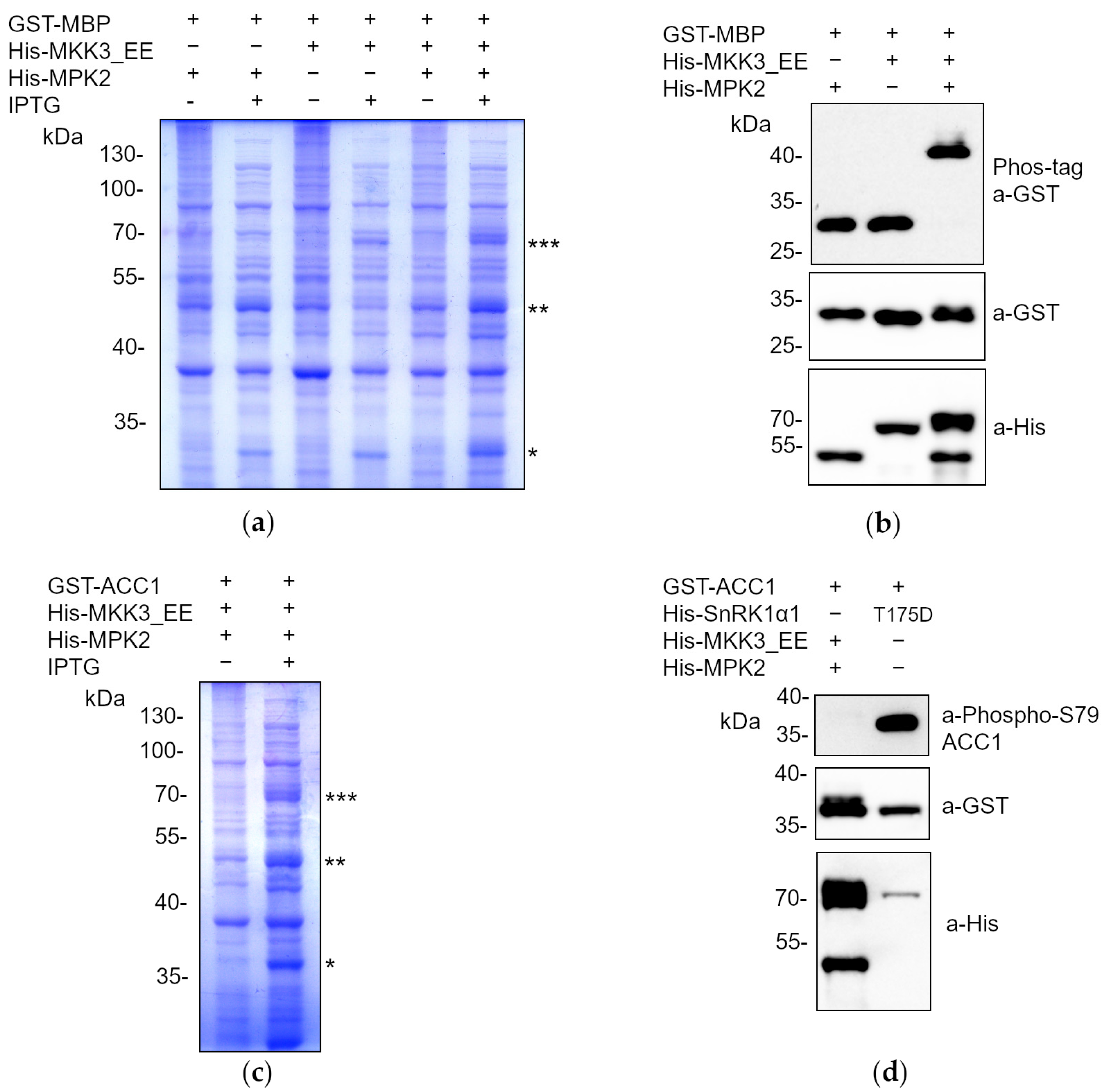
2.2. The Phoscreen System Proves to Be a Valid Tool for Analyzing Phosphorylation Cascades
3. Discussion
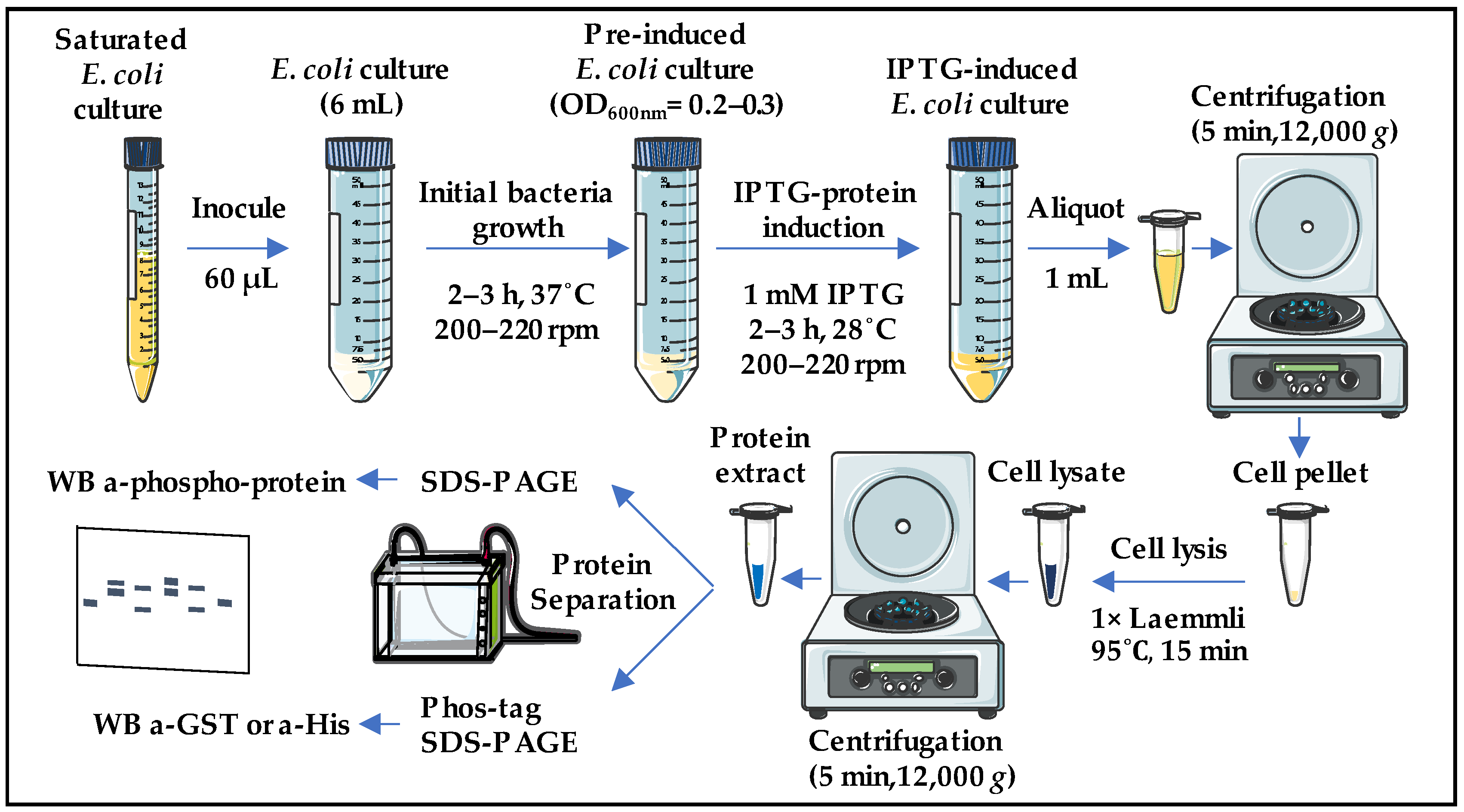
4. Materials and Methods
4.1. Biological Material and Growth Conditions
4.2. Cloning Procedures
4.2.1. Generation of T175D or K48M Mutations in SnRK1α1
4.2.2. Cloning of ACC1 Peptide (S55-V108) and Its Mutated Version (S79A)
4.2.3. Cloning of MPK2 Coding Sequence
4.2.4. Cloning of MBP Peptide (V93-Q102) Coding Sequence
4.2.5. BP Gateway Recombination
4.2.6. Destination Constructs
- 50 ng of the PacI fragment was mixed with 2.5 mM dCTP, 1 U of T4 DNA polymerase (Thermo Fisher Scientific, Waltham, MA, USA), and 3 μL of 5× reaction buffer in a final volume of 15 μL.
