
Potential Therapeutic Targets to Modulate the Endocannabinoid System in Alzheimer’s Disease

 , , , , , and
, , , , , and
Abstract
:1. Introduction
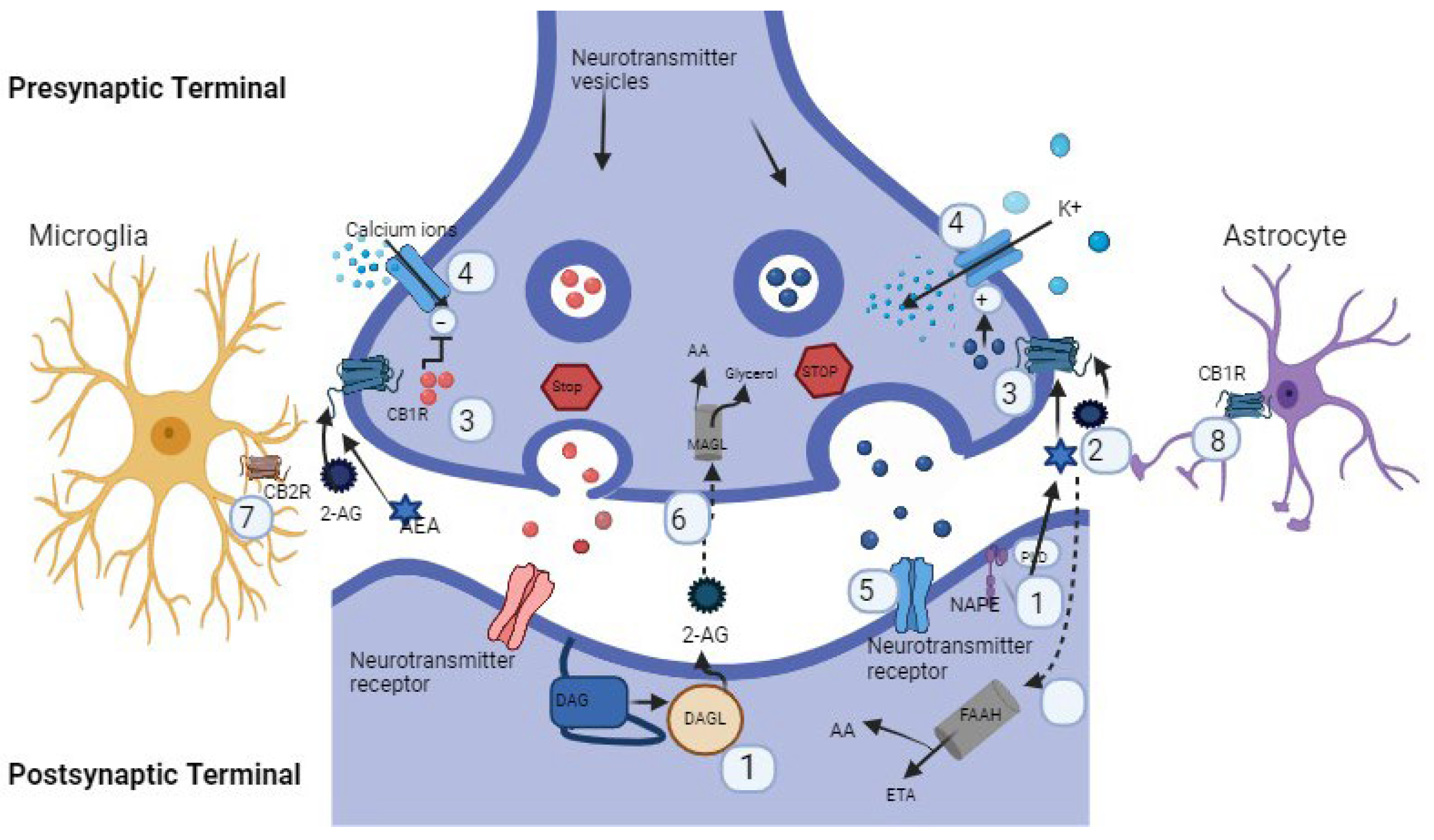
2. Physiology of the Endocannabinoid System
2.1. Components of Endocannabinoid System
2.2. Biosynthesis, Receptor’s Affinity, and Degradation
2.3. ECS Retrograde Signaling
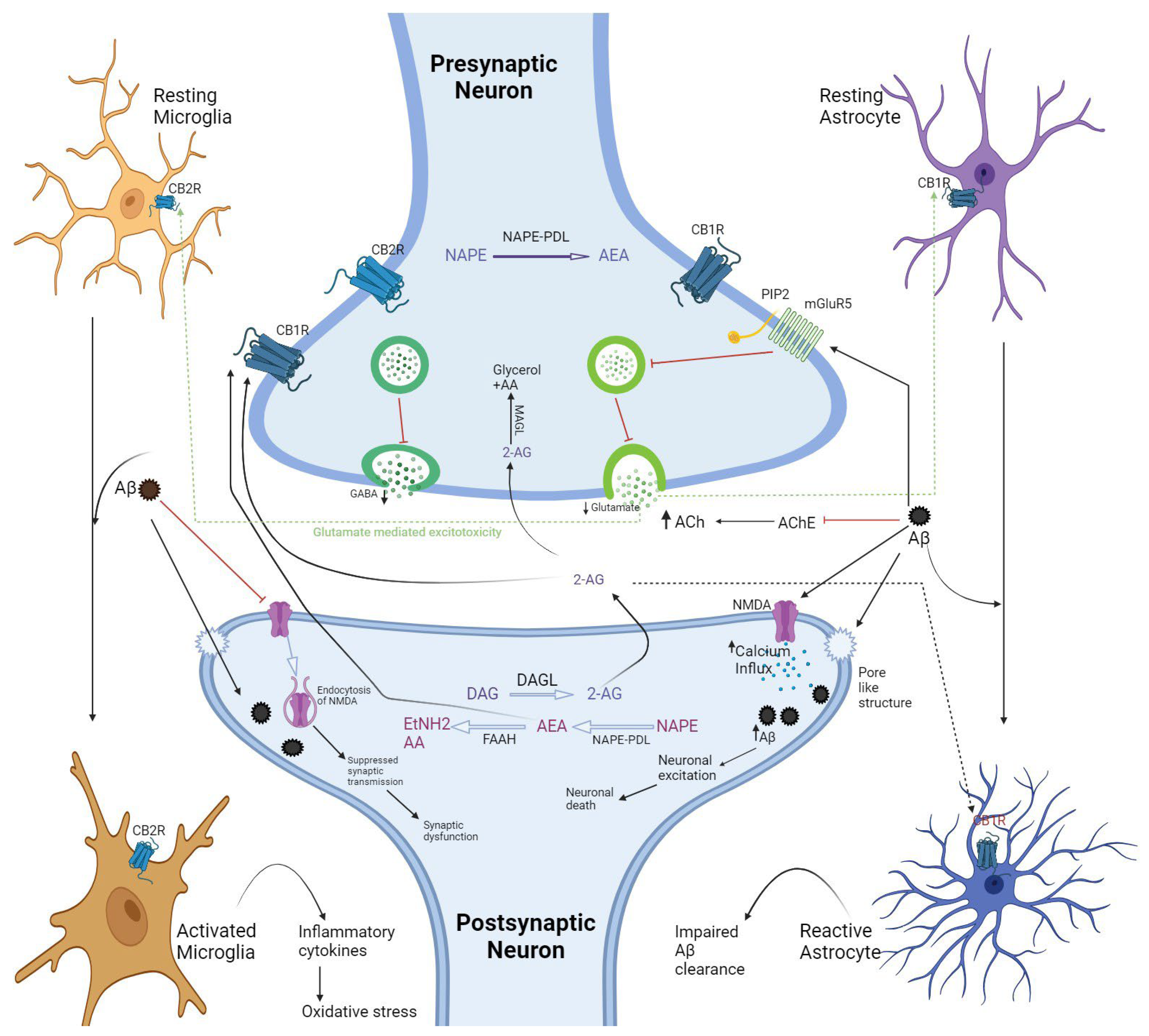
3. Prospective Pathophysiology of the Endocannabinoid System (ECS)
3.1. Functioning of Endocannabinoid Receptors (CBRs)
3.2. Controversial Reviews about the Neuroprotective Roles of CB1R and CB2R
3.3. Alteration in Endocannabinoid’s Expression and Involvement of FAAH and MAGL in AD
3.4. Correlation between CB2R and FAAH
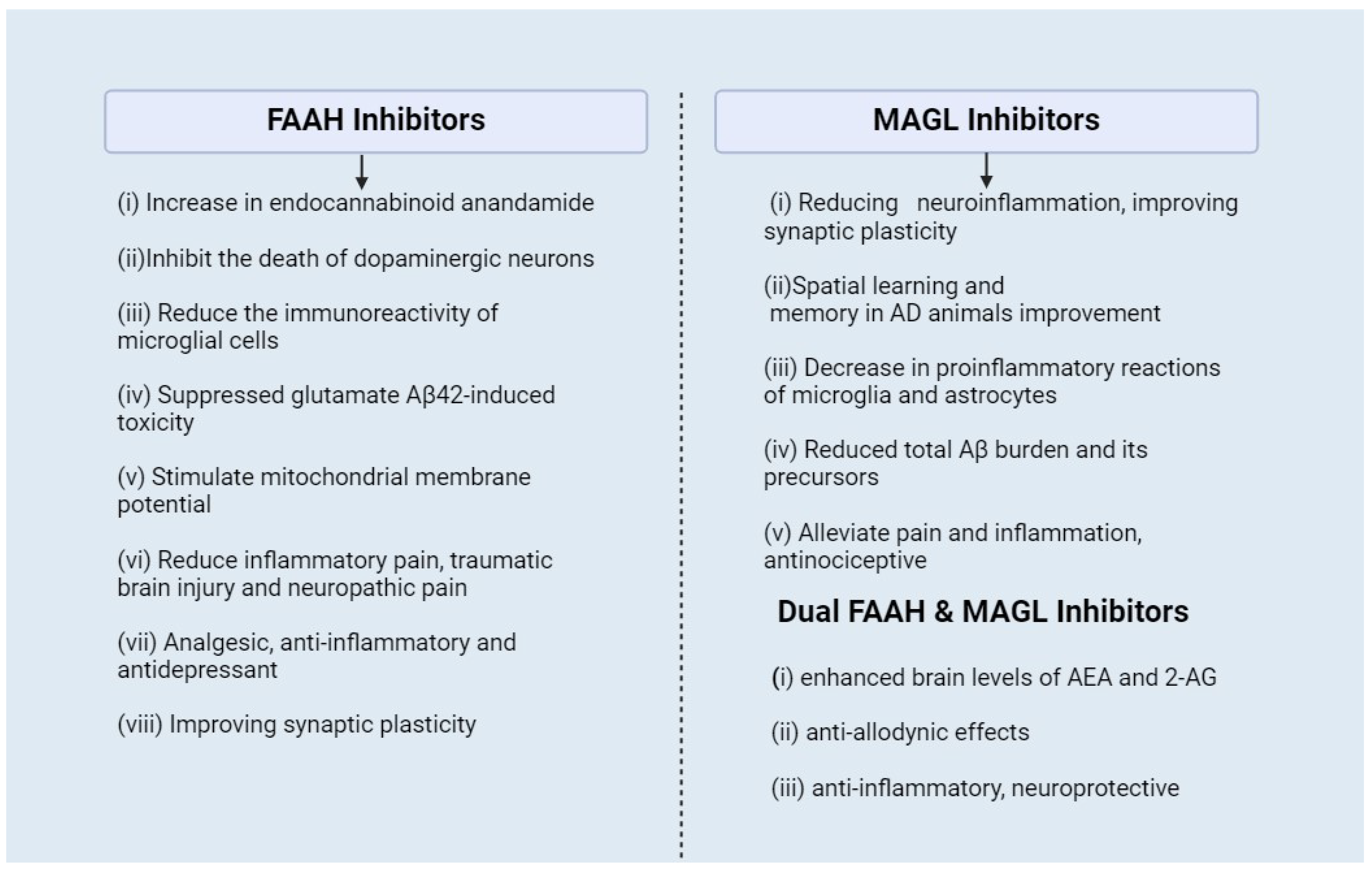
4. Pharmacological Interventions for ECS in Alzheimer’s Disease
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Popa-Wagner, A.; Buga, A.-M.; Popescu, B.; Muresanu, D. Vascular cognitive impairment, dementia, aging and energy demand. A vicious cycle. J. Neural Transm. 2015, 122, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Roubroeks, J.A.; Smith, R.G.; van den Hove, D.L.; Lunnon, K. Epigenetics and DNA methylomic profiling in Alzheimer’s disease and other neurodegenerative diseases. J. Neurochem. 2017, 143, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafò, A.; Bernardini, R.; Cantarella, G. Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca2+ Homeostasis Dysregulation. Cells 2022, 11, 2728. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Madamsetty, V.S.; Bhattacharya, D.; Roy Chowdhury, S.; Paul, M.K.; Mukherjee, A. Recent advancements of nanomedicine in neurodegenerative disorders theranostics. Adv. Funct. Mater. 2020, 30, 2003054. [Google Scholar] [CrossRef]
- Gallelli, C.A.; Calcagnini, S.; Romano, A.; Koczwara, J.B.; De Ceglia, M.; Dante, D.; Villani, R.; Giudetti, A.M.; Cassano, T.; Gaetani, S. Modulation of the oxidative stress and lipid peroxidation by endocannabinoids and their lipid analogues. Antioxidants 2018, 7, 93. [Google Scholar] [CrossRef]
- Bedse, G.; Romano, A.; Lavecchia, A.M.; Cassano, T.; Gaetani, S. The role of endocannabinoid signaling in the molecular mechanisms of neurodegeneration in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 43, 1115–1136. [Google Scholar] [CrossRef]
- Skaper, S.D.; Di Marzo, V. Endocannabinoids in nervous system health and disease: The big picture in a nutshell. Philos. Trans. R Soc. Lond. B Biol. Sci. 2012, 367, 3193–3200. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.; Onaivi, E.S. Endocannabinoid system components: Overview and tissue distribution. Recent Adv. Cannabinoid Physiol. Pathol. 2019, 1162, 1–12. [Google Scholar]
- Fowler, C.J. Transport of endocannabinoids across the plasma membrane and within the cell. FEBS J. 2013, 280, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M. Cannabinoids and gastrointestinal motility: Pharmacology, clinical effects, and potential therapeutics in humans. Neurogastroenterol. Motil. 2018, 30, e13370. [Google Scholar] [CrossRef]
- Simard, M.; Archambault, A.-S.; Lavoie, J.-P.C.; Dumais, É.; Di Marzo, V.; Flamand, N. Biosynthesis and metabolism of endocannabinoids and their congeners from the monoacylglycerol and N-acyl-ethanolamine families. Biochem. Pharmacol. 2022, 205, 115261. [Google Scholar] [CrossRef] [PubMed]
- Baggelaar, M.P.; Maccarrone, M.; van der Stelt, M. 2-Arachidonoylglycerol: A signaling lipid with manifold actions in the brain. Prog. Lipid Res. 2018, 71, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Gomes, T.M.; da Silva, D.D.; Carmo, H.; Carvalho, F.; Silva, J.P. Epigenetics and the endocannabinoid system signaling: An intricate interplay modulating neurodevelopment. Pharmacol. Res. 2020, 162, 105237. [Google Scholar] [CrossRef] [PubMed]
- Murataeva, N.; Straiker, A.; Mackie, K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br. J. Pharmacol. 2014, 171, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiù, V.; Battistini, L.; Maccarrone, M. Endocannabinoid signalling in innate and adaptive immunity. Immunology 2015, 144, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-C.; Mackie, K. An introduction to the endogenous cannabinoid system. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Nicolussi, S.; Gertsch, J. Endocannabinoid transport revisited. Vitam. Horm. 2015, 98, 441–485. [Google Scholar] [PubMed]
- Lu, H.-C.; Mackie, K. Review of the endocannabinoid system. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2021, 6, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Rapaka, D.; Bitra, V.R.; Challa, S.R.; Adiukwu, P.C. Potentiation of microglial endocannabinoid signaling alleviates neuroinflammation in Alzheimer’s disease. Neuropeptides 2021, 90, 102196. [Google Scholar] [CrossRef]
- Chen, F.; Bai, J.; Zhong, S.; Zhang, R.; Zhang, X.; Xu, Y.; Zhao, M.; Zhao, C.; Zhou, Z. Molecular Signatures of Mitochondrial Complexes Involved in Alzheimer’s Disease via Oxidative Phosphorylation and Retrograde Endocannabinoid Signaling Pathways. Oxidative Med. Cell. Longev. 2022, 2022, 9565545. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.; Romero, J.; Ramos, J.A. Endocannabinoids and neurodegenerative disorders: Parkinson’s disease, Huntington’s chorea, Alzheimer’s disease, and others. Endocannabinoids 2015, 231, 233–259. [Google Scholar]
