
Modeling Dynamics of Human Gut Microbiota Derived from Gluten Metabolism: Obtention, Maintenance and Characterization of Complex Microbial Communities

 , , and
, , and
Abstract
1. Introduction
2. Results
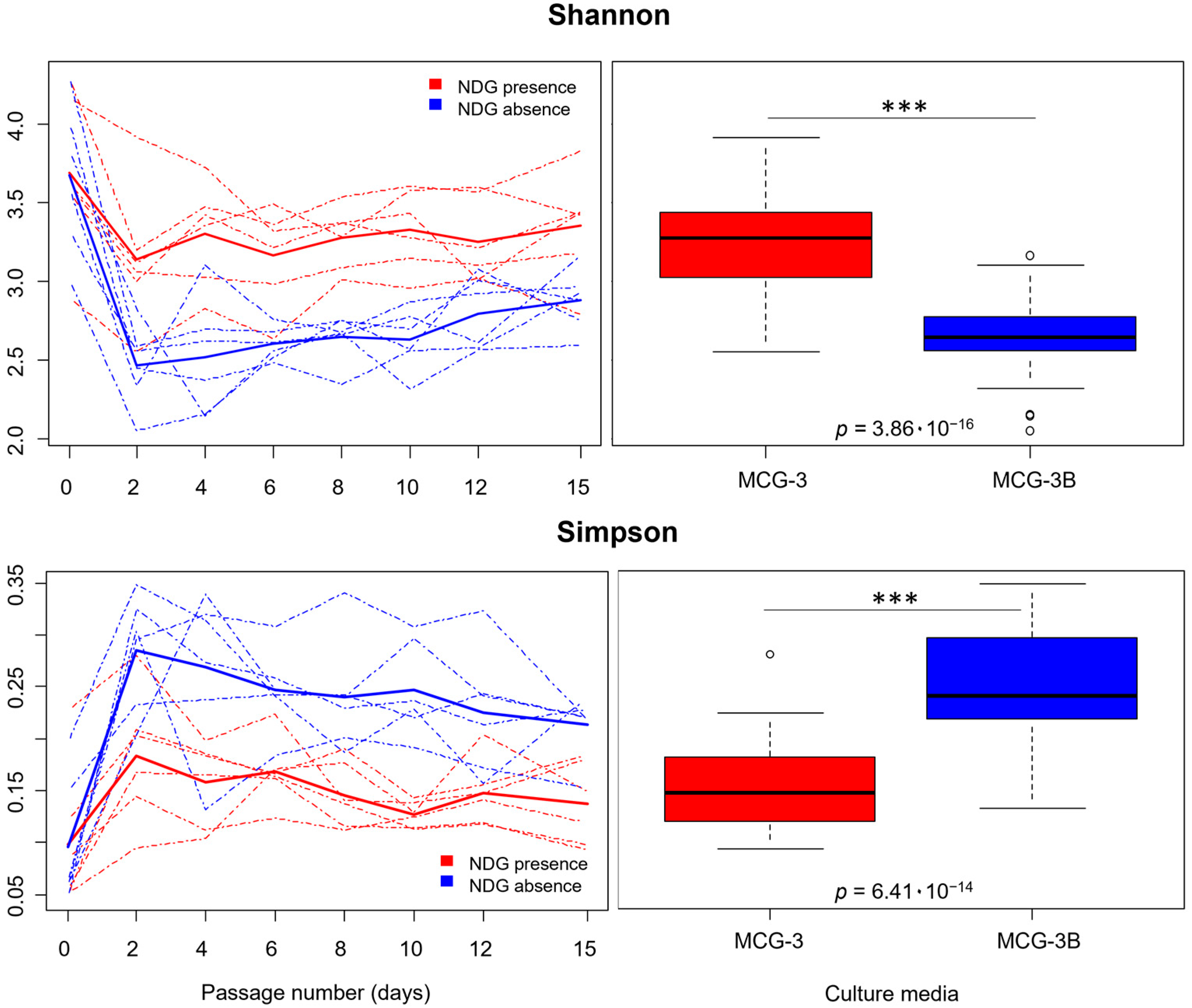
2.1. Alpha Diversity Was Affected by Non-Digested Gluten Presence in the Culture Media
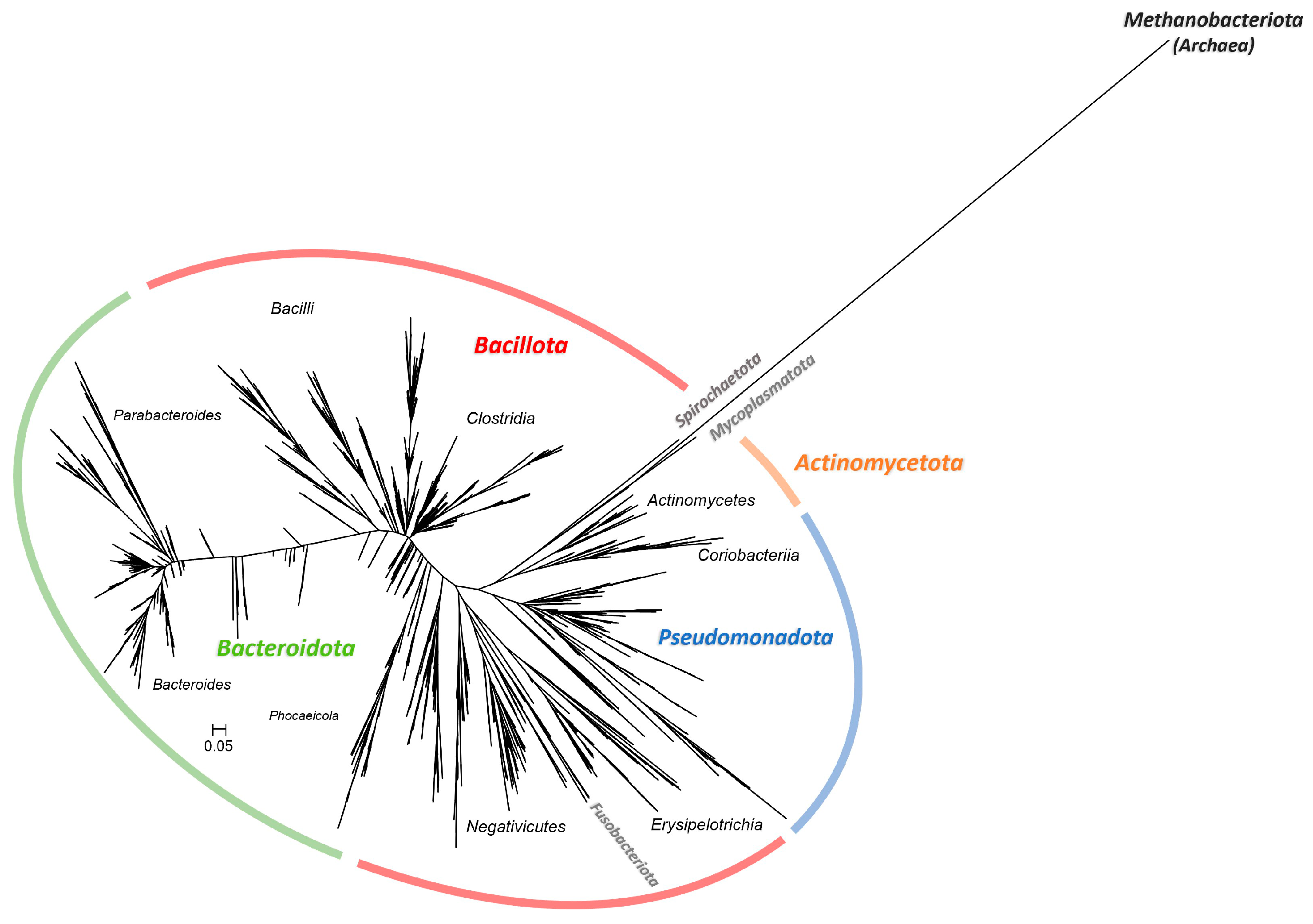
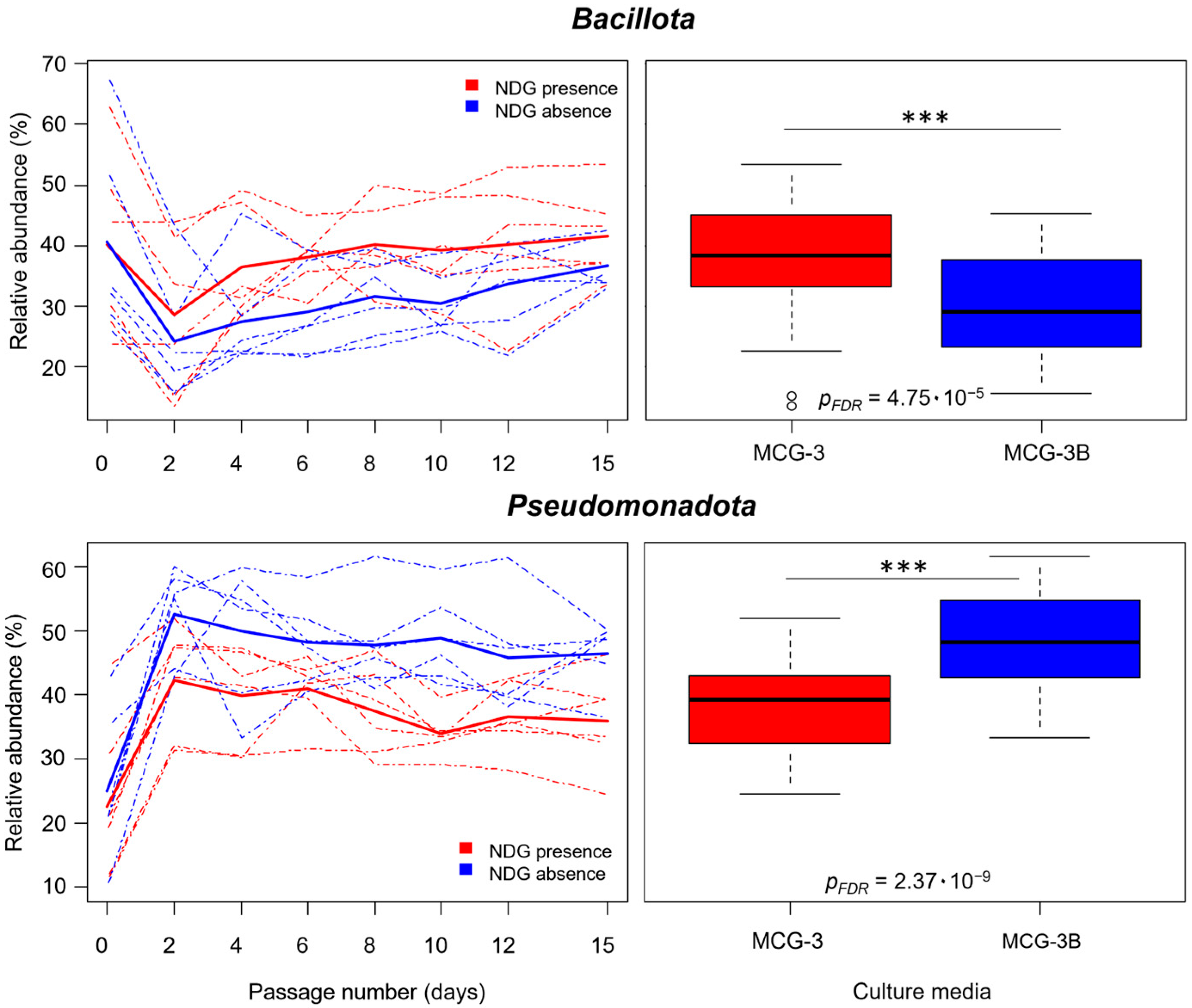
2.2. Non-Digested-Gluten Presence Affected Community Composition at All Taxonomic Levels
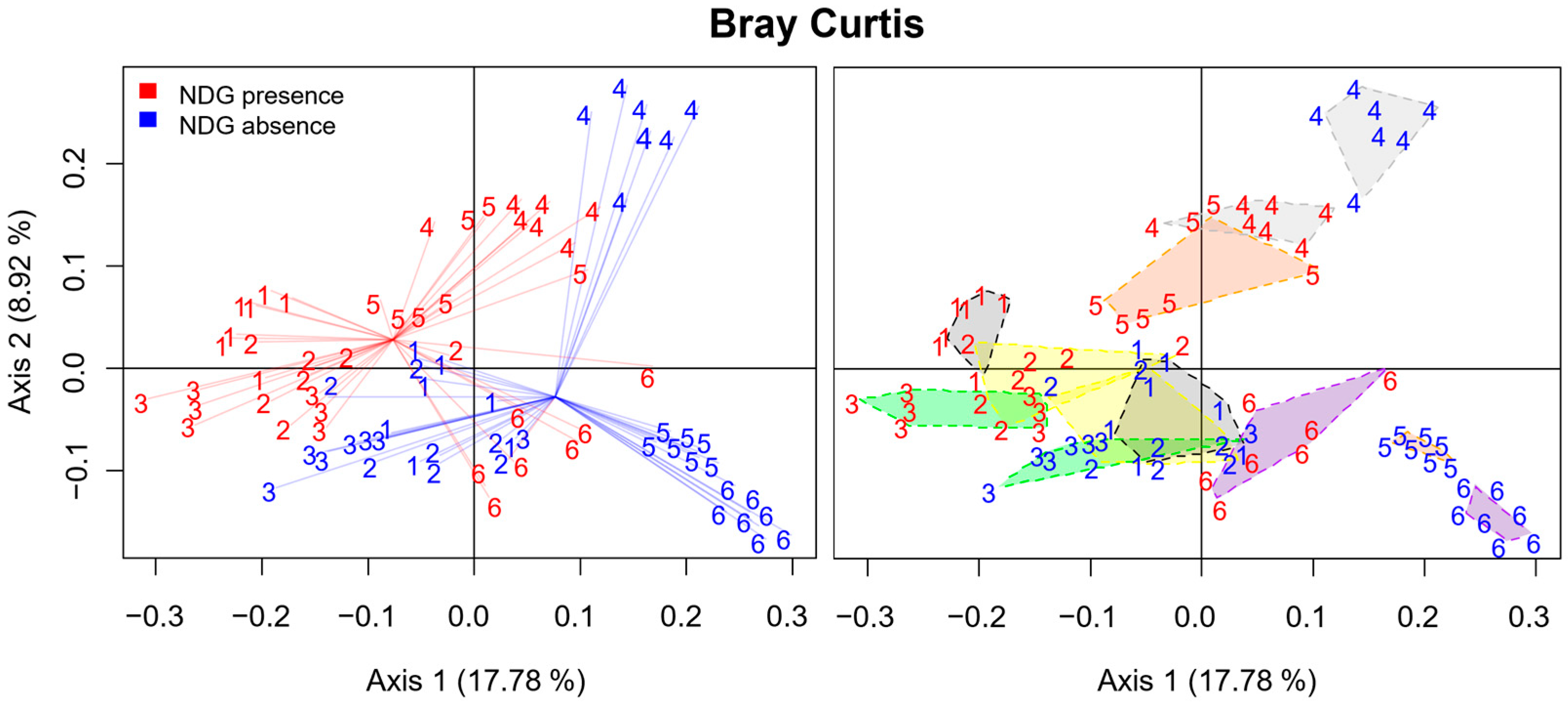
2.3. Community Composition at the OTU Level Was Affected by NDG Presence and Maintained Stability through Subculturing
2.4. Principal Coordinate Analysis Confirmed the Achievement of the Obtention and the Stable Culture of Communities Influenced by NDG Presence
3. Discussion
3.1. The Initial Diversity Loss Observed for Communities Is Common in Top-Down Approaches
3.2. Communities Early Reached Compositional Stability
3.3. NDG Promoted Higher Diversity in Communities Derived from Gluten Metabolism
3.4. Gluten in its Non-Digested Form Affected the Communities at All Taxonomic Levels
3.5. Gluten in Its Non-Digested Form Affected Communities at the OTU Level
3.6. Communities Were Highly Variable among Volunteers, though Common Patterns Were Detected
3.7. Potential Translational Applications of In Vitro Study of Gut Microbial Communities
4. Materials and Methods
4.1. Subjects of Study and Fecal Sampling
4.2. Culture Media
4.3. Obtention and Maintenance of Microbial Communities Derived from Gluten Metabolism
4.4. Microbial Community Composition Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moeller, A.H.; Caro-Quintero, A.; Mjungu, D.; Georgiev, A.V.; Lonsdorf, E.V.; Muller, M.N.; Pusey, A.E.; Peeters, M.; Hahn, B.H.; Ochman, H. Cospeciation of Gut Microbiota with Hominids. Science 2016, 353, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Lagier, J.; Armougom, F.; Million, M.; Hugon, P.; Pagnier, I.; Robert, C.; Bittar, F.; Fournous, G.; Gimenez, G.; Maraninchi, M.; et al. Microbial Culturomics: Paradigm Shift in the Human Gut Microbiome Study. Clin. Microbiol. Infect. 2012, 18, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Lagier, J.-C.; Khelaifia, S.; Alou, M.T.; Ndongo, S.; Dione, N.; Hugon, P.; Caputo, A.; Cadoret, F.; Traore, S.I.; Seck, E.H.; et al. Culture of Previously Uncultured Members of the Human Gut Microbiota by Culturomics. Nat. Microbiol. 2016, 1, 16203. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A. What Is Microbial Community Ecology? ISME J. 2009, 3, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Little, A.E.F.; Robinson, C.J.; Peterson, S.B.; Raffa, K.F.; Handelsman, J. Rules of Engagement: Interspecies Interactions That Regulate Microbial Communities. Annu. Rev. Microbiol. 2008, 62, 375–401. [Google Scholar] [CrossRef] [PubMed]
