Abstract
The nucleophilic addition of 3-(4-cyanopyridin-2-yl)-1,1-dimethylurea (1) to cis-[Pt(CNXyl)2Cl2] (2) gave a new cyclometallated compound 3. It was characterized by NMR spectroscopy (1H, 13C, 195Pt) and high-resolution mass spectrometry, as well as crystallized to obtain two crystalline forms (3 and 3·2MeCN), whose structures were determined by X-ray diffraction. In the crystalline structure of 3, two conformers (3A and 3B) were identified, while the structure 3·2MeCN had only one conformer 3A. The conformers differed by orientation of the N,N-dimethylcarbamoyl moiety relative to the metallacycle plane. In both crystals 3 and 3·2MeCN, the molecules of the Pt(II) complex are associated into supramolecular dimers, either {3A}2 or {3B}2, via stacking interactions between the planes of two metal centers, which are additionally supported by hydrogen bonding. The theoretical consideration, utilizing a number of computational approaches, demonstrates that the C···dz2(Pt) interaction makes a significant contribution in the total stacking forces in the geometrically optimized dimer [3A]2 and reveals the dz2(Pt)→π*(PyCN) charge transfer (CT). The presence of such CT process allowed for marking the C···Pt contact as a new example of a rare studied phenomenon, namely, tetrel bonding, in which the metal site acts as a Lewis base (an acceptor of noncovalent interaction).
1. Introduction
Deprotonated diaminocarbene complexes of late transition metals have been actively studied in the last decade [1,2,3]. From a practical point of view, these complexes have a lot of advantages. They effectively catalyze the processes of C–C cross-coupling [4,5,6,7,8], hydrosilylation [9,10,11], and C≡C triple-bond activation [12,13]. Moreover, several compounds are recognized as promising antitumor agents [14,15], probes of mercury(II) ions in solutions [16], and components of OLED devices [17].
Many studies demonstrate that a solid-state supramolecular structure plays a key role in reactivity [18] and photophysics [2,19,20,21] of such compounds. Their supramolecular organization is determined by combination of various noncovalent interaction: hydrogen bonding (HB) [22], π···π stacking [23], metallophilic interactions [24], halogen bonding [25], chalcogen bonding [26], pnictogen [27], and tetrel bonding (abbreviated as TtB) [28,29] etc. In recent years, special attention has been paid to interactions in which the metal center acts as a Lewis base (a nucleophile) due to a lone pair of electrons in the dz2 orbital [30]. Particularly, metal-involved halogen bonds [31,32,33,34,35,36], chalcogen bonds [37], spodium bonds [20], pnictogen bonds [38], and πh/C···M interactions [39,40,41,42,43] have been recognized. To contrast, in most examples of metal-involved TtB, a metal (commonly, tin or lead) is a σ- or π-hole donor, i.e., electrophilic site [44,45,46,47]. These interactions are the focus of many studies [48,49] due to importance of lead and tin coordination compounds for materials science [50,51,52,53]. At the same time, the TtB, in which the metal site acts as a Lewis base was only recently described. Particularly in our previous work, we experimentally and theoretically evidenced that the C···dz2(Pt) TtB can be a main component of metal-involving stacking interactions between half-lantern PtII2 complexes and electron-deficient arenes [54,55].
In this work, we report a new example of C···dz2(Pt) TtB, which is responsible for supramolecular dimerization of one of two possible conformers of a deprotonated diaminocarbene Pt(II) complex in a solid state. To describe this TtB, X-ray diffraction data of the platinum complex and a combination of quantum chemical methods were used; all these results are given in the sections below.
2. Results and Discussion
2.1. Synthesis and Characterization of 3
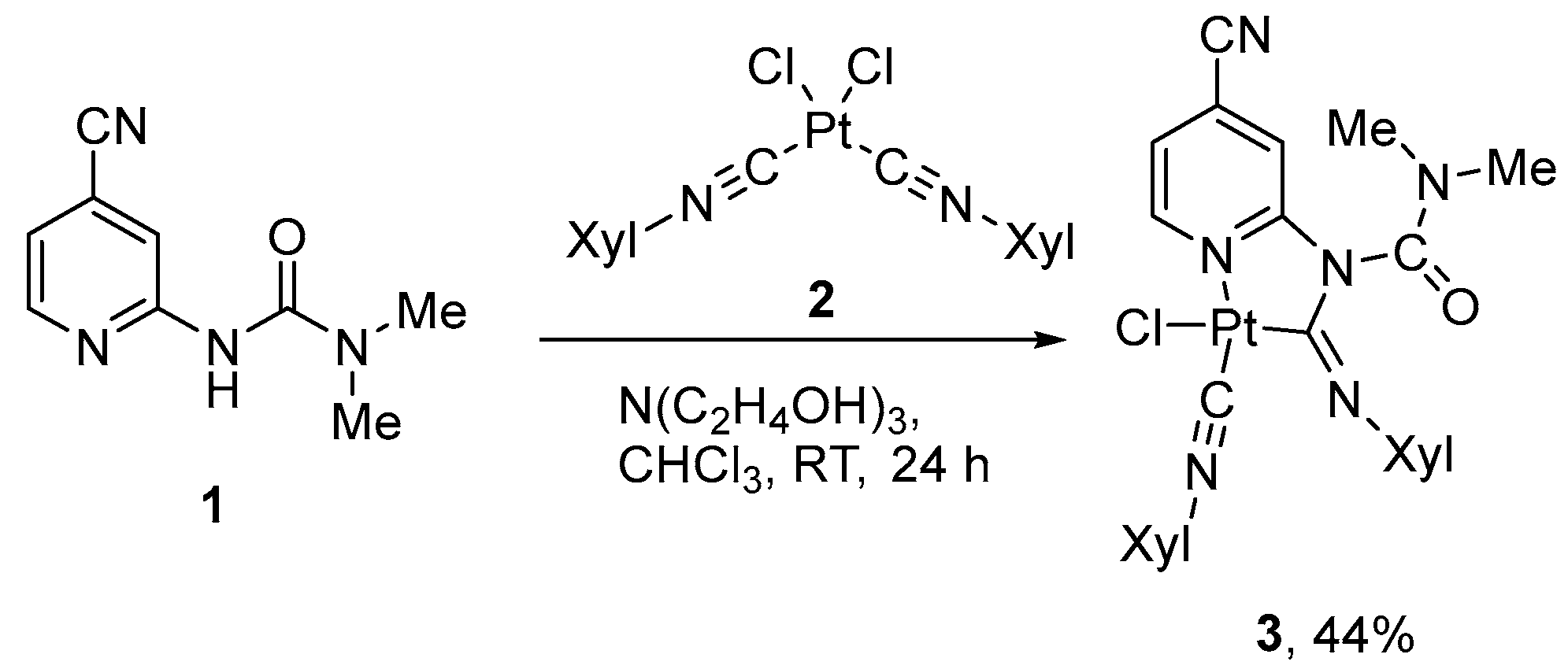
Target complex 3 (Scheme 1) was prepared according to the previously reported methodology [10,56]: the treatment of 3-(4-cyanopyridin-2-yl)-1,1-dimethylurea (1) with equivalent amounts of cis-[Pt(CNXyl)2Cl2] and triethanolamine in chloroform at room temperature (Scheme 1). The complex was studied using NMR (1H, 13C, 195Pt) spectroscopy and HR mass spectrometry. All obtained data are in a full agreement with the supposed structure and display common features of such compounds [15]. In particular, in the 1H and 13C NMR spectra, splitting of the signals of the methyl groups in the carbamoyl and xylyl moieties was observed, which indicates the double-bond character of the carbon–nitrogen bond. In the 1H spectra of both compounds, the α-CH protons of the pyridine rings appear as broad doublets (9.50 ppm), while in the starting urea 1 1H spectrum the corresponding proton resonates at 8.31 ppm. In addition, the chemical shift of 195Pt for 3 (−3801 ppm) is close to those for other Pt(II) metallacycles with N-pyridylurea-based ligands (−3809–3802 ppm) [10,15].
Scheme 1.
Synthesis of the cyclometallated complex 3 with 3-(4-cyanopyridin-2-yl)-1,1-dimethylurea ligand.