- Loera-Valencia, R.; Cedazo-Minguez, A.; Kenigsberg, P.; Page, G.; Duarte, A.; Giusti, P.; Zusso, M.; Robert, P.; Frisoni, G.; Cattaneo, A. Current and emerging avenues for Alzheimer’s disease drug targets. J. Intern. Med. 2019, 286, 398–437. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Shen, R.; Bi, J.; Tian, X.; Hinchliffe, T.; Xia, Y. Catalpol: A potential therapeutic for neurodegenerative diseases. Curr. Med. Chem. 2015, 22, 1278–1291. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Galgani, A.; Puglisi-Allegra, S.; Limanaqi, F.; Busceti, C.L.; Fornai, F. Locus Coeruleus and neurovascular unit: From its role in physiology to its potential role in Alzheimer’s disease pathogenesis. J. Neurosci. Res. 2020, 98, 2406–2434. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Schurman, L.D.; Lu, D.; Kendall, D.A.; Howlett, A.C.; Lichtman, A.H. Molecular mechanism and cannabinoid pharmacology. Subst. Use Disord. Etiol. Treat. 2020, 258, 323–353. [Google Scholar]
- Soubias, O.; Gawrisch, K. The role of the lipid matrix for structure and function of the GPCR rhodopsin. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2012, 1818, 234–240. [Google Scholar] [CrossRef] [PubMed]
- RD, B.; Panu, A.; Ballesteros, J.; Reggio, H.P. Construction of a 3d model of the cannabinoid cb1 receptor: Determination of hellx ends and hellx orientation. Life Sci. 1995, 56, 1971–1982. [Google Scholar]
- Busquets-Garcia, A.; Desprez, T.; Metna-Laurent, M.; Bellocchio, L.; Marsicano, G.; Soria-Gomez, E. Dissecting the cannabinergic control of behavior: The where matters. BioEssays 2015, 37, 1215–1225. [Google Scholar] [CrossRef]
- Basavarajappa, B.S. Endocannabinoid system and alcohol abuse disorders. Recent Adv. Cannabinoid Physiol. Pathol. 2019, 1162, 89–127. [Google Scholar]
- Busquets-Garcia, A.; da Cruz, J.F.O.; Terral, G.; Zottola, A.C.P.; Soria-Gómez, E.; Contini, A.; Martin, H.; Redon, B.; Varilh, M.; Ioannidou, C. Hippocampal CB1 receptors control incidental associations. Neuron 2018, 99, 1247–1259.e7. [Google Scholar] [CrossRef] [PubMed]
- Talani, G.; Licheri, V.; Biggio, F.; Locci, V.; Mostallino, M.C.; Secci, P.P.; Melis, V.; Dazzi, L.; Carta, G.; Banni, S. Enhanced glutamatergic synaptic plasticity in the hippocampal CA1 field of food-restricted rats: Involvement of CB1 receptors. Neuropsychopharmacology 2016, 41, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S.; Shivakumar, M.; Joshi, V.; Subbanna, S. Endocannabinoid system in neurodegenerative disorders. J. Neurochem. 2017, 142, 624–648. [Google Scholar] [CrossRef] [PubMed]
- Bronzuoli, M.R.; Facchinetti, R.; Valenza, M.; Cassano, T.; Steardo, L.; Scuderi, C. Astrocyte function is affected by aging and not Alzheimer’s disease: A preliminary investigation in hippocampi of 3xTg-AD mice. Front. Pharmacol. 2019, 10, 644. [Google Scholar] [CrossRef] [PubMed]
- Farkas, S.; Nagy, K.; Palkovits, M.; Kovács, G.G.; Jia, Z.; Donohue, S.; Pike, V.; Halldin, C.; Máthé, D.; Harkany, T. [125I] SD-7015 reveals fine modalities of CB1 cannabinoid receptor density in the prefrontal cortex during progression of Alzheimer’s disease. Neurochem. Int. 2012, 60, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Karkkainen, E.; Tanila, H.; Laitinen, J.T. Functional autoradiography shows unaltered cannabinoid CB1 receptor signalling in hippocampus and cortex of APP/PS1 transgenic mice. CNS Neurol. Disord. Drug Targets (Former. Curr. Drug Targets-CNS Neurol. Disord.) 2012, 11, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Goffin, K.; Van den Stock, J.; De Winter, F.-L.; Cleeren, E.; Bormans, G.; Tournoy, J.; Persoons, P.; Van Laere, K.; Vandenbulcke, M. In vivo type 1 cannabinoid receptor availability in Alzheimer’s disease. Eur. Neuropsychopharmacol. 2014, 24, 242–250. [Google Scholar] [CrossRef]
- Bedse, G.; Romano, A.; Cianci, S.; Lavecchia, A.M.; Lorenzo, P.; Elphick, M.R.; LaFerla, F.M.; Vendemiale, G.; Grillo, C.; Altieri, F. Altered expression of the CB1 cannabinoid receptor in the triple transgenic mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 40, 701–712. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.; Moro, M.A.; Martínez-Orgado, J. Cannabinoids in neurodegenerative disorders and stroke/brain trauma: From preclinical models to clinical applications. Neurotherapeutics 2015, 12, 793–806. [Google Scholar] [CrossRef]
- Kibret, B.G.; Ishiguro, H.; Horiuchi, Y.; Onaivi, E.S. New insights and potential therapeutic targeting of CB2 cannabinoid receptors in CNS disorders. Int. J. Mol. Sci. 2022, 23, 975. [Google Scholar] [CrossRef]