- Stubbendieck, R.M.; Vargas-Bautista, C.; Straight, P.D. Bacterial Communities: Interactions to Scale. Front. Microbiol. 2016, 7, 1234. [Google Scholar] [CrossRef] [PubMed]
- Freilich, S.; Zarecki, R.; Eilam, O.; Segal, E.S.; Henry, C.S.; Kupiec, M.; Gophna, U.; Sharan, R.; Ruppin, E. Competitive and Cooperative Metabolic Interactions in Bacterial Communities. Nat. Commun. 2011, 2, 589. [Google Scholar] [CrossRef]
- Marx, C.J. Getting in Touch with Your Friends. Science 2009, 324, 1150–1151. [Google Scholar] [CrossRef]
- Ponomarova, O.; Patil, K.R. Metabolic Interactions in Microbial Communities: Untangling the Gordian Knot. Curr. Opin. Microbiol. 2015, 27, 37–44. [Google Scholar] [CrossRef]
- Zelezniak, A.; Andrejev, S.; Ponomarova, O.; Mende, D.R.; Bork, P.; Patil, K.R. Metabolic Dependencies Drive Species Co-Occurrence in Diverse Microbial Communities. Proc. Natl. Acad. Sci. USA 2015, 112, 6449–6454. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.-X.; Chen, X.-Y.; Wang, J.-Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in Health and Diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut Microbiota in Human Metabolic Health and Disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Machado, D.; Maistrenko, O.M.; Andrejev, S.; Kim, Y.; Bork, P.; Patil, K.R.; Patil, K.R. Polarization of Microbial Communities between Competitive and Cooperative Metabolism. Nat. Ecol. Evol. 2021, 5, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of Genome-Scale Metabolic Reconstructions for 773 Members of the Human Gut Microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Venturelli, O.S.; Carr, A.C.; Fisher, G.; Hsu, R.H.; Lau, R.; Bowen, B.P.; Hromada, S.; Northen, T.; Arkin, A.P. Deciphering Microbial Interactions in Synthetic Human Gut Microbiome Communities. Mol. Syst. Biol. 2018, 14, e857. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.J.; Lenski, R.E.; Zinser, E.R. The Black Queen Hypothesis: Evolution of Dependencies through Adaptive Gene Loss. MBio 2012, 3, e00036-12. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.E. The Microbial Exometabolome: Ecological Resource and Architect of Microbial Communities. Philos. Trans. R. Soc. B 2020, 375, 20190250. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.L.; Correia-Melo, C.; Zorrilla, F.; Herrera-Dominguez, L.; Wu, M.Y.; Hartl, J.; Campbell, K.; Blasche, S.; Kreidl, M.; Egger, A.S.; et al. Microbial Communities Form Rich Extracellular Metabolomes That Foster Metabolic Interactions and Promote Drug Tolerance. Nat. Microbiol. 2022, 7, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Van Hoek, M.J.A.; Merks, R.M.H. Emergence of Microbial Diversity Due to Cross-Feeding Interactions in a Spatial Model of Gut Microbial Metabolism. BMC Syst. Biol. 2017, 11, 56. [Google Scholar] [CrossRef]