2.2. Crystals and Their X-ray Structures
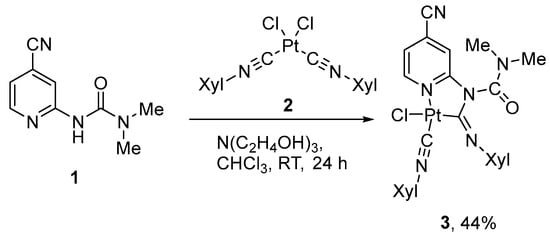
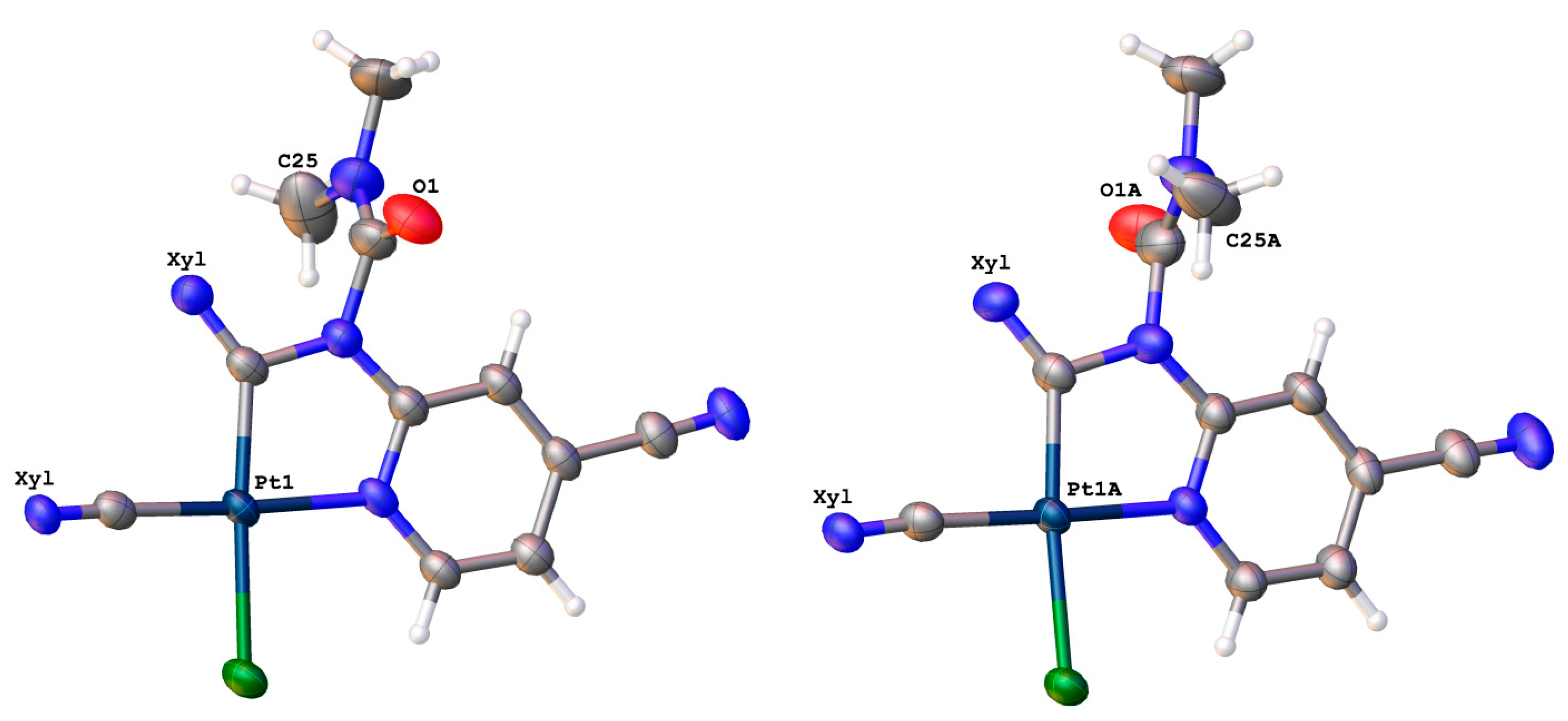
The complex was crystallized from 1,2-dichloroethane and MeCN to give two different crystalline forms: 3 and 3·2MeCN, the structures of which were determined by X-ray diffraction. The crystal structure of 3 includes two crystallographically independent molecules of the metal complex, differing in the orientation of the N,N-dimethylcarbamoyl moiety (Figure 1). Thus, these two types of molecules can be considered as two possible conformers of complex 3, namely, 3A (the carbamoyl moiety is turned by carbonyl group towards a viewer, Figure 1 Left) and 3B (the carbamoyl moiety is turned by methyl group towards a viewer, Figure 1 Right). Notably, among four previously published examples of Pt(II) cyclometallated complexes with N-pyridylureas as a ligand [10,15], three structures (CSD refcodes: CAMPOZ, CAMQUG, PESLEI) represent type A conformers and one structure (CSD refcode CAMPIT) contains a type B conformer. In the structure of 3·2MeCN, only conformer 3A was revealed.

Figure 1.
Two conformers 3A (left) and 3B (right) of the complex 3, realized in its crystal structure. The xylyl moieties are omitted for simplicity.
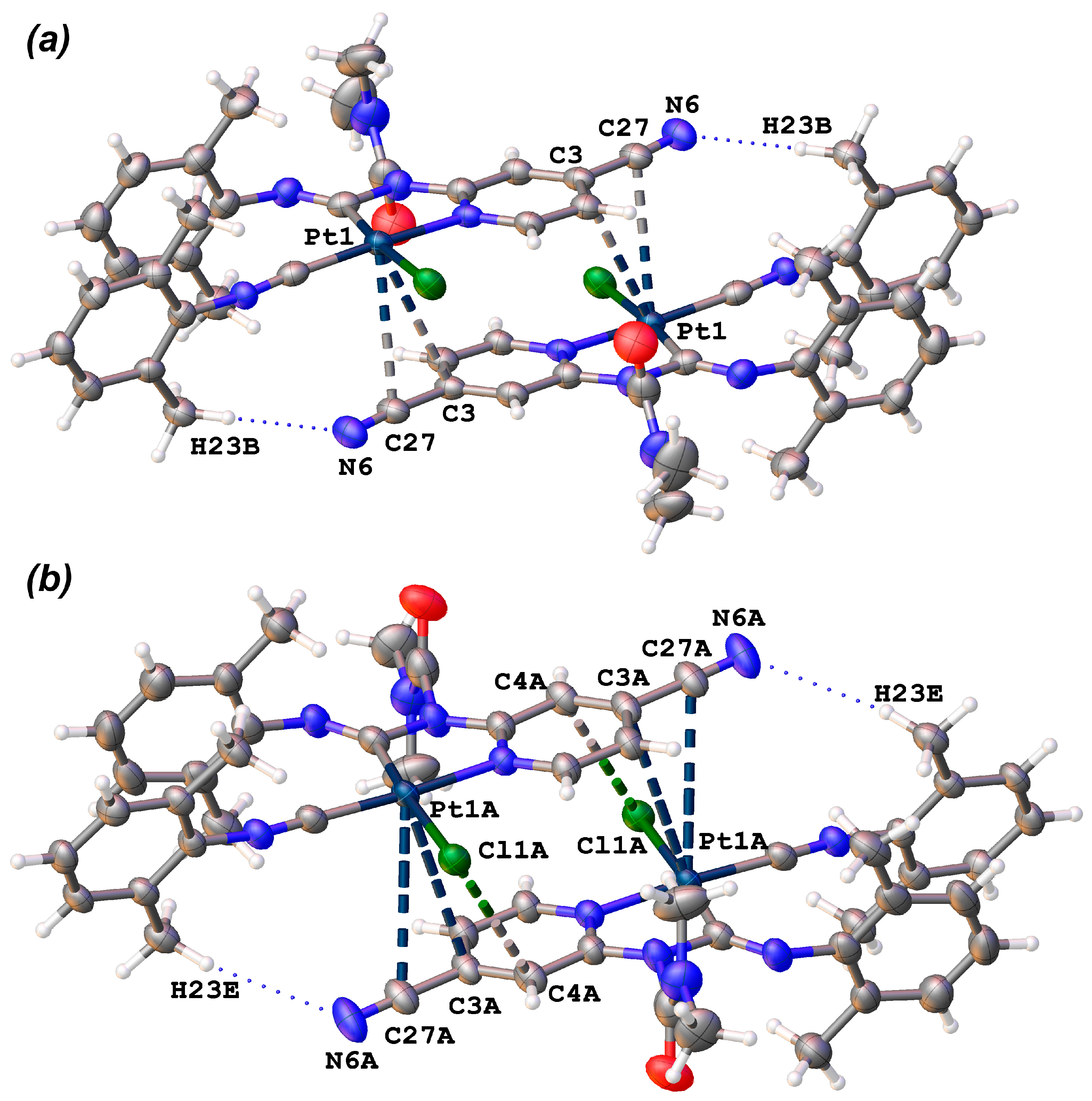
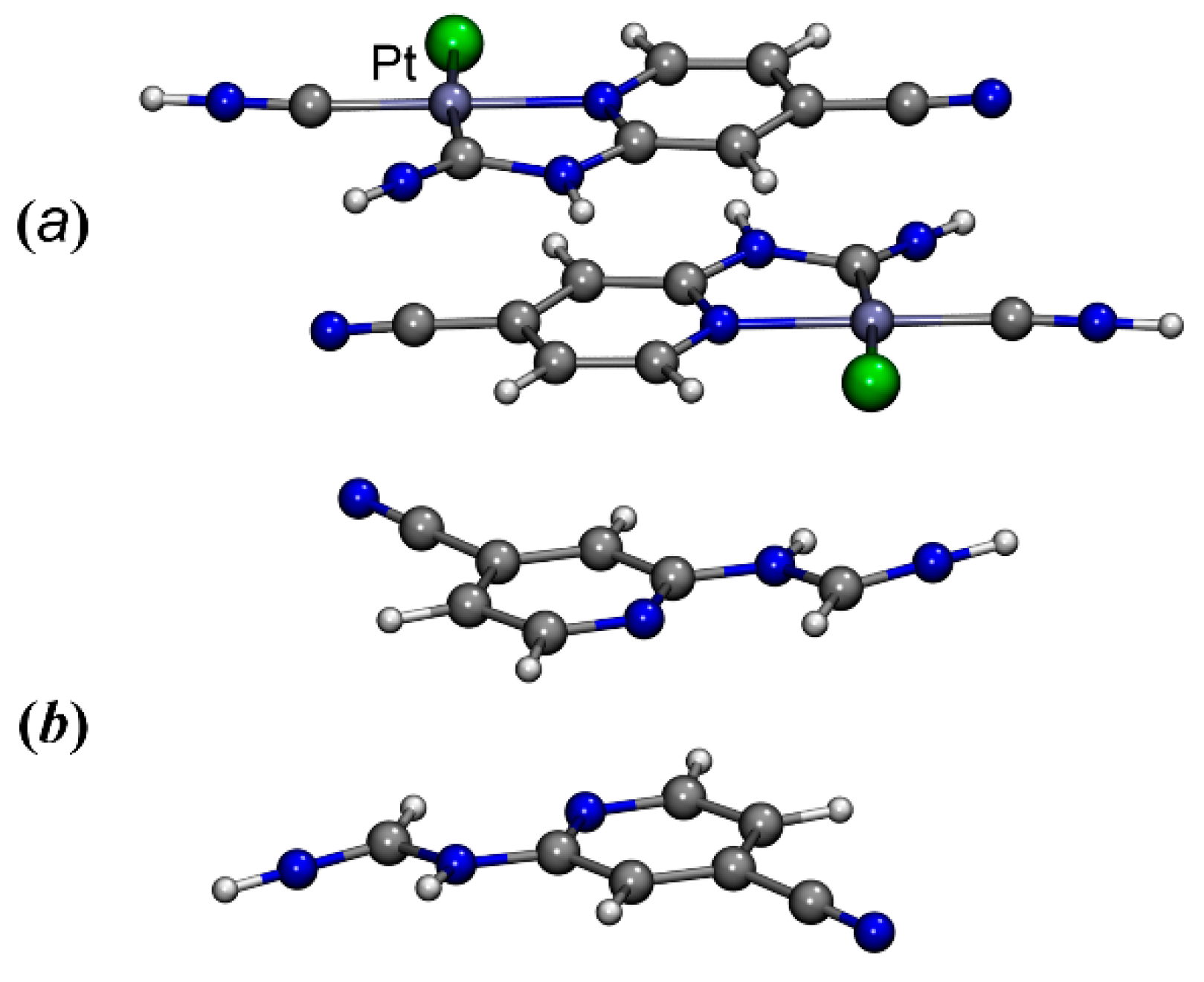
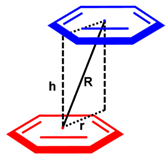
In both structures (3 and 3·2MeCN), the molecules of the metal complex are associated into supramolecular dimers via stacking interactions between two metal square-planes (each of which includes the 4-cyanopyridine moiety and the Pt(II)-based metallocycle, Figure 2 and Figure S1, Table 1 and Table 2), with additional supporting by several hydrogen bonds. In the structure of 3, conformers 3A and 3B form two types of dimers ({3A}2 (Figure 2a) and {3B}2) (Figure 2b), in which the carbamoyl moieties are oriented relative to each other, like “C=O to