- Azam, S.; Haque, M.E.; Jakaria, M.; Jo, S.-H.; Kim, I.-S.; Choi, D.-K. G-protein-coupled receptors in CNS: A potential therapeutic target for intervention in neurodegenerative disorders and associated cognitive deficits. Cells 2020, 9, 506. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Hinden, L.; Drori, A.; Udi, S.; Azar, S.; Baraghithy, S. The therapeutic potential of targeting the peripheral endocannabinoid/CB1 receptor system. Eur. J. Intern. Med. 2018, 49, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Lutz, B.; Marsicano, G.; Maldonado, R.; Hillard, C.J. The endocannabinoid system in guarding against fear, anxiety and stress. Nat. Rev. Neurosci. 2015, 16, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Cooray, R.; Gupta, V.; Suphioglu, C. Current aspects of the endocannabinoid system and targeted THC and CBD phytocannabinoids as potential therapeutics for Parkinson’s and Alzheimer’s diseases: A review. Mol. Neurobiol. 2020, 57, 4878–4890. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Romero, A.; Galera-López, L.; Ortiz-Romero, P.; Llorente-Ovejero, A.; De los Reyes-Ramirez, L.; de Tena, I.B.; Garcia-Elias, A.; Mas-Stachurska, A.; Reixachs-Solé, M.; Pastor, A. Cannabinoid signaling modulation through JZL184 restores key phenotypes of a mouse model for Williams–Beuren syndrome. Elife 2022, 11, e72560. [Google Scholar] [CrossRef] [PubMed]
- Bronzuoli, M.R.; Facchinetti, R.; Romano, A.; Stecca, C.; Passarella, S.; Steardo, L.; Cassano, T.; Scuderi, C. Palmitoylethanolamide dampens reactive astrogliosis and improves neuronal trophic support in a triple transgenic model of Alzheimer’s disease: In vitro and in vivo evidence. Oxidative Med. Cell. Longev. 2018, 2018, 4720532. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Funato, Y.; Meschi, E.; Jovanoski, K.D.; Miki, H.; Waddell, S. Magnesium efflux from Drosophila Kenyon cells is critical for normal and diet-enhanced long-term memory. Elife 2020, 9, e61339. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Mamun, A.A.; Sumsuzzman, D.M.; Ashraf, G.M.; Perveen, A.; Bungau, S.G.; Mousa, S.A.; El-Seedi, H.R.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Emerging promise of cannabinoids for the management of pain and associated neuropathological alterations in Alzheimer’s disease. Front. Pharmacol. 2020, 11, 1097. [Google Scholar] [CrossRef] [PubMed]
- Stempel, A.V.; Stumpf, A.; Zhang, H.-Y.; Özdoğan, T.; Pannasch, U.; Theis, A.-K.; Otte, D.-M.; Wojtalla, A.; Racz, I.; Ponomarenko, A. Cannabinoid type 2 receptors mediate a cell type-specific plasticity in the hippocampus. Neuron 2016, 90, 795–809. [Google Scholar] [CrossRef]
- Huang, Y.; Thathiah, A. Regulation of neuronal communication by G protein-coupled receptors. FEBS Lett. 2015, 589, 1607–1619. [Google Scholar] [CrossRef]
- Kotzadimitriou, D. Altered Function of CCK-Positive Interneurons in Mice Over-Expressing the Schizophrenia Risk Gene Neuregulin 1; University of Oxford: Oxford, UK, 2016. [Google Scholar]
- Janssen, B.; Vugts, D.J.; Funke, U.; Molenaar, G.T.; Kruijer, P.S.; van Berckel, B.N.; Lammertsma, A.A.; Windhorst, A.D. Imaging of neuroinflammation in Alzheimer’s disease, multiple sclerosis and stroke: Recent developments in positron emission tomography. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Aso, E.; Ferrer, I. CB2 cannabinoid receptor as potential target against Alzheimer’s disease. Front. Neurosci. 2016, 10, 243. [Google Scholar] [CrossRef] [PubMed]
- Komorowska-Müller, J.A.; Schmöle, A.-C. CB2 receptor in microglia: The guardian of self-control. Int. J. Mol. Sci. 2020, 22, 19. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Michaelides, M.; Baler, R. The neuroscience of drug reward and addiction. Physiol. Rev. 2019, 99, 2115–2140. [Google Scholar] [CrossRef] [PubMed]
- Colizzi, M. Stefano Pallanti 1, 2*, Anna Marras 1, 3 and Nikolaos Makris 4. The Endocannabinoid System: Filling the Translational Gap between Neuroscience and Psychiatry. Front. Psychiatry 2022, 12, 771442. [Google Scholar]
- Chen, L.; Yan, Y.; Chen, T.; Zhang, L.; Gao, X.; Du, C.; Du, H. Forsythiaside prevents β-amyloid-induced hippocampal slice injury by upregulating 2-arachidonoylglycerol via cannabinoid receptor 1-dependent NF-κB pathway. Neurochem. Int. 2019, 125, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Lim, C.-S. Understanding the modulatory effects of cannabidiol on Alzheimer’s disease. Brain Sci. 2021, 11, 1211. [Google Scholar] [CrossRef]
- Aso, E.; Ferrer, I. Cannabinoids for treatment of Alzheimer’s disease: Moving toward the clinic. Front. Pharmacol. 2014, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Ledesma, J.C.; Rodríguez-Arias, M.; Gavito, A.L.; Sánchez-Pérez, A.M.; Viña, J.; Medina Vera, D.; Rodríguez de Fonseca, F.; Miñarro, J. Adolescent binge-ethanol accelerates cognitive impairment and β-amyloid production and dysregulates endocannabinoid signaling in the hippocampus of APP/PSE mice. Addict. Biol. 2021, 26, e12883. [Google Scholar] [CrossRef]
- Gowran, A.; Noonan, J.; Campbell, V.A. The multiplicity of action of cannabinoids: Implications for treating neurodegeneration. CNS Neurosci. Ther. 2011, 17, 637–644. [Google Scholar] [CrossRef]
- Merelli, A.; Repetto, M.; Lazarowski, A.; Auzmendi, J. Hypoxia, oxidative stress, and inflammation: Three faces of neurodegenerative diseases. J. Alzheimer’s Dis. 2021, 82, S109–S126. [Google Scholar] [CrossRef]
- Robbe, D.; Alonso, G.; Duchamp, F.; Bockaert, J.; Manzoni, O.J. Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J. Neurosci. 2001, 21, 109–116. [Google Scholar] [CrossRef]