- Morris, B.E.L.; Henneberger, R.; Huber, H.; Moissl-Eichinger, C. Microbial Syntrophy: Interaction for the Common Good. FEMS Microbiol. Rev. 2013, 37, 384–406. [Google Scholar] [CrossRef]
- D’Onofrio, A.; Crawford, J.M.; Stewart, E.J.; Witt, K.; Gavrish, E.; Epstein, S.; Clardy, J.; Lewis, K. Siderophores from Neighboring Organisms Promote the Growth of Uncultured Bacteria. Chem. Biol. 2010, 17, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, T.; Lewis, K.; Epstein, S.S. Isolating “Uncultivable” Microorganisms in Pure Culture in a Simulated Natural Environment. Science 2002, 296, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.; Lewis, K.; Orjala, J.; Mo, S.; Ortenberg, R.; O’Connor, P.; Zhao, C.; Vouros, P.; Kaeberlein, T.; Epstein, S.S. Short Peptide Induces an “Uncultivable” Microorganism to Grow in vitro. Appl. Environ. Microbiol. 2008, 74, 4889–4897. [Google Scholar] [CrossRef] [PubMed]
- Madsen, J.S.; Sørensen, S.J.; Burmølle, M. Bacterial Social Interactions and the Emergence of Community-Intrinsic Properties. Curr. Opin. Microbiol. 2018, 42, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Nesme, J.; Røder, H.L.; Li, X.; Zuo, Z.; Petersen, M.; Burmølle, M.; Sørensen, S.J. Emergent Bacterial Community Properties Induce Enhanced Drought Tolerance in Arabidopsis. NPJ Biofilms Microbiomes 2021, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Lisa, R.; Faust, K. From Hairballs to Hypotheses—Biological Insights from Microbial Networks. FEMS Microbiol. Rev. 2018, 42, 761–780. [Google Scholar] [CrossRef]
- Reimer, R.A. Establishing the Role of the Diet in the Microbiota—Disease Axis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 86–87. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Cintoni, M.; Raoul, P.; Lopetuso, L.R.; Scaldaferri, F.; Pulcini, G.; Donato Miggiano, G.A.; Gasbarrini, A.; Mele, M.C. Food Components and Dietary Habits: Keys for a Healthy Gut Microbiota Composition. Nutrients 2019, 11, 2393. [Google Scholar] [CrossRef] [PubMed]
- Spor, A.; Koren, O.; Ley, R. Unravelling the Effects of the Environment and Host Genotype on the Gut Microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef]