C=O” ({3A}2) and like “CH3 to CH3” ({3B}2). In dimer {3A}2, the molecules of the metal complex are located much closer to each other in comparison with {3B}2. Particularly in {3A}2, the shortest interplanar distance is the distance between two pyridine rings (3.479(3) Å; the shortest C···C contact is 3.386(5) Å; Table 1) and it has an appropriate value for typical π···π stacking (3.41–3.61 Å) [57,58]. In contrast, in {3B}2, the metal square-planes are more parallel displaced in comparison with {3A}2, which is illustrated by the “r” value (the distance between the centroid of one of the ring and the projection of the centroid of a π···π stacked ring to the first plane, Table 1). They are 0.948(5) Å and 1.805(6) Å for {3A}2 and {3B}2, respectively. The interplanar distance in {3B}2 is significantly larger (3.987(3) Å) than in {3A}2. The shortest C···C contact in {3B}2 is 3.613(6) Å, which is at the end of the range of typical π···π stacking interactions (3.41–3.61 Å) [57,58]. According to our observation, this difference is caused by the steric repulsion of two methyl groups of the carbomoyl moieties facing each other in the case of the dimer {3B}2.
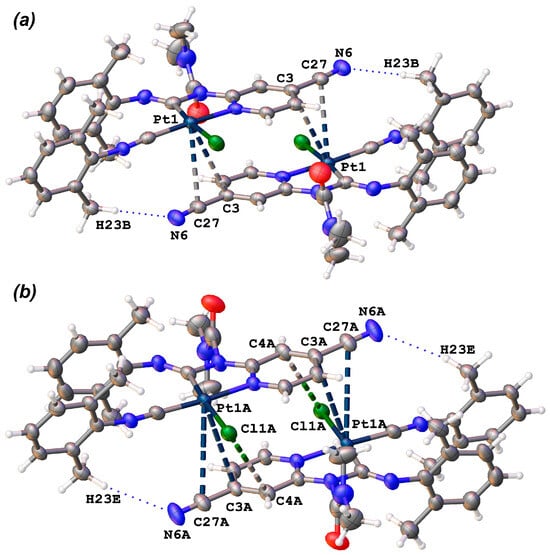
Figure 2.
Structures of supramolecular dimers {3A}2 (a) and {3B}2 (b).

Table 1.
Geometrical parameters of the π···π stacking interactions in crystal structures of 3 and 3·2MeCN.
Table 2.
Geometrical parameters of the observed C···Pt contacts in supramolecular dimers {3A}2, {3A’}2, {3B}2.
In the structure of 3·2MeCN, conformer 3A also provides the dimer with “C=O to C=O” arrangement of the carbamoyl moieties; however, its geometry is slightly different ({3A’}2, Figure S1). In this case, the shortest interplanar distance is between the pyridine ring and the metallacycle (3.547(2) Å; the shortest C···C contact is 3.275(5) Å), which also belongs to the range of typical π···π stacking interactions (3.41–3.61 Å) [57,58].