- 50 ng of the SwaI fragment was mixed with 2.5 mM dGTP, 1 U of T4 DNA polymerase (Thermo Fisher Scientific, Waltham, MA, USA), and 3 μL of 5× reaction buffer in a final volume of 15 μL.
4.3. Protein Analysis and Manipulation Techniques
4.3.1. Heterologous Expression
4.3.2. Protein Extraction
4.3.3. SDS-PAGE and Coomassie Staining
4.3.4. Mn+2-Phos-Tag SDS-PAGE
4.3.5. Western Blot (WB)
- 1 h incubation at room temperature with blocking buffer [5% w/v non-fat dry milk in 1× TBS (Tris buffered saline, 0.05 M Tris and 0.15 M sodium chloride, pH 7.6), 0.05% Tween®] in agitation.
- Overnight incubation at 4 °C on rocking platform with the primary antibody of interest (Table 4) diluted in blocking buffer.
- 3 washes of 10 min with 1× TBS containing 0.05% Tween®.
- 1 h incubation at room temperature on rocking platform with the corresponding secondary antibody (Table 4).
- 3 washes of 10 min with 1× TBS containing 0.05% Tween®.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Millar, A.H.; Heazlewood, J.L.; Giglione, C.; Holdsworth, M.J.; Bachmair, A.; Schulze, W.X. The Scope, Functions, and Dynamics of Posttranslational Protein Modifications. Annu. Rev. Plant Biol. 2019, 70, 119–151. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Kelleher, N.L.; Linial, M.; Goodlett, D.; Langridge-Smith, P.; Goo, Y.A.; Safford, G.; Bonilla, L.; Kruppa, G.; Zubarev, R.; et al. Proteoform: A Single Term Describing Protein Complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Ramazi, S.; Zahiri, J. Post-Translational Modifications in Proteins: Resources, Tools and Prediction Methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef] [PubMed]
- Ubersax, J.A.; Ferrell, J.E. Mechanisms of Specificity in Protein Phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Muzio, L.L. The Crucial Role of Protein Phosphorylation in Cell Signaling and Its Use as Targeted Therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Amanchy, R.; Periaswamy, B.; Mathivanan, S.; Reddy, R.; Tattikota, S.G.; Pandey, A. A Curated Compendium of Phosphorylation Motifs. Nat. Biotechnol. 2007, 25, 285–286. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved Gene Annotation and New Tools. Nucleic Acids Res. 2012, 40, D1202–D1210. [Google Scholar] [CrossRef]
- Zulawski, M.; Schulze, G.; Braginets, R.; Hartmann, S.; Schulze, W.X. The Arabidopsis Kinome: Phylogeny and Evolutionary Insights into Functional Diversification. BMC Genom. 2014, 15, 548. [Google Scholar] [CrossRef]
- Fang, Y.; Jiang, J.; Hou, X.; Guo, J.; Li, X.; Zhao, D.; Xie, X. Plant Protein-Coding Gene Families: Their Origin and Evolution. Front. Plant Sci. 2022, 13, 995746. [Google Scholar] [CrossRef]
- Johnson, S.A.; Hunter, T. Kinomics: Methods for Deciphering the Kinome. Nat. Methods 2004, 2, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Von Stechow, L. Phospho-Proteomics: Methods and Protocols (Methods in Molecular Biology, 1355), 2nd ed.; Humana Press (Springer): New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Wu, X.N. Plant Phosphoproteomics: Methods and Protocols (Methods in Molecular Biology, 2358), 1st ed.; Humana Press (Springer): New York, NY, USA, 2021. [Google Scholar] [CrossRef]