- Karimi, S.A.; Kazemi, F.; Komaki, H.; Kourosh Arami, M.; Shahidi, S.; Komaki, A. Electrophysiological study of the interactive role of the cannabinoid breakdown inhibitors and L-type calcium channels on granular neurons in the hippocampal dentate gyrus in rats. Neurol. Res. 2022, 44, 446–454. [Google Scholar] [CrossRef]
- Jain, S.; Bisht, A.; Verma, K.; Negi, S.; Paliwal, S.; Sharma, S. The role of fatty acid amide hydrolase enzyme inhibitors in Alzheimer’s disease. Cell Biochem. Funct. 2022, 40, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Haghani, M.; Shabani, M.; Javan, M.; Motamedi, F.; Janahmadi, M. CB1 cannabinoid receptor activation rescues amyloid β-induced alterations in behaviour and intrinsic electrophysiological properties of rat hippocampal CA1 pyramidal neurones. Cell. Physiol. Biochem. 2012, 29, 391–406. [Google Scholar] [CrossRef]
- Ren, S.-y.; Wang, Z.-z.; Zhang, Y.; Chen, N.-h. Potential application of endocannabinoid system agents in neuropsychiatric and neurodegenerative diseases—Focusing on FAAH/MAGL inhibitors. Acta Pharmacol. Sin. 2020, 41, 1263–1271. [Google Scholar] [CrossRef]
- Bajaj, S.; Jain, S.; Vyas, P.; Bawa, S.; Vohora, D. The role of endocannabinoid pathway in the neuropathology of Alzheimer’s disease: Can the inhibitors of MAGL and FAAH prove to be potential therapeutic targets against the cognitive impairment associated with Alzheimer’s disease? Brain Res. Bull. 2021, 174, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Pasquini, S.; Contri, C.; Gemma, S.; Campiani, G.; Butini, S.; Varani, K.; Vincenzi, F. Polypharmacological Approaches for CNS Diseases: Focus on Endocannabinoid Degradation Inhibition. Cells 2022, 11, 471. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.S.-J.; Mackie, K. Distribution of the endocannabinoid system in the central nervous system. Endocannabinoids 2015, 231, 59–93. [Google Scholar]
- Hillard, C.J. The endocannabinoid signaling system in the CNS: A primer. Int. Rev. Neurobiol. 2015, 125, 1–47. [Google Scholar]
- Tuo, W.; Leleu-Chavain, N.; Spencer, J.; Sansook, S.; Millet, R.; Chavatte, P. Therapeutic potential of fatty acid amide hydrolase, monoacylglycerol lipase, and N-acylethanolamine acid amidase inhibitors. J. Med. Chem. 2017, 60, 4–46. [Google Scholar] [CrossRef] [PubMed]
- Lowe, H.; Toyang, N.; Steele, B.; Bryant, J.; Ngwa, W. The endocannabinoid system: A potential target for the treatment of various diseases. Int. J. Mol. Sci. 2021, 22, 9472. [Google Scholar] [CrossRef] [PubMed]
- Schlosburg, J.E.; Kinsey, S.G.; Lichtman, A.H. Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation. AAPS J. 2009, 11, 39–44. [Google Scholar] [CrossRef]
- Biernacki, M.; Ambrożewicz, E.; Gęgotek, A.; Toczek, M.; Skrzydlewska, E. Long-term administration of fatty acid amide hydrolase inhibitor (URB597) to rats with spontaneous hypertension disturbs liver redox balance and phospholipid metabolism. Adv. Med. Sci. 2019, 64, 15–23. [Google Scholar] [CrossRef]
- Adamson Barnes, N.S.; Mitchell, V.A.; Kazantzis, N.P.; Vaughan, C.W. Actions of the dual FAAH/MAGL inhibitor JZL195 in a murine neuropathic pain model. Br. J. Pharmacol. 2016, 173, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Kinsey, S.G.; Liu, Q.-s.; Hruba, L.; McMahon, L.R.; Grim, T.W.; Merritt, C.R.; Wise, L.E.; Abdullah, R.A.; Selley, D.E. Full fatty acid amide hydrolase inhibition combined with partial monoacylglycerol lipase inhibition: Augmented and sustained antinociceptive effects with reduced cannabimimetic side effects in mice. J. Pharmacol. Exp. Ther. 2015, 354, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Pasquarelli, N.; Porazik, C.; Bayer, H.; Buck, E.; Schildknecht, S.; Weydt, P.; Witting, A.; Ferger, B. Contrasting effects of selective MAGL and FAAH inhibition on dopamine depletion and GDNF expression in a chronic MPTP mouse model of Parkinson’s disease. Neurochem. Int. 2017, 110, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Rusek, M.; Pluta, R.; Ułamek-Kozioł, M.; Czuczwar, S.J. Ketogenic diet in Alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 3892. [Google Scholar] [CrossRef] [PubMed]
- Durrenberger, P.F.; Fernando, F.S.; Kashefi, S.N.; Bonnert, T.P.; Seilhean, D.; Nait-Oumesmar, B.; Schmitt, A.; Gebicke-Haerter, P.J.; Falkai, P.; Grünblatt, E.; et al. Common mechanisms in neurodegeneration and neuroinflammation: A BrainNet Europe gene expression microarray study. J. Neural Transm. 2015, 122, 1055–1068. [Google Scholar] [CrossRef]
- Montarolo, F.; Perga, S.; Martire, S.; Navone, D.N.; Marchet, A.; Leotta, D.; Bertolotto, A. Altered NR4A Subfamily Gene Expression Level in Peripheral Blood of Parkinson’s and Alzheimer’s Disease Patients. Neurotox. Res. 2016, 30, 338–344. [Google Scholar] [CrossRef]
- Wu, M.; Fang, K.; Wang, W.; Lin, W.; Guo, L.; Wang, J. Identification of key genes and pathways for Alzheimer’s disease via combined analysis of genome-wide expression profiling in the hippocampus. Biophys. Rep. 2019, 5, 98–109. [Google Scholar] [CrossRef]
- Tang, Y.; Han, Y.; Yu, H.; Zhang, B.; Li, G. Increased GABAergic development in iPSC-derived neurons from patients with sporadic Alzheimer’s disease. Neurosci. Lett. 2020, 735, 135208. [Google Scholar] [CrossRef]