The metal centers in all dimers form contacts with the 4-cyanopyridine moieties (PyCN), namely, the CCN–CPy bonds are located above the Pt atoms. The shortest PtII-involved contacts are CCN···Pt (3.419(4) Å), CPy···Pt (3.414(3) Å), and CPy···Pt (3.677(4) Å) for {3A}2, {3A’}2, and {3B}2, respectively (Figure 2 and Figure S1, Table 2). In the case of {3A}2 and {3A’}2, both C···Pt distances are less than the sum of the Bondi vdW radii (ΣBvdW (C + Pt) = 3.45 Å) [59,60], and all three C···Pt distances for {3A}2, {3A’}2, and {3B}2 are less than the sum of the Alvarez vdW radii (ΣAvdW (C + Pt) = 4.06 Å) [61,62]. It should be mentioned that in the previous work [55], describing Pt···C tetrel bond, the C···Pt distances were larger that the corresponding ΣBvdW. Moreover, according to results of the early performed CSD search [55], the C···Pt contacts in our structures belong to top 15% of the shortest known C···Pt contacts.
As it was mentioned above, the stacking interactions are additionally supported by hydrogen bonds (H2C–H···N for all dimers and CPy–H···O for {3A}2 and {3A’}2) (Figure 2). Geometrical parameters of these hydrogen bonds are collected in Table S3. Moreover, in the dimer {3B}2, a CPy···Cl short contact was observed (3.579(4) Å vs. ΣBvdW [59] (C + Cl) = 3.45 Å and Alvarez radii sum ΣAvdW (C + Cl) = 3.59 Å) [61,62].
2.3. Theoretical Considerations of the Dimers [3A]2 and [3B]2
Several forces, including C···Pt TtB, C···Cl TtB, and π···π interactions, as well as CPy–H···O and H2C–H···N HBs, are responsible for the formation of supramolecular dimers {3A}2 and {3B}2. The nature of these interactions was studied theoretically by the DFT (PBE0-D3BJ) method. Since the crystal packing may significantly affect the intermolecular interactions, we carried out the geometry optimization of three isolated bimolecular clusters with starting geometries {3A}2 and {3A’}2 and {3B}2 and obtained two geometry optimized dimes abbreviated as [3A]2 and [3B]2. It should be noted that the geometry of the optimized dimer [3A]2 was the same regardless of the starting dimer ({3A}2 or {3A’}2) and only slightly differed from the starting X-ray dimers. The geometry-optimized dimer [3B]2 demonstrated changes that led to elongation of the C···Pt contact with simultaneous shortening of the C···Cl contact as compared to the X-ray dimer {3B}2.
The nature of C···Pt and C···Cl contacts that occurred between molecules of 3 was investigated by the wave analysis including the quantum theory of atoms in molecules (QTAIM) [63,64], independent gradient model based on Hirshfeld partition (IGMH) [65], electron localization function (ELF) [66], the charge displacement function (CDF) [67,68], methods combined with the extended transition state natural orbital for chemical valence theory (ETS–NOCV) [69], and natural bond orbital (NBO) [70] methods. Moreover, interaction energies were estimated according to super-molecular (SM) approach and the generalized Kohn–Sham energy decomposition analysis (GKS-EDA) [71].
2.3.1. Molecular Electrostatic Potential
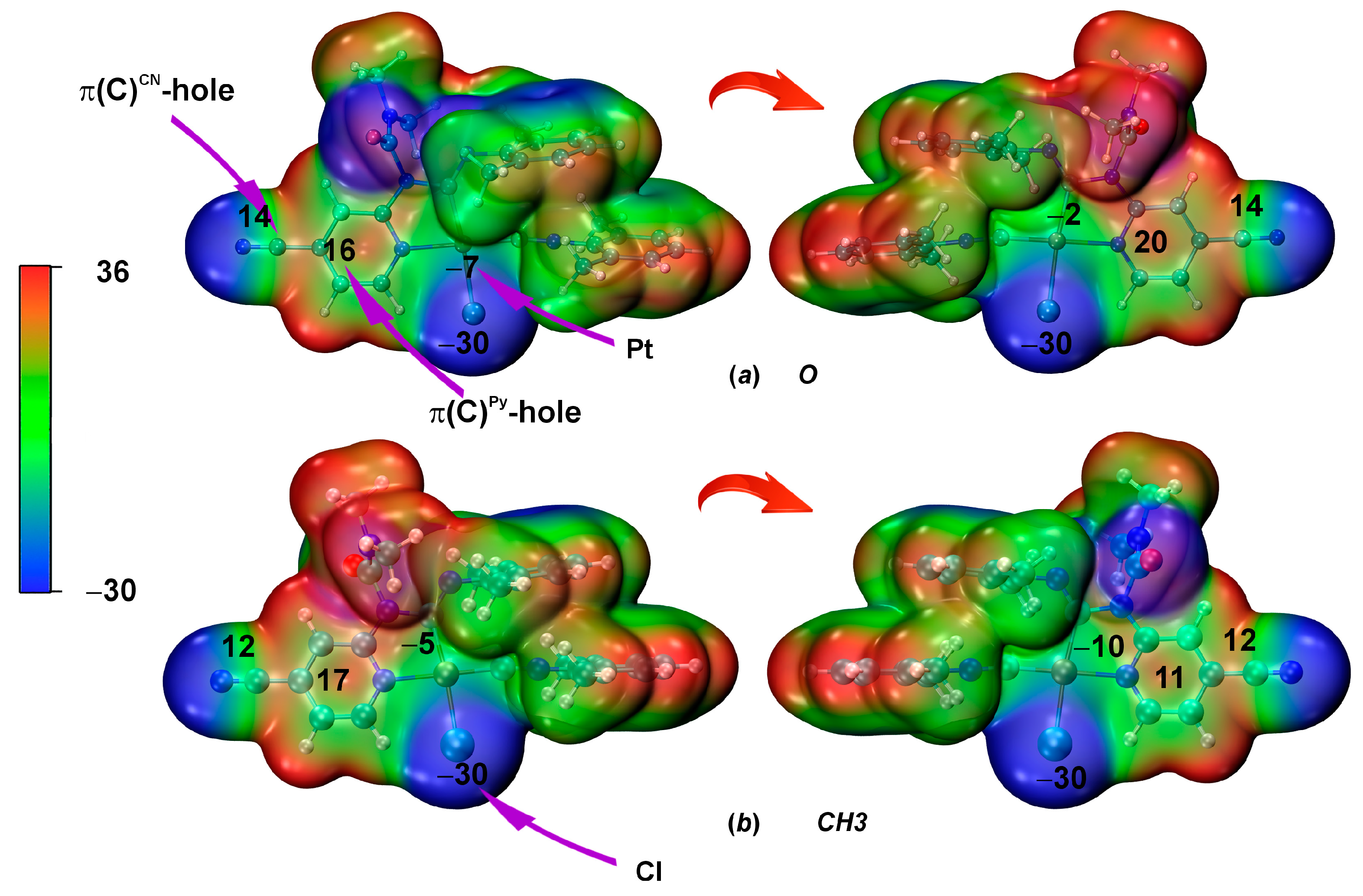
As the first step of the theoretical study, the MEP of both conformers 3A and 3B was computed at the DFT (PBE0-D3BJ/ZORA-def2-TZVP) level of theory (Figure 3). Platinum atoms for both 3A and 3B are characterized by electronegative potential (Vs,min = (−6)–(−2) kcal/mol (3A) and (−10)–(−5) kcal/mol (3B)). For both conformers, two positive regions were revealed: first, on the center of the pyridine ring on both sides of the cycle (Vs,max = 16–20 kcal/mol (3A) and 11–17 kcal/mol (3B)) and second, a belt between the C and N atoms of the cyano group ring (Vs,max = 14 for 3A and 12 kcal/mol for 3B). Such belt is typical for cyano groups [72].

Figure 3.