- Peck, S.C. Analysis of Protein Phosphorylation: Methods and Strategies for Studying Kinases and Substrates. Plant J. 2006, 45, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, A.; Koncz-Kálmán, Z.; Farràs, R.; Tiburcio, A.; Schell, J.; Koncz, C. Detection of in Vivo Protein Interactions between Snf1-Related Kinase Subunits with Intron-Tagged Epitope-Labelling in Plants Cells. Nucleic Acids Res. 2001, 29, 3685–3693. [Google Scholar] [CrossRef] [PubMed]
- Skalak, J.; Nicolas, K.L.; Vankova, R.; Hejatko, J. Signal Integration in Plant Abiotic Stress Responses via Multistep Phosphorelay Signaling. Front. Plant Sci. 2021, 12, 196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, S. Mitogen-Activated Protein Kinase Cascades in Plant Signaling. J. Integr. Plant Biol. 2022, 64, 301–341. [Google Scholar] [CrossRef] [PubMed]
- Hrabak, E.M.; Chan, C.W.M.; Gribskov, M.; Harper, J.F.; Choi, J.H.; Halford, N.; Kudla, J.; Luan, S.; Nimmo, H.G.; Sussman, M.R.; et al. The Arabidopsis CDPK-SnRK Superfamily of Protein Kinases. Plant Physiol. 2003, 132, 666–680. [Google Scholar] [CrossRef] [PubMed]
- van Zelm, E.; Zhang, Y.; Testerink, C. Salt Tolerance Mechanisms of Plants. Annu. Rev. Plant Biol. 2020, 71, 403–433. [Google Scholar] [CrossRef] [PubMed]
- Kamiyama, Y.; Katagiri, S.; Umezawa, T. Growth Promotion or Osmotic Stress Response: How SNF1-Related Protein Kinase 2 (SnRK2) Kinases Are Activated and Manage Intracellular Signaling in Plants. Plants 2021, 10, 1443. [Google Scholar] [CrossRef] [PubMed]
- Jamsheer, K.M.; Kumar, M.; Srivastava, V. SNF1-Related Protein Kinase 1: The Many-Faced Signaling Hub Regulating Developmental Plasticity in Plants. J. Exp. Bot. 2021, 72, 6042–6065. [Google Scholar] [CrossRef]
- Baena-González, E.; Rolland, F.; Thevelein, J.M.; Sheen, J. A Central Integrator of Transcription Networks in Plant Stress and Energy Signalling. Nature 2007, 448, 938–942. [Google Scholar] [CrossRef]
- Margalha, L.; Confraria, A.; Baena-González, E. SnRK1 and TOR: Modulating Growth–Defense Trade-Offs in Plant Stress Responses. J. Exp. Bot. 2019, 70, 2261–2274. [Google Scholar] [CrossRef] [PubMed]
- Belda-Palazón, B.; Adamo, M.; Valerio, C.; Ferreira, L.J.; Confraria, A.; Reis-Barata, D.; Rodrigues, A.; Meyer, C.; Rodriguez, P.L.; Baena-González, E. A Dual Function of SnRK2 Kinases in the Regulation of SnRK1 and Plant Growth. Nat. Plants 2020, 6, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Belda-Palazón, B.; Costa, M.; Beeckman, T.; Rolland, F.; Baena-González, E. ABA Represses TOR and Root Meristem Activity through Nuclear Exit of the SnRK1 Kinase. Proc. Natl. Acad. Sci. USA 2022, 119, e2204862119. [Google Scholar] [CrossRef] [PubMed]
- Margalha, L.; Valerio, C.; Baena-González, E. Plant SnRK1 Kinases: Structure, Regulation, and Function. AMP-Act. Protein Kinase 2016, 107, 403–438. [Google Scholar]