- Wen, L.; Duffy, A. Factors Influencing the Gut Microbiota, Inflammation and Type 2 Diabetes. J. Nutr. 2017, 147, 1468S–1475S. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, M.; Wang, S.; Han, R.; Cao, Y.; Hua, W.; Mao, Y.; Zhang, X.; Pang, X.; Wei, C.; et al. Interactions between Gut Microbiota, Host Genetics and Diet Relevant to Development of Metabolic Syndromes in Mice. ISME J. 2010, 4, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Croall, I.D.; Aziz, I.; Trott, N.; Tosi, P.; Hoggard, N.; Sanders, D.S. Gluten Does Not Induce Gastrointestinal Symptoms in Healthy Volunteers: A Double-Blind Randomized Placebo Trial. Gastroenterology 2019, 157, 881–883. [Google Scholar] [CrossRef] [PubMed]
- Niland, B.; Cash, B.D. Health Benefits and Adverse Effects of a Gluten-Free Diet in Non-Celiac Disease Patients. Gastroenterol. Hepatol. 2018, 14, 82–91. [Google Scholar]
- Caminero, A.; Herrán, A.R.; Nistal, E.; Pérez-Andrés, J.; Vaquero, L.; Vivas, S.; Ruiz de Morales, J.M.G.; Albillos, S.M.; Casqueiro, J. Diversity of the Cultivable Human Gut Microbiome Involved in Gluten Metabolism: Isolation of Microorganisms with Potential Interest for Coeliac Disease. FEMS Microbiol. Ecol. 2014, 88, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Herrán, A.R.; Pérez-Andrés, J.; Caminero, A.; Nistal, E.; Vivas, S.; Ruiz de Morales, J.M.; Casqueiro, J. Gluten-Degrading Bacteria Are Present in the Human Small Intestine of Healthy Volunteers and Celiac Patients. Res. Microbiol. 2017, 168, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Senicar, T.; Kukovicic, A.; Tkalec, V.; Mahnic, A.; Dolinsek, J.; Rupnik, M. Comparison of Microbial Populations in Saliva and Feces from Healthy and Celiac Adolescents with Conventional and Molecular Approaches after Cultivation on Gluten-Containing Media: An Exploratory Study. Microorganisms 2021, 9, 2375. [Google Scholar] [CrossRef] [PubMed]
- Zamakhchari, M.; Wei, G.; Dewhirst, F.; Lee, J.; Schuppan, D.; Oppenheim, F.G.; Helmerhorst, E.J. Identification of Rothia Bacteria as Gluten-Degrading Natural Colonizers of the Upper Gastro-Intestinal Tract. PLoS ONE 2011, 6, e24455. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Nandhra, G.K.; Mark, E.B.; Di Tanna, G.L.; Haase, A.M.; Poulsen, J.; Christodoulides, S.; Kung, V.; Klinge, M.W.; Knudsen, K.; Borghammer, P.; et al. Normative Values for Region-Specific Colonic and Gastrointestinal Transit Times in 111 Healthy Volunteers Using the 3D-Transit Electromagnet Tracking System: Influence of Age, Gender, and Body Mass Index. Neurogastroenterol. Motil. 2020, 32, e13734. [Google Scholar] [CrossRef]
- Poeker, S.A.; Lacroix, C.; De Wouters, T.; Spalinger, M.R.; Scharl, M.; Geirnaert, A. Stepwise Development of an in vitro Continous Fermentation Model for the Murine Caecal Microbiota. Front. Microbiol. 2019, 10, 1166. [Google Scholar] [CrossRef]