MEP distributions for the optimized structures of conformers 3A (a) and 3B (b); Vs,max and Vs,min values in kcal/mol, electron density isosurfaces 0.001 a.u. The potential values are given for the Pt, C, Cl atoms, as well as for the C of the pyridine ring.
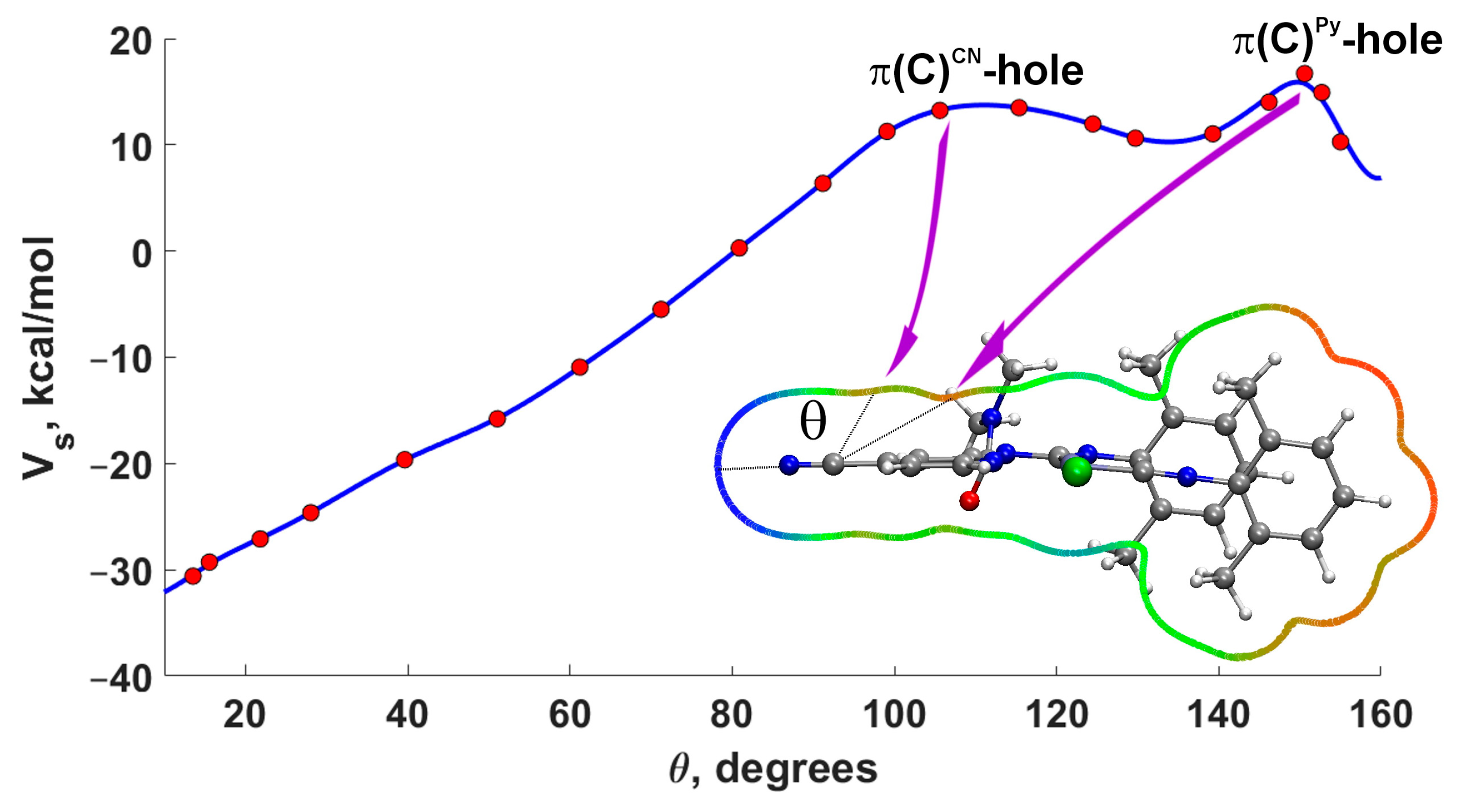
For a more precise analysis of the presence of π-holes on carbon atoms, we constructed the dependence of the MEP (0.001) value on the angle in the plane passing through the CN group and the pyridine ring [73]. Examination of the plot (Figure 4) for 3A indeed confirms the presence of maxima at angles of 108° and 150°. The first maximum corresponds to the π-hole on the carbon atom of the CN group, while the second maximum relates to the π-hole on the carbon atom of the pyridine ring. It should be noted that the π-hole is shifted towards the center of the ring, which is characteristic of aromatic rings [74].
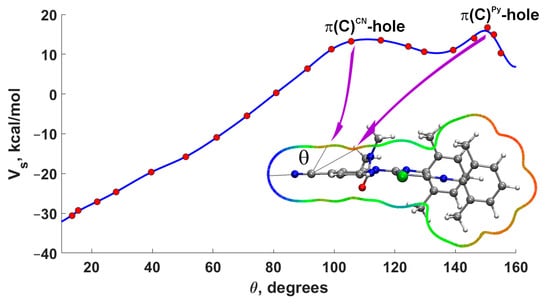
Figure 4.
Dependence of the MEP value vs. angle θ for 3A (0.001 a.u. isosurface of the electron density). The MEP plot is shown around the molecule in the plane passing through the CN atoms and the middle point of the C–C bond of the pyridine cycle (blue–green–red color scale −30 < Vs < 36 kcal mol−1).
2.3.2. QTAIM and IGMH
Analysis of both optimized bimolecular clusters ([3A]2 and [3B]2) using the QTAIM method allowed identification of bond critical points (BCPs, maxima of the density along bond paths) and bond paths, indicating interatomic interactions C···Pt, C···Cl, C···C, and HBs (Table 3 and Figure 5). Since the clusters are symmetrical, only unique contacts will be discussed in the manuscript to avoid a repetition. The small electron density values (ρb = 0.003–0.012 a.u.), positive Laplacian (∇2ρb = 0.012–0.042 a.u.), and positive total energy densities (Hb = 0.001–0.002 a.u.) indicate all these contacts are weak closed-shell noncovalent interactions. The electronic density at the BCPs for the C···Pt contact in [3A]2 is greater than in [3B]2, while the opposite trend is observed for the C···Cl contact. These results indicate that the C···Pt interaction is stronger in [3A]2, whereas the C···Cl interaction is stronger in [3B]2. The ELF value at the C···Pt and C···Cl bond critical points is 0.04, indicating efficient bonding between Pt or Cl and the C centers.
Table 3.
Electron density (ρb), its Laplacian (∇2ρb), potential and kinetic energy densities (Vb and Gb), second eigenvalue of the Hessian matrix (λ2) (in a.u.), electron localization function at BCPs calculated.