- Baena-González, E.; Hanson, J. Shaping Plant Development through the SnRK1–TOR Metabolic Regulators. Curr. Opin. Plant Biol. 2017, 35, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Van Leene, J.; Eeckhout, D.; Gadeyne, A.; Matthijs, C.; Han, C.; De Winne, N.; Persiau, G.; Van De Slijke, E.; Persyn, F.; Mertens, T.; et al. Mapping of the Plant SnRK1 Kinase Signalling Network Reveals a Key Regulatory Role for the Class II T6P Synthase-like Proteins. Nat. Plants 2022, 8, 1245–1261. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, S. Mitogen-Activated Protein Kinase Cascades in Signaling Plant Growth and Development. Trends Plant Sci. 2015, 20, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Wu, J.; Jiang, M.; Wang, Y. Plant Mitogen-Activated Protein Kinase Cascades in Environmental Stresses. Int. J. Mol. Sci. 2021, 22, 1543. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, K.; Shinozaki, K.; Tena, G.; Sheen, J.; Henry, Y.; Champion, A.; Kreis, M.; Zhang, S.; Hirt, H.; Wilson, C.; et al. Mitogen-Activated Protein Kinase Cascades in Plants: A New Nomenclature. Trends Plant Sci. 2002, 7, 301–308. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, Y.; Li, P.; Jian, J.; Zhao, C.; Wen, G. Mitogen-Activated Protein Kinase and Substrate Identification in Plant Growth and Development. Int. J. Mol. Sci. 2022, 23, 2744. [Google Scholar] [CrossRef]
- Danquah, A.; De Zélicourt, A.; Boudsocq, M.; Neubauer, J.; Frei Dit Frey, N.; Leonhardt, N.; Pateyron, S.; Gwinner, F.; Tamby, J.P.; Ortiz-Masia, D.; et al. Identification and Characterization of an ABA-Activated MAP Kinase Cascade in Arabidopsis Thaliana. Plant J. 2015, 82, 232–244. [Google Scholar] [CrossRef]
- de Zelicourt, A.; Colcombet, J.; Hirt, H. The Role of MAPK Modules and ABA during Abiotic Stress Signaling. Trends Plant Sci. 2016, 21, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, T.; Sugiyama, N.; Takahashi, F.; Anderson, J.C.; Ishihama, Y.; Peck, S.C.; Shinozaki, K. Genetics and Phosphoproteomics Reveal a Protein Phosphorylation Network in the Abscisic Acid Signaling Pathway in Arabidopsis Thaliana. Sci. Signal 2013, 6, rs8. [Google Scholar] [CrossRef]
- Wang, P.; Xue, L.; Batelli, G.; Lee, S.; Hou, Y.J.; van Oosten, M.J.; Zhang, H.; Tao, W.A.; Zhu, J.K. Quantitative Phosphoproteomics Identifies SnRK2 Protein Kinase Substrates and Reveals the Effectors of Abscisic Acid Action. Proc. Natl. Acad. Sci. USA 2013, 110, 11205–11210. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Takiyama, K.; Koike, T. Phosphate-Binding Tag, a New Tool to Visualize Phosphorylated Proteins. Mol. Cell. Proteom. 2006, 5, 749–757. [Google Scholar] [CrossRef]
- Crozet, P.; Margalha, L.; Confraria, A.; Rodrigues, A.; Martinho, C.; Adamo, M.; Elias, C.A.; Baena-González, E. Mechanisms of Regulation of SNF1/AMPK/SnRK1 Protein Kinases. Front. Plant Sci. 2014, 5, 190. [Google Scholar] [CrossRef]
- Sanagi, M.; Aoyama, S.; Kubo, A.; Lu, Y.; Sato, Y.; Ito, S.; Abe, M.; Mitsuda, N.; Ohme-Takagi, M.; Kiba, T.; et al. Low Nitrogen Conditions Accelerate Flowering by Modulating the Phosphorylation State of FLOWERING BHLH 4 in Arabidopsis. Proc. Natl. Acad. Sci. USA 2021, 118, e2022942118. [Google Scholar] [CrossRef]