- Van Den Abbeele, P.; Grootaert, C.; Marzorati, M.; Possemiers, S.; Verstraete, W.; Gérard, P.; Rabot, S.; Bruneau, A.; Aidy Ei, S.; Derrien, M.; et al. Microbial Community Development in a Dynamic Gut Model Is Reproducible, Colon Region Specific, and Selective for Bacteroidetes and Clostridium Cluster IX. Appl. Environ. Microbiol. 2010, 76, 5237–5246. [Google Scholar] [CrossRef] [PubMed]
- Bayer, G.; Ganobis, C.M.; Allen-Vercoe, E.; Philpott, D.J. Defined Gut Microbial Communities: Promising Tools to Understand and Combat Disease. Microbes Infect. 2021, 23, 104816. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, K.; Parreira, V.R.; Cochrane, K.; Allen-Vercoe, E. Drivers of Human Gut Microbial Community Assembly: Coadaptation, Determinism and Stochasticity. ISME J. 2019, 13, 3080–3092. [Google Scholar] [CrossRef] [PubMed]
- Yen, S.; Mcdonald, J.A.K.; Schroeter, K.; Oliphant, K.; Sokolenko, S.; Blondeel, E.J.M.; Allen-vercoe, E.; Aucoin, M.G. Metabolomic Analysis of Human Fecal Microbiota: A Comparison of Feces-Derived Communities and Defined Mixed Communities. J. Proteome Res. 2015, 14, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.A.K.; Schroeter, K.; Fuentes, S.; Heikamp-deJong, I.; Khursigara, C.M.; de Vos, W.M.; Allen-Vercoe, E. Evaluation of Microbial Community Reproducibility, Stability and Composition in a Human Distal Gut Chemostat Model. J. Microbiol. Methods 2013, 95, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Rubio, L.A.; Duncan, S.H.; Donachie, G.E.; Holtrop, G.; Lo, G.; Farquharson, F.M.; Wagner, J.; Parkhill, J.; Louis, P.; et al. Pivotal Roles for pH, Lactate, and Lactate-Utilizing Bacteria in the Stability of a Human Colonic Microbial Ecosystem. mSystems 2020, 5, e00645-20. [Google Scholar] [CrossRef]
- Macfarlane, G.T.; Macfarlane, S.; Gibson, G.R. Validation of a Three-Stage Compound Continuous Culture System for Investigating the Effect of Retention Time on the Ecology and Metabolism of Bacteria in the Human Colon. Microb. Ecol. 1998, 35, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Possemiers, S.; Verthé, K.; Uyttendaele, S.; Verstraete, W. PCR-DGGE-Based Quantification of Stability of the Microbial Community in a Simulator of the Human Intestinal Microbial Ecosystem. FEMS Microbiol. Ecol. 2004, 49, 495–507. [Google Scholar] [CrossRef]
- Wang, J.; Carper, D.L.; Burdick, L.H.; Shrestha, H.K.; Appidi, M.R.; Abraham, P.E.; Timm, C.M.; Hettich, R.L.; Pelletier, D.A.; Doktycz, M.J. Formation, Characterization and Modeling of Emergent Synthetic Microbial Communities. Comput. Struct. Biotechnol. J. 2021, 19, 1917–1927. [Google Scholar] [CrossRef]
- Yao, T.; Chen, M.H.; Lindemann, S.R. Structurally Complex Carbohydrates Maintain Diversity in Gut-Derived Microbial Consortia under High Dilution Pressure. FEMS Microbiol. Ecol. 2020, 96, fiaa158. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet Rapidly and Reproducibly Alters the Human Gut Microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [PubMed]