- Abyadeh, M.; Tofigh, N.; Hosseinian, S.; Hasan, M.; Amirkhani, A.; Fitzhenry, M.J.; Gupta, V.; Chitranshi, N.; Salekdeh, G.H.; Haynes, P.A. Key genes and biochemical networks in various brain regions affected in Alzheimer’s disease. Cells 2022, 11, 987. [Google Scholar] [CrossRef] [PubMed]
- Echeverry, S.; Rodriguez, M.J.; Torres, Y.P. Transient Receptor Potential Channels in Microglia: Roles in Physiology and Disease. Neurotox. Res. 2016, 30, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Du, Y.; Zhao, X.; Tang, Q.; Su, W.; Hu, Y.; Yu, P. Cannabidiol enhances microglial Beta-amyloid peptide phagocytosis and clearance via Vanilloid family type 2 channel activation. Int. J. Mol. Sci. 2022, 23, 5367. [Google Scholar] [CrossRef]
- Hassan, S.; Eldeeb, K.; Millns, P.J.; Bennett, A.J.; Alexander, S.P.; Kendall, D.A. Cannabidiol enhances microglial phagocytosis via transient receptor potential (TRP) channel activation. Br. J. Pharmacol. 2014, 171, 2426–2439. [Google Scholar] [CrossRef]
- Sun, J.-X.; Zhu, K.-Y.; Wang, Y.-M.; Wang, D.-J.; Zhang, M.-Z.; Sarlus, H.; Benito-Cuesta, I.; Zhao, X.-Q.; Zou, Z.-F.; Zhong, Q.-Y. Activation of TRPV1 receptor facilitates myelin repair following demyelination via the regulation of microglial function. Acta Pharmacol. Sin. 2022, 44, 766–779. [Google Scholar] [CrossRef]
- Reid, H.M.; Chen-Mack, N.; Snowden, T.; Christie, B.R. Understanding changes in hippocampal interneurons subtypes in the pathogenesis of Alzheimer’s disease: A systematic review. Brain Connect. 2021, 11, 159–179. [Google Scholar] [CrossRef]
- Tanaka, M.; Sackett, S.; Zhang, Y. Endocannabinoid modulation of microglial phenotypes in neuropathology. Front. Neurol. 2020, 11, 87. [Google Scholar] [CrossRef]
- Leo, L.M.; Abood, M.E. CB1 cannabinoid receptor signaling and biased signaling. Molecules 2021, 26, 5413. [Google Scholar] [CrossRef]
- Hryhorowicz, S.; Kaczmarek-Ryś, M.; Andrzejewska, A.; Staszak, K.; Hryhorowicz, M.; Korcz, A.; Słomski, R. Allosteric modulation of cannabinoid receptor 1—Current challenges and future opportunities. Int. J. Mol. Sci. 2019, 20, 5874. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Li, J.X.; Thomas, B.F.; Wiley, J.L.; Kenakin, T.P.; Zhang, Y. Allosteric modulation: An alternate approach targeting the cannabinoid CB1 receptor. Med. Res. Rev. 2017, 37, 441–474. [Google Scholar] [CrossRef] [PubMed]
- Laprairie, R.B.; Kulkarni, P.M.; Deschamps, J.R.; Kelly, M.E.; Janero, D.R.; Cascio, M.G.; Stevenson, L.A.; Pertwee, R.G.; Kenakin, T.P.; Denovan-Wright, E.M. Enantiospecific allosteric modulation of cannabinoid 1 receptor. ACS Chem. Neurosci. 2017, 8, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Polini, B.; Cervetto, C.; Carpi, S.; Pelassa, S.; Gado, F.; Ferrisi, R.; Bertini, S.; Nieri, P.; Marcoli, M.; Manera, C. Positive allosteric modulation of CB1 and CB2 cannabinoid receptors enhances the neuroprotective activity of a dual CB1R/CB2R orthosteric agonist. Life 2020, 10, 333. [Google Scholar] [CrossRef] [PubMed]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Abate, G.; Uberti, D.; Tambaro, S. Potential and limits of cannabinoids in alzheimer’s disease therapy. Biology 2021, 10, 542. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, E.D.; Dutra, R.C. Cannabinoid receptors as therapeutic targets for autoimmune diseases: Where do we stand? Drug Discov. Today 2019, 24, 1845–1853. [Google Scholar] [CrossRef]
- Cassano, T.; Villani, R.; Pace, L.; Carbone, A.; Bukke, V.N.; Orkisz, S.; Avolio, C.; Serviddio, G. From Cannabis sativa to cannabidiol: Promising therapeutic candidate for the treatment of neurodegenerative diseases. Front. Pharmacol. 2020, 11, 124. [Google Scholar] [CrossRef]
- Morris, G.; Walder, K.; Kloiber, S.; Amminger, P.; Berk, M.; Bortolasci, C.C.; Maes, M.; Puri, B.K.; Carvalho, A.F. The endocannabinoidome in neuropsychiatry: Opportunities and potential risks. Pharmacol. Res. 2021, 170, 105729. [Google Scholar] [CrossRef]
- Duffy, S.S.; Hayes, J.P.; Fiore, N.T.; Moalem-Taylor, G. The cannabinoid system and microglia in health and disease. Neuropharmacology 2021, 190, 108555. [Google Scholar] [CrossRef]
- Cassano, T.; Calcagnini, S.; Pace, L.; De Marco, F.; Romano, A.; Gaetani, S. Cannabinoid receptor 2 signaling in neurodegenerative disorders: From pathogenesis to a promising therapeutic target. Front. Neurosci. 2017, 11, 30. [Google Scholar] [CrossRef]
- Chen, D.-J.; Gao, M.; Gao, F.-F.; Su, Q.-X.; Wu, J. Brain cannabinoid receptor 2: Expression, function and modulation. Acta Pharmacol. Sin. 2017, 38, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Wu, J. Cannabis, cannabinoid receptors, and endocannabinoid system: Yesterday, today, and tomorrow. Acta Pharmacol. Sin. 2019, 40, 297–299. [Google Scholar] [CrossRef]
- Abd El-Rahman, S.S.; Fayed, H.M. Improved cognition impairment by activating cannabinoid receptor type 2: Modulating CREB/BDNF expression and impeding TLR-4/NFκBp65/M1 microglia signaling pathway in D-galactose-injected ovariectomized rats. PLoS ONE 2022, 17, e0265961. [Google Scholar] [CrossRef] [PubMed]
- Elsouri, K. Amyloid Cascade Hypothesis Perspective on Alzheimer’s Disease. Ph.D. Thesis, Florida Atlantic University, Boca Raton, FL, USA.