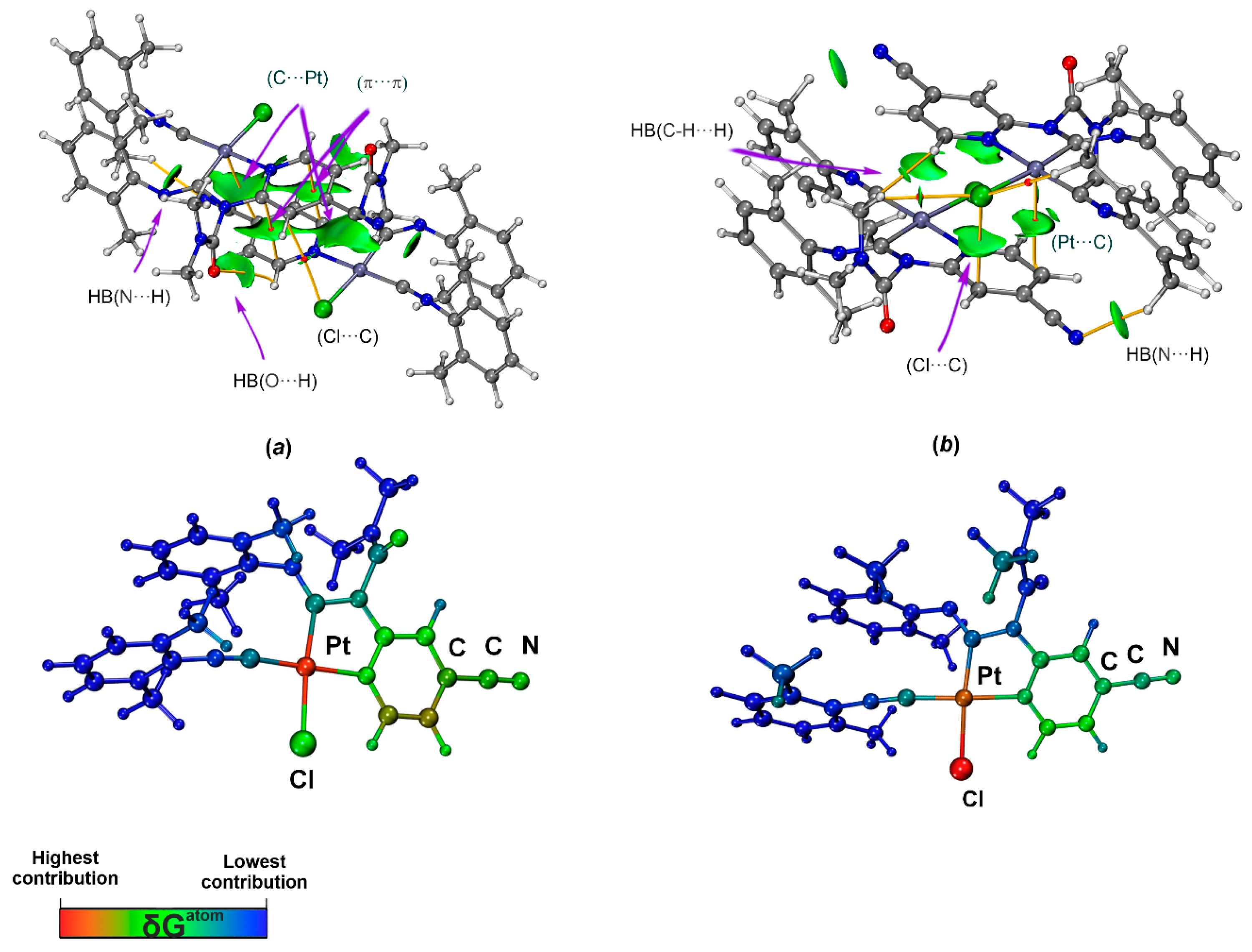
Figure 5.
Sign(λ2)ρ(r) function mapped on the δginter isosurface (δginter = 0.006 a.u. and blue–cyan–green–yellow–red color scale −0.01 < sign(λ2)ρ(r) < 0.01; top) and the δGatom colored structure (BCPs are shown as red dot, selected bond paths are shown as orange lines; bottom) for the structure of [3A]2 (a) and [3B]2 (b).
The QTAIM analysis also reveals the presence of various HBs that are involved in the association of 3A and 3B molecules into corresponding dimers. To estimate HBs energy we used the Espinosa–Molins–Lecomte formula [75] (Eint(HB) ≈ 0.5Vb). Values of HBs energy were calculated, with energies ranging from −0.5 to −2.1 kcal/mol. The HB energy values are given in the Table 3 for each dimer.
In addition, two BCPs and bond paths corresponding to C···C interactions, which confirm π···π stacking between the arene rings, were found in the calculated structure [3A]2. The ρb values indicate that these C···C interactions are weaker than the corresponding C···Pt contacts and HBs.
The IGMH isosurface for [3A]2 shows large areas of attractive interactions between the π-systems of two 3A molecules, corresponding to the C···Pt and C···C contacts (Figure 5b). At the same time, for the C···Cl contact, the isosurface at values of δg(inter) = 0.006 is not detected, indicating a negligible contribution of C···Cl to dimer bonding. The investigation of the δGatoms distribution [76] (Figure 5a) shows that the Pt atom indeed makes the largest contribution to the interactions between the π-systems, demonstrating the key role of the metal in supramolecular dimerization of 3A. Negative sign(λ2)ρ(r) functions were also found between N or O atoms and H atoms, indicating intermolecular HB N···H and O···H.
For the IGMH isosurface (Figure 5b) of the [3B]2 dimer, green disk-shaped surfaces were found between the atoms (Pt or Cl) and C atom, confirming the presence of C···Cl and C···Pt contacts. However, the Cl atom dominates in dimer bonding, as evidenced by the δGatom contribution distribution scheme. The presence of HB in the dimer structure is also confirmed by the IGMH method.
2.3.3. ETC-NOCV/CDF and NBO
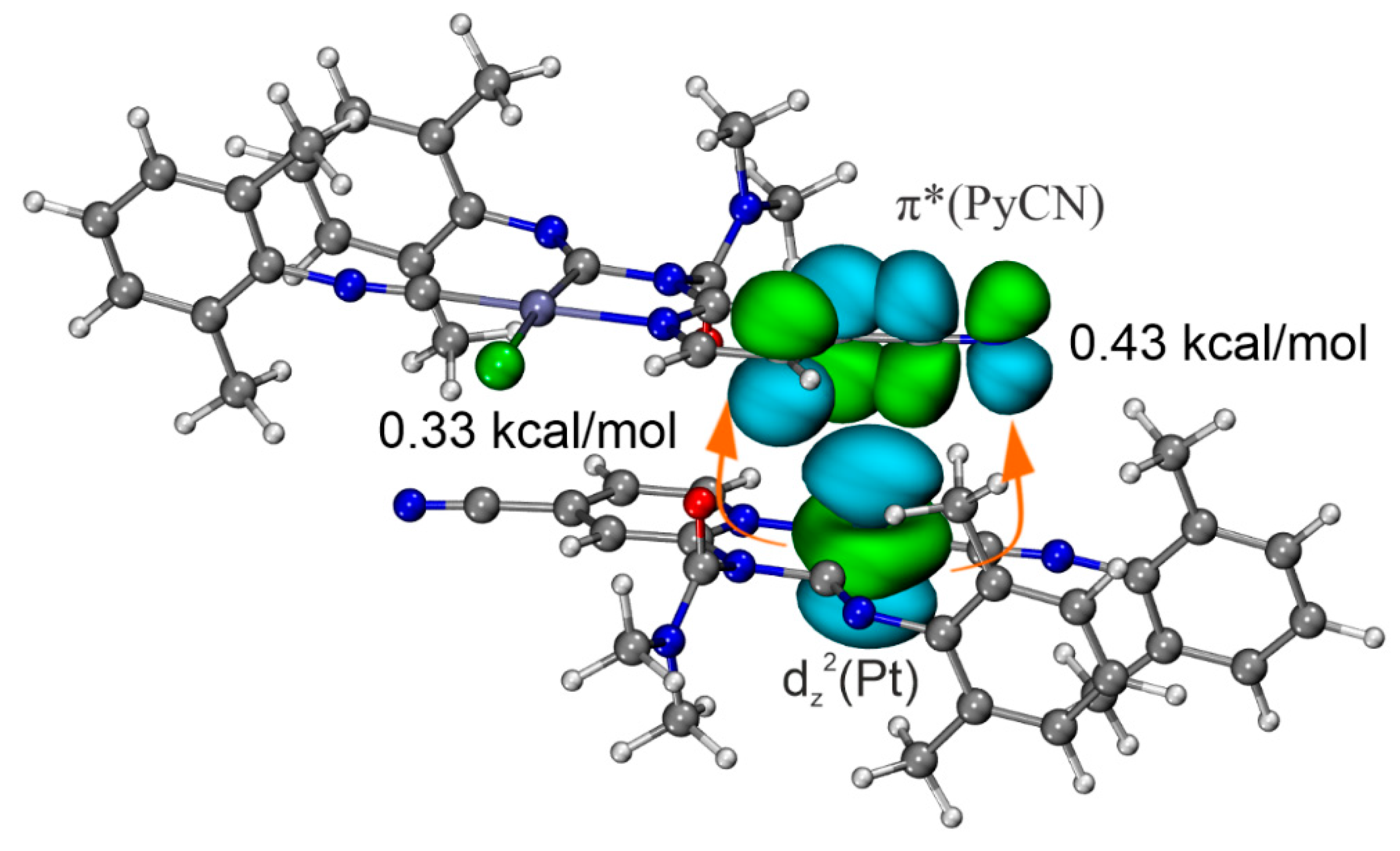
The charge transfer (CT) effect is critical for categorizing a noncovalent interaction into a specific type. As has previously been shown, CT significantly contributes to the metal-involving TtB in cocrystals of [Pt(pbt)(μ-S∩N)]2 (S∩N = 2-thiopyridine, pbt = 2-phenylbenzothiazole) with electron-deficient arenes [55]. We analyzed the CT in [3A]2 and [3B]2 using the CDF and ETS-NOCV methods. ETS-NOCV enables estimation of individual orbital contributions to the electron density difference between isolated molecules and their resultant bimolecular cluster (Figure 6). Meanwhile, CDF visualizes and quantifies the CT at the moment of bond formation.