- Germerodt, S.; Bohl, K.; Lück, A.; Pande, S.; Schröter, A.; Kaleta, C.; Schuster, S.; Kost, C. Pervasive Selection for Cooperative Cross-Feeding in Bacterial Communities. PLoS Comput. Biol. 2016, 12, e1004986. [Google Scholar] [CrossRef] [PubMed]
- Pande, S.; Merker, H.; Bohl, K.; Reichelt, M.; Schuster, S.; De Figueiredo, L.F.; Kaleta, C.; Kost, C. Fitness and Stability of Obligate Cross-Feeding Interactions That Emerge upon Gene Loss in Bacteria. ISME J. 2014, 8, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Kiewiet, M.B.G.; Elderman, M.E.; El Aidy, S.; Burgerhof, J.G.M.; Visser, H.; Vaughan, E.E.; Faas, M.M.; de Vos, P. Flexibility of Gut Microbiota in Ageing Individuals during Dietary Fiber Long-Chain Inulin Intake. Mol. Nutr. Food Res. 2021, 65, 2000390. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Bihan, M.; Yooseph, S.; Methé, B.A. Analyses of the Microbial Diversity across the Human Microbiome. PLoS ONE 2012, 7, e32118. [Google Scholar] [CrossRef]
- Zhou, Y.; Gao, H.; Mihindukulasuriya, K.A.; La Rosa, P.S.; Wylie, K.M.; Vishnivetskaya, T.; Podar, M.; Warner, B.; Tarr, P.I.; Nelson, D.E.; et al. Biogeography of the Ecosystems of the Healthy Human Body. Genome Biol. 2013, 14, R1. [Google Scholar] [CrossRef]
- Canfora, E.E.; Meex, R.C.R.; Venema, K.; Blaak, E.E. Gut Microbial Metabolites in Obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 2019, 15, 261–273. [Google Scholar] [CrossRef]
- Davila, A.M.; Blachier, F.; Gotteland, M.; Andriamihaja, M.; Benetti, P.H.; Sanz, Y.; Tomé, D. Re-Print of “Intestinal Luminal Nitrogen Metabolism: Role of the Gut Microbiota and Consequences for the Host”. Pharmacol. Res. 2013, 69, 114–126. [Google Scholar] [CrossRef]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.J.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and Associated Dietary Extremes Impact on Gut Microbial Diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef]
- Kõiv, V.; Adamberg, K.; Adamberg, S.; Sumeri, I.; Kasvandik, S.; Kisand, V.; Maiväli, Ü.; Tenson, T. Microbiome of Root Vegetables—A Source of Gluten-Degrading Bacteria. Appl. Microbiol. Biotechnol. 2020, 104, 8871–8885. [Google Scholar] [CrossRef] [PubMed]
- Hugenholtz, F.; Davids, M.; Schwarz, J.; Müller, M.; Tomé, D.; Schaap, P.; Hooiveld, G.J.E.J.; Smidt, H.; Kleerebezem, M. Metatranscriptome Analysis of the Microbial Fermentation of Dietary Milk Proteins in the Murine Gut. PLoS ONE 2018, 13, e0194066. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.-L.; Wu, G.; Zhu, W.-Y. Amino Acid Metabolism in Intestinal Bacteria: Links between Gut Ecology and Host Health. Front. Biosci. 2011, 16, 1768–1786. [Google Scholar] [CrossRef] [PubMed]