- Muralidhara, P.; Weiste, C.; Collani, S.; Krischke, M.; Kreisz, P.; Draken, J.; Feil, R.; Mair, A.; Teige, M.; Müller, M.J.; et al. Perturbations in Plant Energy Homeostasis Prime Lateral Root Initiation via SnRK1-BZIP63-ARF19 Signaling. Proc. Natl. Acad. Sci. USA 2021, 118, e2106961118. [Google Scholar] [CrossRef]
- Scheich, C.; Kümmel, D.; Soumailakakis, D.; Heinemann, U.; Büssow, K. Vectors for Co-Expression of an Unrestricted Number of Proteins. Nucleic Acids Res. 2007, 35, e43. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Li, B.; Huang, S.; Li, H.; Cao, W.; Chen, Y.; Liu, G.; Li, Z.; Yang, C.; Feng, L.; et al. The Plant Unique ESCRT Component FREE1 Regulates Autophagosome Closure. Nat. Commun. 2023, 14, 1768. [Google Scholar] [CrossRef]
- DeMarco, A.G.; Hall, M.C. Phosphoproteomic Approaches for Identifying Phosphatase and Kinase Substrates. Molecules 2023, 28, 3675. [Google Scholar] [CrossRef] [PubMed]
- Arora, D.; van Damme, D. Motif-Based Endomembrane Trafficking. Plant Physiol. 2021, 186, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Kinoshita-Kikuta, E. Improved Phos-Tag SDS-PAGE under Neutral PH Conditions for Advanced Protein Phosphorylation Profiling. Proteomics 2011, 11, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T. Zn(II)-Phos-Tag SDS-PAGE for Separation and Detection of a DNA Damage-Related Signaling Large Phosphoprotein. Methods Mol. Biol. 2017, 1599, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G. A Simple Method for Detecting Phosphorylation of Proteins by Using Zn2+-Phos-Tag SDS-PAGE at Neutral PH. Methods Mol. Biol. 2018, 1853, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.S.; Kliegman, J.I.; Shokat, K.M. The Logic and Design of Analog-Sensitive Kinases and Their Small Molecule Inhibitors. Methods Enzymol. 2014, 548, 189–213. [Google Scholar] [CrossRef] [PubMed]
- Carlson, S.M.; White, F.M. Labeling and Identification of Direct Kinase Substrates. Sci. Signal 2012, 5, pl3. [Google Scholar] [CrossRef] [PubMed]
- Hertz, N.T.; Wang, B.T.; Allen, J.J.; Zhang, C.; Dar, A.C.; Burlingame, A.L.; Shokat, K.M. Chemical Genetic Approach for Kinase-Substrate Mapping by Covalent Capture of Thiophosphopeptides and Analysis by Mass Spectrometry. Curr. Protoc. Chem. Biol. 2010, 2, 15–36. [Google Scholar] [CrossRef] [PubMed]
- Banko, M.R.; Allen, J.J.; Schaffer, B.E.; Wilker, E.W.; Tsou, P.P.; White, J.L.; Villén, J.; Wang, B.; Kim, S.R.; Sakamoto, K.; et al. Chemical Genetic Screen for AMPKα2 Substrates Uncovers a Network of Proteins Involved in Mitosis. Mol. Cell 2011, 44, 878–892. [Google Scholar] [CrossRef]
- Bai, B. Identification of Components and Substrates of Snf1-Related Kinase 1 (SnRK1) Complexes in Arabidopsis thaliana. Ph.D. Thesis, Rheinische Friedrich-Wilhelms-Universität Bonn, Bonn, Germany, 2019. Available online: https://nbn-resolving.org/urn:nbn:de:hbz:5-53816 (accessed on 4 April 2019).