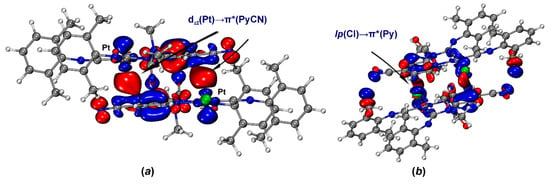
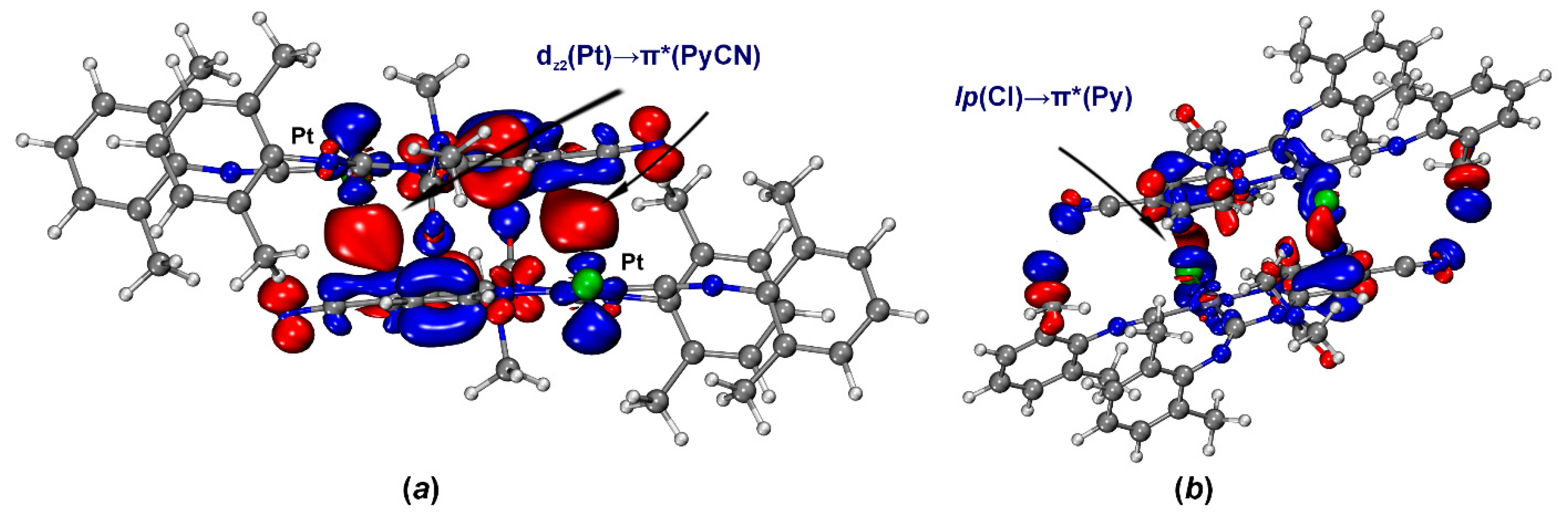
Figure 6.
ETS-NOCV deformation densities for [3A]2 (a) and [3B]2 (b); isovalues 0.0005 a.u., electrons transfer occurs from the decreased electron density regions (blue) to the increased electron density regions (red).
According to ETS-NOCV data of [3A]2 (Figure 6a), dz2(Pt)→π*(PyCN) (PyCN is 4-cyanopyridine moiety) orbital interaction results in the electron density accumulation in the intermolecular space between fragments. This interaction provides 60% of the total energy of the orbital interaction (Eintorb) between of two molecules of 3A in the bimolecular cluster [3A]2. Analysis of the CDF curve for this pair of NOCV demonstrates the presence of the CT, which is estimated at 10 millielectron (me) (Figure 7a). In other cases, orbital interactions associate with HBs and intramolecular polarization in the absence of effective intermolecular CT. The involvement of charge transfer with platinum is also confirmed by the increase in Hirshfeld charges charge on the Pt atom by 4 me.
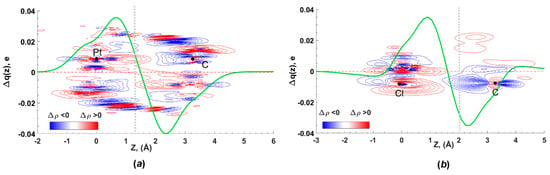
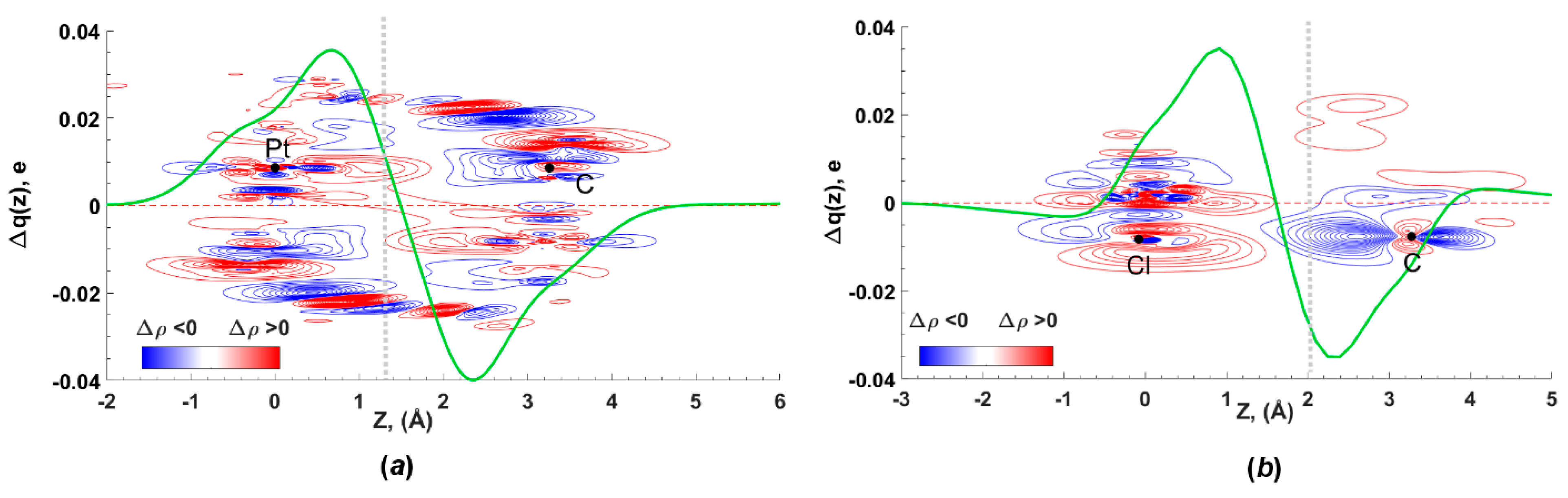
Figure 7.
Total EDD contour plot (red—charge concentration, blue—charge depletion, range—0.01 to 0.04 a.u., step 0.0002 a.u.) and CDF functions for the C···dz2(Pt) interaction in [3A]2 (a) and the C···lp(Cl) interaction in [3B]2 (b); black dots indicate positions of the atomic nuclei, grey vertical lines identify the boundaries between the C, Pt, and Cl atoms, which are placed along the z axis.