- Ezaki, T.; Kawamura, Y.; Li, N.; Li, Z.-Y.; Zhao, L.; Shu, S. Proposal of the Genera Anaerococcus Gen. Nov., Peptoniphilus Gen. Nov. and Gallicola Gen. Nov for Members of the Genus Peptostreptococcus. Int. J. Syst. Evol. Microbiol. 2001, 51, 1521–1528. [Google Scholar] [CrossRef]
- Smith, E.A.; MacFarlane, G.T. Enumeration of Amino Acid Fermenting Bacteria in the Human Large Intestine: Effects of pH and Starch on Peptide Metabolism and Dissimilation of Amino Acids. FEMS Microbiol. Ecol. 1998, 25, 355–368. [Google Scholar] [CrossRef]
- Laparra, J.M.; Sanz, Y. Bifidobacteria Inhibit the Inflammatory Response Induced by Gliadins in Intestinal Epithelial Cells via Modifications of Toxic Peptide Generation during Digestion. J. Cell Biochem. 2010, 109, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, E.; Laparra, J.M.; Sanz, Y. Discerning the Role of Bacteroides fragilis in Celiac Disease Pathogenesis. Appl. Environ. Microbiol. 2012, 78, 6507–6515. [Google Scholar] [CrossRef] [PubMed]
- Stojanov, S.; Berlec, A.; Štrukelj, B. The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio in the Treatment of Obesity and Inflammatory Bowel Disease. Microorganisms 2020, 8, 1715. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Lopetuso, L.R.; Rizzatti, G.; Gibiino, G.; Cennamo, V.; Gasbarrini, A. Actinobacteria: A Relevant Minority for the Maintenance of Gut Homeostasis. Dig. Liver Dis. 2018, 50, 421–428. [Google Scholar] [CrossRef]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial Signature of Dysbiosis in Gut Microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Salonen, A.; Lahti, L.; Salojärvi, J.; Holtrop, G.; Korpela, K.; Duncan, S.H.; Date, P.; Farquharson, F.; Johnstone, A.M.; Lobley, G.E.; et al. Impact of Diet and Individual Variation on Intestinal Microbiota Composition and Fermentation Products in Obese Men. ISME J. 2014, 8, 2218–2230. [Google Scholar] [CrossRef]
- Basson, A.R.; Zhou, Y.; Seo, B.; Rodriguez-Palacios, A.; Cominelli, F. Autologous Fecal Microbiota Transplantation for the Treatment of Inflammatory Bowel Disease. Transl. Res. 2020, 226, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nugent, S.G.; Kumar, D.; Rampton, D.S.; Evans, D.F. Intestinal Luminal pH in Inflammatory Bowel Disease: Possible Determinants and Implications for Therapy with Aminosalicylates and Other Drugs. Gut 2001, 48, 571–577. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and Tools for High Throughput rRNA Analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA Alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-5. 2019. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 10 July 2019).