- Sánchez-Sarasúa, S.; Fernández-Pérez, I.; Espinosa-Fernández, V.; Sánchez-Pérez, A.M.; Ledesma, J.C. Can we treat neuroinflammation in Alzheimer’s disease? Int. J. Mol. Sci. 2020, 21, 8751. [Google Scholar] [CrossRef]
- Mahdi, O.; Baharuldin, M.T.; Nor, N.H.M.; Chiroma, S.M.; Jagadeesan, S.; Moklas, M.A. The Neuroprotective Properties, Functions, and Roles of Cannabis sativa in Selected Diseases Related to the Nervous System. Cent. Nerv. Syst. Agents Med. Chem. (Former. Curr. Med. Chem. Cent. Nerv. Syst. Agents) 2021, 21, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Lodola, A.; Castelli, R.; Mor, M.; Rivara, S. Fatty acid amide hydrolase inhibitors: A patent review (2009–2014). Expert Opin. Ther. Pat. 2015, 25, 1247–1266. [Google Scholar] [CrossRef]
- Di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Gao, J. Macrophage polarization: A key event in the secondary phase of acute spinal cord injury. J. Cell. Mol. Med. 2017, 21, 941–954. [Google Scholar] [CrossRef]
- Wang, D.; Lin, Q.; Su, S.; Liu, K.; Wu, Y.; Hai, J. URB597 improves cognitive impairment induced by chronic cerebral hypoperfusion by inhibiting mTOR-dependent autophagy. Neuroscience 2017, 344, 293–304. [Google Scholar] [CrossRef]
- Ogawa, S.; Kunugi, H. Inhibitors of fatty acid amide hydrolase and monoacylglycerol lipase: New targets for future antidepressants. Curr. Neuropharmacol. 2015, 13, 760–775. [Google Scholar] [CrossRef] [PubMed]
- Karanian, D.A.; Karim, S.L.; Wood, J.T.; Williams, J.S.; Lin, S.; Makriyannis, A.; Bahr, B.A. Endocannabinoid enhancement protects against kainic acid-induced seizures and associated brain damage. J. Pharmacol. Exp. Ther. 2007, 322, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Gunduz-Cinar, O.; MacPherson, K.P.; Cinar, R.; Gamble-George, J.; Sugden, K.; Williams, B.; Godlewski, G.; Ramikie, T.; Gorka, A.; Alapafuja, S. Convergent translational evidence of a role for anandamide in amygdala-mediated fear extinction, threat processing and stress-reactivity. Mol. Psychiatry 2013, 18, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Booker, L.; Kinsey, S.G.; Abdullah, R.A.; Blankman, J.L.; Long, J.Z.; Ezzili, C.; Boger, D.L.; Cravatt, B.F.; Lichtman, A.H. The fatty acid amide hydrolase (FAAH) inhibitor PF-3845 acts in the nervous system to reverse LPS-induced tactile allodynia in mice. Br. J. Pharmacol. 2012, 165, 2485–2496. [Google Scholar] [CrossRef]
- Battista, N.; Maccarrone, M. Basic mechanisms of synthesis and hydrolysis of major endocannabinoids. In The Endocannabinoid System; Elsevier: London, UK, 2017; pp. 1–23. [Google Scholar]
- Ortar, G.; Morera, E.; De Petrocellis, L.; Ligresti, A.; Moriello, A.S.; Morera, L.; Nalli, M.; Ragno, R.; Pirolli, A.; Di Marzo, V. Biaryl tetrazolyl ureas as inhibitors of endocannabinoid metabolism: Modulation at the N-portion and distal phenyl ring. Eur. J. Med. Chem. 2013, 63, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Lemos, C.; Valério-Fernandes, Â.; Ghisleni, G.C.; Ferreira, S.G.; Ledent, C.; de Ceballos, M.L.; Köfalvi, A. Impaired hippocampal glucoregulation in the cannabinoid CB1 receptor knockout mice as revealed by an optimized in vitro experimental approach. J. Neurosci. Methods 2012, 204, 366–373. [Google Scholar] [CrossRef]
- Petrosino, S.; Ligresti, A.; Di Marzo, V. Endocannabinoid chemical biology: A tool for the development of novel therapies. Curr. Opin. Chem. Biol. 2009, 13, 309–320. [Google Scholar] [CrossRef]
- Ates, M.; Hamza, M.; Seidel, K.; Kotalla, C.E.; Ledent, C.; Gühring, H. Intrathecally applied flurbiprofen produces an endocannabinoid-dependent antinociception in the rat formalin test. Eur. J. Neurosci. 2003, 17, 597–604. [Google Scholar] [CrossRef]
- Grace, M.; Lieu, T.; Darby, B.; Abogadie, F.C.; Veldhuis, N.; Bunnett, N.W.; McIntyre, P. The tyrosine kinase inhibitor bafetinib inhibits PAR 2-induced activation of TRPV 4 channels in vitro and pain in vivo. Br. J. Pharmacol. 2014, 171, 3881–3894. [Google Scholar] [CrossRef]
- Minkkilä, A.; Savinainen, J.R.; Käsnänen, H.; Xhaard, H.; Nevalainen, T.; Laitinen, J.T.; Poso, A.; Leppänen, J.; Saario, S.M. Screening of various hormone-sensitive lipase inhibitors as endocannabinoid-hydrolyzing enzyme inhibitors. ChemMedChem 2009, 4, 1253–1259. [Google Scholar] [CrossRef]
- Owens, R.A.; Mustafa, M.A.; Ignatowska-Jankowska, B.M.; Damaj, M.I.; Beardsley, P.M.; Wiley, J.L.; Niphakis, M.J.; Cravatt, B.F.; Lichtman, A.H. Inhibition of the endocannabinoid-regulating enzyme monoacylglycerol lipase elicits a CB1 receptor-mediated discriminative stimulus in mice. Neuropharmacology 2017, 125, 80–86. [Google Scholar] [CrossRef]
- Wang, L.; Yui, J.; Wang, Q.; Zhang, Y.; Mori, W.; Shimoda, Y.; Fujinaga, M.; Kumata, K.; Yamasaki, T.; Hatori, A. Synthesis and preliminary PET imaging studies of a FAAH radiotracer ([11C] MPPO) based on α-ketoheterocyclic scaffold. ACS Chem. Neurosci. 2016, 7, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Feledziak, M.; Lambert, D.; Marchand-Brynaert, J.; Muccioli, G.G. Inhibitors of the endocannabinoid-degrading enzymes, or how to increase endocannabinoid’s activity by preventing their hydrolysis. Recent Pat. CNS Drug Discov. (Discontin.) 2012, 7, 49–70. [Google Scholar] [CrossRef] [PubMed]
- Myllymäki, M. Synthesis of 3-heterocycle phenyl N-alkyl Carbamates and Their Activity as FAAH Inhibitors. Ph.D. Thesis, Helsinki University of Technology, Department of Chemistry, Helsinki, Finland, 2009. [Google Scholar]
- Chicca, A.; Arena, C.; Manera, C. Beyond the direct activation of cannabinoid receptors: New strategies to modulate the endocannabinoid system in CNS-related diseases. Recent Pat. CNS Drug Discov. (Discontin.) 2015, 10, 122–141. [Google Scholar] [CrossRef] [PubMed]
- Molinski, S.V.; Shahani, V.M.; MacKinnon, S.S.; Morayniss, L.D.; Laforet, M.; Woollard, G.; Kurji, N.; Sanchez, C.G.; Wodak, S.J.; Windemuth, A. Computational proteome-wide screening predicts neurotoxic drug-protein interactome for the investigational analgesic BIA 10–2474. Biochem. Biophys. Res. Commun. 2017, 483, 502–508. [Google Scholar] [CrossRef]
- Patel, J.Z.; Parkkari, T.; Laitinen, T.; Kaczor, A.A.; Saario, S.M.; Savinainen, J.R.; Navia-Paldanius, D.; Cipriano, M.; Leppanen, J.; Koshevoy, I.O. Chiral 1, 3, 4-oxadiazol-2-ones as highly selective FAAH inhibitors. J. Med. Chem. 2013, 56, 8484–8496. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.Z. The development of potent and selective inhibitors of enzymes involved in endocannabinoid inactivation. Ph.D. Thesis, University of Eastern Finland, Kuopio, Finland, 2015. [Google Scholar]