- Belda-Palazón, B.; Nohales, M.A.; Rambla, J.L.; Aceña, J.L.; Delgado, O.; Fustero, S.; Martínez, M.C.; Granell, A.; Carbonell, J.; Ferrando, A. Biochemical Quantitation of the EIF5A Hypusination in Arabidopsis Thaliana Uncovers ABA-Dependent Regulation. Front. Plant Sci. 2014, 5, 202. [Google Scholar] [CrossRef]
- Belda-Palazón, B.; Ruiz, L.; Martí, E.; Tárraga, S.; Tiburcio, A.F.; Culiáñez, F.; Farràs, R.; Carrasco, P.; Ferrando, A. Aminopropyltransferases Involved in Polyamine Biosynthesis Localize Preferentially in the Nucleus of Plant Cells. PLoS ONE 2012, 7, e46907. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Name | Source |
|---|---|
| pENTR 3C-KIN10 | Belda-Palazón et al. (2012) [54] |
| pDONR/ZEO-KIN10_T175D | This work |
| pDONR_ZEO-ACC_S55-V108 | This work |
| pDONR_ZEO-ACC_S55-V108_S79A | This work |
| pDONR207-MKK3_S235E_T241E | Danquah et al. (2015) [33] |
| pDONR/ZEO-MPK2 | This work |
| pDONR_ZEO-MBP_V93-Q102 | This work |
| Name | Sequence |
|---|---|
| MPK2_attB1 | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGGCGACTCCTGTTGAT |
| MPK2_attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTATCAAAACTCAGAGACCTCATT |
| MBP_V93-Q102_attB1 | GGGACAAGTTTGTACAAAAAAGCAGGCTTCGTGACCCCGCGCACCCCGCCGCCGAGCCAG |
| MBP_V93-Q102_attB2 | GGGACCACTTTGTACAAGAAAGCTGGGTACTGGCTCGGCGGCGGGGTGCGCGGGGTCAC |
| ACC1_S55-V108_attB1 | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGTCAGATACACTTTCTGATTT |
| ACC1_S55-V108_attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTAAACAAATTCTGCTGGCGAAGC |
| KIN10_Fw_attB1 | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGGATGGATCAGGCACAGG |
| KIN10_Rv_attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTATCAGAGGACTCGGAGCTGAGCA |
| KIN10_K48M_Rv | CGACGATTGAGGATCATGATAGCAACCTTAT |
| KIN10_K48M_Fw | ATAAGGTTGCTATCATGATCCTCAATCGTCG |
| KIN10_T175D_Rv | GGACTTCCACAACTATCCTTCAAAAAATGAC |
| KIN10_T175D_Fw | GTCATTTTTTGAAGGATAGTTGTGGAAGTCC |
| Name | Cloning Method |
|---|---|
| pQLinkH-KIN10_T175D | LR reaction: linearized pDONR/ZEO-KIN10_T175D + pQLinKH |
| pQLinkH-KIN10_K48M | LR reaction: linearized pDONR/ZEO-KIN10_K48M + pQLinKH |
| pQLinkG-ACC_S55-V108 | LR reaction: linearized pDONR/ZEO-ACC_S55-V108D + pQLinKG |
| pQLinkG-ACC_S55-V108_S79A | LR reaction: linearized pDONR/ZEO-ACC_S55-V108D_S79A + pQLinKG |
| pQLinkG-MBP_V93-Q102 | LR reaction: linearized pDONR/ZEO-MBP_V93-Q102 + pQLinKG |
| pQLinkH-MKK3_S235E_T241E | LR reaction: linearized pDONR/ZEO-MKK3_S235E_T241E + pQLinKH |
| pQLinkH-MPK2 | LR reaction: linearized pDONR/ZEO-MPK2 + pQLinKH |
| pQLinkH-KIN10_T175D + G-ACC_S55-V108 | LIC reaction: SwaI fragment pQLinkH-KIN10_T175D + PacI fragment G-ACC_S55-V108 |