- Kembel, S.W.; Cowan, P.D.; Helmus, M.R.; Cornwell, W.K.; Morlon, H.; Ackerly, D.D.; Blomberg, S.P.; Webb, C.O. Picante: R Tools for Integrating Phylogenies and Ecology. Bioinformatics 2010, 26, 1463–1464. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, A.; Brockhoff, P.B.; Christensen, R.H.B. LmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw. 2017, 82, 1–26. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. 2021. Available online: https://www.R-project.org (accessed on 2 March 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linear Model (y~NDG 1 Presence) Degrees of Freedom: 1, 82 | |||||
|---|---|---|---|---|---|
| Digested Gluten + NDG 1 (MCG-3) | Digested Gluten (MCG-3B) | MSB | F | Significance (p) 2 | |
| Shannon diversity (H’) | 3.26 ± 0.30 | 2.65 ± 0.25 | 7.89 | 102.93 | 3.86 × 10−16 *** |
| Simpson dominance (D) | 0.15 ± 0.04 | 0.25 ± 0.05 | 0.19 | 81.46 | 6.41 × 10−14 *** |
| NRI 3 | −5.60 ± 1.87 | −7.40 ± 1.81 | 67.75 | 20.07 | 2.40 × 10−5 *** |
| Taxon | Relative Abundance (% Reads) in MCG-3 | Relative Abundance (% Reads) in MCG-3B | LFC | Significance (pFDR) 1 |
|---|---|---|---|---|
| Enterobacteriaceae | 38.17 | 48.98 | −0.36 | 8.19 × 10−10 *** |
| Lachnospiraceae | 13.47 | 10.90 | 0.31 | 6.19 × 10−4 *** |
| Clostridiaceae 1 | 8.42 | 3.28 | 1.36 | 6.70 × 10−7 *** |
| Streptococcaceae | 1.26 | 0.09 | 3.88 | 4.87 × 10−5 *** |
| Bifidobacteriaceae | 1.06 | 0.27 | 2.00 | 1.43 × 10−3 ** |
| Peptoniphilaceae | 0.75 | 0.24 | 1.64 | 2.02 × 10−3 ** |
| Prevotellaceae | 0.56 | 0.08 | 2.84 | 3.98 × 10−3 ** |
| Porphyromonadaceae | 0.50 | 0.10 | 2.37 | 3.58 × 10−17 *** |
| Sutterellaceae | 0.39 | 0.08 | 2.27 | 4.17 × 10−4 *** |
| Erysipelotrichaceae | 0.30 | 0.10 | 1.65 | 3.41 × 10−2 * |
| Eggerthellaceae | 0.12 | 0.05 | 1.34 | 8.77 × 10−3 ** |
| Odoribacteraceae | 3.81 × 10−3 | 4.78 × 10−4 | 3.00 | 1.74 × 10−5 *** |
| Selenomonadaceae | 1.52 × 10−2 | 4.74 × 10−3 | 1.68 | 6.63 × 10−3 ** |
| Escherichia/Shigella | 40.69 | 51.12 | −0.33 | 8.19 × 10−10 *** |
| Bacteroides | 18.84 | 19.30 | −0.03 | 5.18 × 10−1 |
| Clostridium sensu stricto | 8.97 | 3.38 | 1.41 | 7.74 × 10−7 *** |
| Enterococcus | 4.94 | 5.54 | −0.17 | 2.82 × 10−1 |
| Veillonella | 4.69 | 5.55 | −0.24 | 2.54 × 10−1 |
| Roseburia | 2.81 | 1.26 | 1.16 | 6.56 × 10−3 ** |
| Enterocloster | 2.31 | 3.34 | −0.53 | 8.31 × 10−8 *** |
| Phocaeicola | 2.27 | 1.03 | 1.14 | 7.88 × 10−4 *** |
| Paraclostridium | 1.57 | 2.05 | −0.38 | 9.24 × 10−2 |
| Streptococcus | 1.41 | 0.09 | 3.92 | 4.87 × 10−5 *** |
| Collinsella | 1.21 | 0.63 | 0.96 | 2.88 × 10−1 |
| Bifidobacterium | 1.02 | 0.25 | 2.04 | 1.43 × 10−3 ** |
| Peptoniphilus | 0.77 | 0.25 | 1.62 | 6.22 × 10−3 ** |
| Parabacteroides | 0.56 | 0.10 | 2.52 | 5.67 × 10−18 *** |
| Clostridium XIVa | 0.29 | 0.18 | 0.70 | 9.68 × 10−3 ** |
| Mediterraneibacter | 2.99 × 10−2 | 7.14 × 10−3 | 2.06 | 8.77 × 10−3 ** |
| Intestinimonas | 2.38 × 10−2 | 6.44 × 10−3 | 1.88 | 6.19 × 10−4 *** |
| Intestinibacillus | 8.20 × 10−3 | 2.23 × 10−3 | 1.88 | 6.12 × 10−5 *** |
| Butyricimonas | 1.35 × 10−3 | 1.56 × 10−4 | 3.11 | 9.17 × 10−3 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carnicero-Mayo, Y.; Sáenz de Miera, L.E.; Ferrero, M.Á.; Navasa, N.; Casqueiro, J. Modeling Dynamics of Human Gut Microbiota Derived from Gluten Metabolism: Obtention, Maintenance and Characterization of Complex Microbial Communities. Int. J. Mol. Sci. 2024, 25, 4013. https://doi.org/10.3390/ijms25074013
Carnicero-Mayo Y, Sáenz de Miera LE, Ferrero MÁ, Navasa N, Casqueiro J. Modeling Dynamics of Human Gut Microbiota Derived from Gluten Metabolism: Obtention, Maintenance and Characterization of Complex Microbial Communities. International Journal of Molecular Sciences. 2024; 25(7):4013. https://doi.org/10.3390/ijms25074013
Chicago/Turabian StyleCarnicero-Mayo, Yaiza, Luis E. Sáenz de Miera, Miguel Ángel Ferrero, Nicolás Navasa, and Javier Casqueiro. 2024. "Modeling Dynamics of Human Gut Microbiota Derived from Gluten Metabolism: Obtention, Maintenance and Characterization of Complex Microbial Communities" International Journal of Molecular Sciences 25, no. 7: 4013. https://doi.org/10.3390/ijms25074013
APA StyleCarnicero-Mayo, Y., Sáenz de Miera, L. E., Ferrero, M. Á., Navasa, N., & Casqueiro, J. (2024). Modeling Dynamics of Human Gut Microbiota Derived from Gluten Metabolism: Obtention, Maintenance and Characterization of Complex Microbial Communities. International Journal of Molecular Sciences, 25(7), 4013. https://doi.org/10.3390/ijms25074013