- Pertwee, R.G. Cannabinoid Receptor Ligands; Tocris Bioscience Scientific Review Series; University of Aberdeen: Aberdeen, Scotland, 2020. [Google Scholar]
- Keith, J.M.; Jones, W.M.; Tichenor, M.; Liu, J.; Seierstad, M.; Palmer, J.A.; Webb, M.; Karbarz, M.; Scott, B.P.; Wilson, S.J. Preclinical characterization of the FAAH inhibitor JNJ-42165279. ACS Med. Chem. Lett. 2015, 6, 1204–1208. [Google Scholar] [CrossRef]
- van Heerden, M.; Roosen, W.; Lachau-Durand, S.; Bailey, G.; Ndifor, A. Exacerbation of Background Nuclear Cataracts in Sprague-Dawley Rats in Embryo-Fetal Development Studies With JNJ-42165279, a Fatty Acid Amide Hydrolase Inhibitor. Toxicol. Pathol. 2021, 49, 1193–1205. [Google Scholar] [CrossRef]
- Mor, M.; Lodola, A.; Rivara, S.; Vacondio, F.; Duranti, A.; Tontini, A.; Sanchini, S.; Piersanti, G.; Clapper, J.R.; King, A.R. Synthesis and QSAR of fatty acid amide hydrolase inhibitors: Modulation at the N-portion of biphenyl-3-yl alkylcarbamates. J. Med. Chem. 2008, 51, 3487. [Google Scholar] [CrossRef]
- Bisogno, T.; Ortar, G.; Petrosino, S.; Morera, E.; Palazzo, E.; Nalli, M.; Maione, S.; Di Marzo, V.; Group, E.R. Development of a potent inhibitor of 2-arachidonoylglycerol hydrolysis with antinociceptive activity in vivo. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2009, 1791, 53–60. [Google Scholar] [CrossRef]
- Xie, S.; Borazjani, A.; Hatfield, M.J.; Edwards, C.C.; Potter, P.M.; Ross, M.K. Inactivation of lipid glyceryl ester metabolism in human THP1 monocytes/macrophages by activated organophosphorus insecticides: Role of carboxylesterases 1 and 2. Chem. Res. Toxicol. 2010, 23, 1890–1904. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Li, W. Monoacylglycerol lipase inhibitors: Modulators for lipid metabolism in cancer malignancy, neurological and metabolic disorders. Acta Pharm. Sin. B 2020, 10, 582–602. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, J.; Kuusisto, A.; van Bruchem, J.; Patel, J.Z.; Laitinen, T.; Navia-Paldanius, D.; Laitinen, J.T.; Savinainen, J.R.; Parkkari, T.; Nevalainen, T.J. Piperazine and piperidine carboxamides and carbamates as inhibitors of fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL). Bioorganic Med. Chem. 2014, 22, 6694–6705. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, N.; Savinainen, J.R.; Ribas, C.R.; Rönkkö, J.; Kuusisto, A.; Korhonen, J.; Navia-Paldanius, D.; Häyrinen, J.; Takabe, P.; Käsnänen, H. Piperazine and piperidine triazole ureas as ultrapotent and highly selective inhibitors of monoacylglycerol lipase. Chem. Biol. 2013, 20, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.W.; Niphakis, M.J.; Lum, K.M.; Cognetta, A.B.; Wang, C.; Matthews, M.L.; Niessen, S.; Buczynski, M.W.; Parsons, L.H.; Cravatt, B.F. Highly selective inhibitors of monoacylglycerol lipase bearing a reactive group that is bioisosteric with endocannabinoid substrates. Chem. Biol. 2012, 19, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Long, J.Z.; Li, W.; Booker, L.; Burston, J.J.; Kinsey, S.G.; Schlosburg, J.E.; Pavón, F.J.; Serrano, A.M.; Selley, D.E.; Parsons, L.H. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 2009, 5, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.Z.; Ahenkorah, S.; Vaara, M.; Staszewski, M.; Adams, Y.; Laitinen, T.; Navia-Paldanius, D.; Parkkari, T.; Savinainen, J.R.; Walczyński, K. Loratadine analogues as MAGL inhibitors. Bioorganic Med. Chem. Lett. 2015, 25, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, V.; Karanian, D.A.; Vadivel, S.K.; Locklear, J.R.; Wood, J.T.; Nasr, M.; Quizon, P.M.P.; Graves, E.E.; Shukla, V.; Makriyannis, A. Equipotent inhibition of fatty acid amide hydrolase and monoacylglycerol lipase–dual targets of the endocannabinoid system to protect against seizure pathology. Neurotherapeutics 2012, 9, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Zvonok, N.; Pandarinathan, L.; Williams, J.; Johnston, M.; Karageorgos, I.; Janero, D.R.; Krishnan, S.C.; Makriyannis, A. Covalent inhibitors of human monoacylglycerol lipase: Ligand-assisted characterization of the catalytic site by mass spectrometry and mutational analysis. Chem. Biol. 2008, 15, 854–862. [Google Scholar] [CrossRef]
- Cheng, R.; Fujinaga, M.; Yang, J.; Rong, J.; Haider, A.; Ogasawara, D.; Van, R.S.; Shao, T.; Chen, Z.; Zhang, X. A novel monoacylglycerol lipase-targeted 18F-labeled probe for positron emission tomography imaging of brown adipose tissue in the energy network. Acta Pharmacol. Sin. 2022, 43, 3002–3010. [Google Scholar] [CrossRef]
- Gil-Ordóñez, A.; Martín-Fontecha, M.; Ortega-Gutiérrez, S.; López-Rodríguez, M.L. Monoacylglycerol lipase (MAGL) as a promising therapeutic target. Biochem. Pharmacol. 2018, 157, 18–32. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aspects | Summary |
|---|---|
| eCBs and alzheimer’s | no conclusive evidence that cannabis or eCBs can stop, reverse, or prevent dementia. |
| Behavioral symptoms | some studies suggest eCBs may help manage agitation and aggression in dementia patients. However, long-term effects are unclear. |
| Lab studies | endocannabinoids appear to remove amyloid protein (Alzheimer’s hallmark) from nerve cells in some lab studies. |
| Clinical trials | no specific clinical trials on endocannabinoids in dementia patients. |
| ECS and CB1R | ECS plays a role in neurotransmission and neuroimmune systems. CB1R are abundant in the brain and hypothalamus, modulating endocrine axes. |
| ECS and CB2R | CB2R are mainly expressed in immune cells. |
| effects of CB1R agonists | may help manage symptoms such as agitation and anxiety in dementia. |
| Effects of eCBs antagonists | antagonists can inhibit CB1R activation by endogenously released eCBs. They may also act as inverse agonists, shifting CB1R activity from an active state to an inactive state. |
| Upregulated or downregulated | moderate activation of CB1R by selective agonists or eCBs may have acute beneficial therapeutic actions, such as pain relief and anti-psychotic effects. Elevation of eCBs levels by inhibiting degrading enzymes (such as FAAH and MAGL) can also be beneficial. |
| Neuroprotective properties | ECs may have neuroprotective properties, potentially reducing the risk of neurodegenerative diseases associated with dementia. |
| AEA and 2-AG Modulators | CB1R Specific Agonists | CB1R Specific Antagonists | Allosteric Modulators of CB1R | CB2R Specific Agonists | CB2R Specific Antagonists | Mixed Receptors Agonist |
|---|---|---|---|---|---|---|