| pQLinkH-KIN10_T175D + G-ACC_S55-V108_S79A | LIC reaction: SwaI fragment pQLinkH-KIN10_T175D + PacI fragment G-ACC_S55-V108_S79A |
| pQLinkH-KIN10_K48M + G-ACC_S55-V108 | LIC reaction: SwaI fragment pQLinkH-KIN10_K48M + PacI fragment G-ACC_S55-V108 |
| pQLinkH-MPK2 + G-MBP_V93-Q103 | LIC reaction: SwaI fragment pQLinkH-MPK2 + PacI fragment G-MBP_V93-Q103 |
| pQLinkH-MKK3_S235E_T241E + G-MBP_V93-Q104 | LIC reaction: SwaI fragment pQLinkH-MKK3_S235E_T241E + PacI fragment G-MBP_V93-Q104 |
| pQLinkH-MPK2 + H-MKK3_S235E_T241E + G-MBP_V93-Q105 | LIC reaction: SwaI fragment pQLinkH-MPK2 + PacI fragment H-MKK3_S235E_T241E + G-MBP_V93-Q105 |
| pQLinkH-MKK3_S235E_T241E + G-ACC_S55-V108 | LIC reaction: SwaI fragment pQLinkH-MKK3_S235E_T241E + PacI fragment G-ACC_S55-V108 |
| pQLinkH-MPK2 + H-MKK3_S235E_T241E + G-ACC_S55-V108 | LIC reaction: SwaI fragment pQLinkH-MPK2 + PacI fragment H-MKK3_S235E_T241E + G-ACC_S55-V108 |
| Name | Source | Animal | Dilution |
|---|---|---|---|
| Anti-GST | GeneCopoeia (Rockville, MD, USA) CGAB-GST-0050 | Mouse | 1:2000 |
| Anti-His | Merck (Darmstadt, Alemania) 70796-3 | Mouse | 1:2000 |
| Anti-Phospho-S79-ACC1 | Cell Signaling (Danvers, MA, USA) #1673661S | Rabbit | 1:1000 |
| Anti-rabbit IgG-HRP | Jackson ImmunoResearch (West Grove, PA, USA) #111035144 | Goat | 1:20,000 |
| Anti-mouse IgG-HRP | Jackson ImmunoResearch (West Grove, PA, USA) #115035146 | Goat | 1:20,000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cayuela, A.; Villasante-Fernández, A.; Corbalán-Acedo, A.; Baena-González, E.; Ferrando, A.; Belda-Palazón, B. An Escherichia coli-Based Phosphorylation System for Efficient Screening of Kinase Substrates. Int. J. Mol. Sci. 2024, 25, 3813. https://doi.org/10.3390/ijms25073813
Cayuela A, Villasante-Fernández A, Corbalán-Acedo A, Baena-González E, Ferrando A, Belda-Palazón B. An Escherichia coli-Based Phosphorylation System for Efficient Screening of Kinase Substrates. International Journal of Molecular Sciences. 2024; 25(7):3813. https://doi.org/10.3390/ijms25073813
Chicago/Turabian StyleCayuela, Andrés, Adela Villasante-Fernández, Antonio Corbalán-Acedo, Elena Baena-González, Alejandro Ferrando, and Borja Belda-Palazón. 2024. "An Escherichia coli-Based Phosphorylation System for Efficient Screening of Kinase Substrates" International Journal of Molecular Sciences 25, no. 7: 3813. https://doi.org/10.3390/ijms25073813
APA StyleCayuela, A., Villasante-Fernández, A., Corbalán-Acedo, A., Baena-González, E., Ferrando, A., & Belda-Palazón, B. (2024). An Escherichia coli-Based Phosphorylation System for Efficient Screening of Kinase Substrates. International Journal of Molecular Sciences, 25(7), 3813. https://doi.org/10.3390/ijms25073813