| URB597 (symptoms of schizophrenia) | ACEA (anti-inflammatory) | Rimonabant (SR141716) (obesity, type 2 diabetes, dyslipidemia) | Org27569-positive allosteric modulator (PAM) | JWH-133 (reduction in inflammatory components) | SR144528 (anti-nociceptive) | THC (inflammatory pain, metabolic syndrome, obesity)) |
| PF-04457845 (osteoarthritic pain) | ACPA (anti-inflammatory) | AM-251 (decrease excitotoxicity) | Pepcan-12-negative allosteric modulator (NAMs) | JWH-051 (relative lack of psychotropic effects compared to CB1R agonist) | AM-630 (pain) | CBD (neuroinflammatory diseases, chronic and inflammatory pain) |
| JZL184 (anti-nociceptive) | Methanandamide (neuroprotection) | AM-281 (neuroprotection) | PM-226 | WIN-55, 212–2 | ||
| JZL195 (anti-nociceptive) | O-1812 (Neuroprotection) | Taranabant (MK-0364) | HU-308 | Nabilone (prevent memory loss) | ||
| JD5037-peripheral CB1R inverse agonist (neither affects behavioral responses mediated by CB1Rs in the brain) | GW-405833 (pain) | JWH-018 | ||||
| AM4113 | L-759, 633 | HU-210 (anti-inflammatory, anti-psychotic) | ||||
| LH-21 | L-759, 656 | CP-55, 940 (anti-emetic) | ||||
| PIMSR | O-1966 AM1241 Xie2–64 AM1710 JHW015 (specific CB2R agonists in CNS disorder) | |||||
| MDA7 |
| Class | Compounds | IC50 (nM) | Therapeutic Effects | References |
|---|---|---|---|---|
| FAAH inhibitor (irreversible covalent) | URB597 | 33.5 (h) 3.8 (r) 45 (m) | (i) increase in endocannabinoid anandamide, (ii) suppressed glutamate Aβ42-induced toxicity (iii) stimulate mitochondrial membrane potential (iv) reduction in interleukin (IL)-1β (v) tumor necrosis factor-α (TNFα) expression (vi) neuropathic pain | [111,112] |
| URB937 | 26.8 ± 4.9 (r) | (i) increase in endocannabinoid anandamide, (ii) inflammatory pain (iii) neuropathic pain | [113] | |
| AM374 | 13 (r) | (i) increase in endocannabinoid anandamide | [114] | |
| AM3506 | 2.8 ± 0.3 (r) | (i) hypertension (ii) posttraumatic stress disorder | [115] | |
| PF-3845 | 18 (h) | (i) inflammatory pain (ii) traumatic brain injury (iii) neuropathic pain (iv) visceral pain (v) anxiety-related disorders | [116] | |
| PF-04457845 (PF-7845) | 7.2–50.4 (h) 7.4–43.1 (r) 2.5 (m) | (i) analgesic and anxiolytic effects (ii) 25-fold higher than URB597 for high in vivo efficacy (iii) long duration of action for inflammatory pain (iv) very well tolerated in healthy subjects | [117] | |
| PF-750 | 16.2-595 | (i) analgesic, (ii) anti-inflammatory, (iii) anti-depressant | [114,118] | |
| LY2183240 | 37.3 ± 5.4 (h) 2.1 (r) 12.4 (m) | (i) analgesic | [118,119,120] | |
| MAFP | 0.33 ± 0.07 (h) 2.5 (m) 1–3 (r, m) | [121,122,123] | ||
| SA-57 | 1.9 (h) 3.2 (m) | [124,125] | ||
| FAAH inhibitor (reversible binding) | OL-135 | 206 (h) 2.1 (m) | (i) neuropathic pain (ii) inflammatory pain (iii) visceral pain | [114,122,126] |
| OL-92 | 0.28 (m) | [127] | ||
| MK-4409 | 11 (h) 11 (r) | (i) neuropathic pain | [110,128,129] | |
| ST4070 | 9 (m) | [130] | ||
| FAAH inhibitors (slowly reversible binding) | JZP-327A | 11 (h) | [73,131] | |
| FAAH inhibitor (partial reversible binding) | JNJ-1661010 | 33 ± 2.1 (h) 34 ± 6.5 (r) | (i) neuropathic pain (ii) inflammatory pain | [132,133] |
| FAAH inhibitor (partial irreversible inhibitor) | JNJ-42165279 | 313 ± 28 (r) | (i) analgesic and anxiolytic effects | [114,134] |
| FAAH inhibitor (reversibility not available) | URB880 | 0.63 ± 0.04 (r) | (i) analgesic (ii) anti-depressant | [135] |
| irreversible binding with MAGL | N-arachidonoyl maleimide | 2180 ± 620 (h) >10,000 (r) | (i) reducing neuroinflammation (ii) improving synaptic plasticity (iii) spatial learning (iv) memory in AD animals | [136,137] |
| Disulfiram | 360 (h) (surrogate substrate assay) | (i) reducing neuroinflammation (ii) improving synaptic plasticity (iii) spatial learning (iv) memory in AD animals | [138] | |
| SAR-629 | 0.9 (h) 0.22 (m) | (i) reducing neuroinflammation (ii) improving synaptic plasticity (iii) spatial learning (iv) memory in AD animals | [139] | |
| JJKK-006 | 0.6 (h) | (i) reducing neuroinflammation (ii) improving synaptic plasticity (iii) spatial learning (iv) memory in AD animals | [140] | |
| JJKK-048 | 0.363 (h) 0.214 (r) 0.275 (m) | (i) reducing neuroinflammation (ii) improving synaptic plasticity (iii) spatial learning (iv) memory in AD animals | [140,141] | |
| ML-30 | 0.54 (h) 4.4 (r) 1.9 (m) | (i) reducing neuroinflammation (ii) improving synaptic plasticity (iii) spatial learning (iv) memory in AD animals | [139,140] | |
| KML-29 | 5.9 (h) 43 (r) 15 (m) | (i) reducing neuroinflammation (ii) improving synaptic plasticity (iii) spatial learning (iv) memory in AD animals | [141] | |
| JZL-184 | 3.9 (h) 262 (r) 10 (m) | (i) decrease in proinflammatory reactions of microglia and astrocytes. (ii) reduced total Aβ burden and its precursors | [142,143] | |
| JW642 | 4.7 (h) 14 (r) 7.6 (m) | (i) anti-hyperalgesia (ii) anti-anxiety/depression | [142] | |
| reversible inhibitor of MAGL | JZP-361 | 46 (h) | (i) alleviate pain and inflammation | [142] |
| slowly reversible with MAGL | AM6701 | 1.2 ± 0.35 (h) 1.7 (r) | (i) neurodegenerative cascade, Behavioral deficits linked to Seizure’s damage | [144,145] |
| partially reversible binding with MAGL | URB-602 | 360 (h) 28,000 (r) | (i) anti-nociceptive, anxiolytic, anti-inflammatory | [146] |
| MAGL/FAAH dual inhibitor | JZL-195 | 4 (hMAGL) 2 (hFAAH) 19 (mMAGL) 13 (mFAAH) | (i) enhanced brain levels of anandamide and 2-AG (ii) anti-allodynic effects | [139,142,147] |
| irreversible inhibitor of both MAGL and FAAH enzymes | CAY10499 | 76 (h) 86 (r) | (i) anti-inflammatory (ii) neuroprotective | [147] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanwal, H.; Sangineto, M.; Ciarnelli, M.; Castaldo, P.; Villani, R.; Romano, A.D.; Serviddio, G.; Cassano, T. Potential Therapeutic Targets to Modulate the Endocannabinoid System in Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 4050. https://doi.org/10.3390/ijms25074050
Kanwal H, Sangineto M, Ciarnelli M, Castaldo P, Villani R, Romano AD, Serviddio G, Cassano T. Potential Therapeutic Targets to Modulate the Endocannabinoid System in Alzheimer’s Disease. International Journal of Molecular Sciences. 2024; 25(7):4050. https://doi.org/10.3390/ijms25074050
Chicago/Turabian StyleKanwal, Hina, Moris Sangineto, Martina Ciarnelli, Pasqualina Castaldo, Rosanna Villani, Antonino Davide Romano, Gaetano Serviddio, and Tommaso Cassano. 2024. "Potential Therapeutic Targets to Modulate the Endocannabinoid System in Alzheimer’s Disease" International Journal of Molecular Sciences 25, no. 7: 4050. https://doi.org/10.3390/ijms25074050