Second-order perturbation theory (E2), based on NBO analysis, also reveals the presence of dz2(Pt)→π*(PyCN) CT. NBO analysis shows that the direct CT in the C···Pt contact is associated with lone pair transitions from dz2(Pt) to π*-orbital of PyCN moiety with total second-order perturbation energies E(2) −0.76 kcal/mol (Figure 8). Thus, the dz2(Pt)→π*(PyCN) CT is an important component of the studied C···dz2(Pt) interaction, and, therefore, this noncovalent contact should be attributed to TtB.
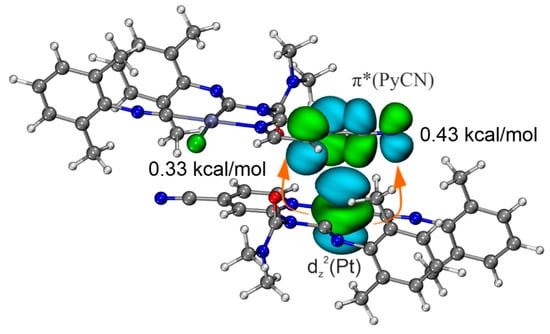
Figure 8.
Natural bond orbitals corresponding to the dz2(Pt)→π*(PyCN) lone pair transitions in [3A]2; PyCN is a 4-cyanopyridine moiety.
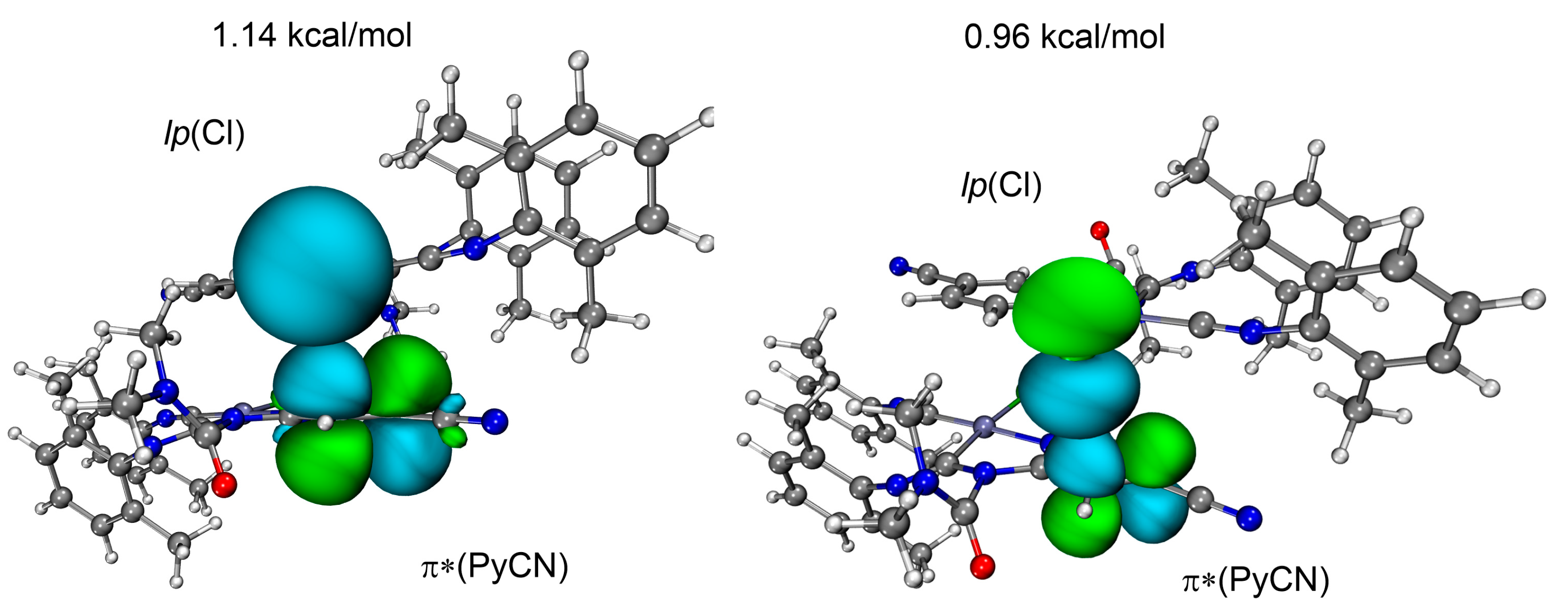
NOCV results indicate significant orbital interactions (50% of the total Eintorb) between the Cl atom and π*(PyCN) for the dimer [3B]2. According to the CDF curve, the charge transfer amounts to 28 me, considerably exceeding the analogous value for the [3A]2 dimer (Figure 7b). This indicates pronounced CT from the chloride orbitals to the π*(PyCN) orbital. The Hirshfeld charges on the chlorine atom decrease from −0.328e to −0.309e, while the charges on the platinum atom remain practically unchanged. Due to the CT, the C···Cl interaction in the [3B]2 dimer satisfies the criteria of a TtB. In contrast to the dimer [3A]2, the dz2(Pt)→π*(PyCN) orbital interaction is absent in the dimer [3B]2. The HB, orbital polarization, and intramolecular electron distribution are responsible for the residual part of the total Eintorb.
NBO analysis results also emphasize the definitive role of the chloride ligand in enabling orbital interactions between complexes in the [3B]2 dimer. The total energy of the lp(Cl)→π*(PyCN) donor–acceptor interaction is 2.1 kcal/mol, exceeding the analogous value for dz2(Pt)→π*(PyCN) in the dimer [3A]2 (Figure 9). Thus, in the dimer [3A]2, the dz2(Pt)→π*(PyCN) interaction mediated by charge transfer is dominant, while in the [3B]2 dimer, the lp(Cl) interaction with π*(PyCN) prevails over the analogous dz2(Pt)→π*(PyCN) interaction, serving as the primary center of orbital interactions. At the same time, the C···dz2(Pt) interaction in the [3B]2 cannot be classified as a tetrel bond due to the lack of CT involving platinum. Based on the significant charge transfer, the dz2(Pt)→π*(PyCN) interaction in the dimer [3A]2 and the lp(Cl)→π*(PyCN) interaction in the dimer [3B]2 can be attributed to tetrel bonds.

Figure 9.
Natural bond orbitals corresponding to the lp(Cl)→π*(PyCN) lone pair transitions in [3B]2; PyCN is a 4-cyanopyridine moiety.
2.3.4. Interaction Energies
The overall interaction and binding (ΣEintSM and ΣEbSM) energies for the formation of bimolecular clusters [3A]2 and [3B]2 were calculated using the SM approach. The corrected interaction energies for the BSSE error are given in the Table 4. These energies have values of more than 20 kcal/mol, which correspond to strong noncovalent interaction. As indicated in Section 2.3.2, two types of noncovalent force are responsible for the association of 3A into the bimolecular cluster [3A]2, i.e., the stacking interaction (with participation of C···dz2(Pt) TtB) between two planes, each of which consists of the 4-cyanopyridine moiety and the Pt(II)-based metallocycle, and several HBs. In order to quantitatively estimate the energy of the stacking interaction and C···dz2(Pt) TtB in [3A]2, interaction energy calculations were performed on specifically constructed model systems (M[3A]2 and M1[3A]2; (Figure 10a). To eliminate the influence of auxiliary HBs, in the first simplified model of the M[3A]2 the dimethylcarbamoyl and xylyl moieties were replaced with hydrogen atoms located at a distance of 1 Å. In the second model of the M1[3A]2, to eliminate the coordination bond with the metal, the {PtClCNH} fragment was removed from the M[3A]2 structure (Figure 10b), which made it possible to calculate the energy of the pure π···π interaction between the aromatic systems. Thus, by subtracting the interaction energy of the second model from the first one, it is possible to estimate the contribution of the tetrel bond C···dz2(Pt) (according to the formula: Eint(C···dz2(Pt))SM= (Eint (M [3A]2SM) − Eint(M1[3A]2)SM)). The obtained values of the energies of π···π stacking and C···dz2[Pt] were −12 kcal/mol and −5 kcal/mol, respectively. The remaining part of the energy −9.2 kcal/mol is accounted for by intermolecular hydrogen bonds, which is consistent with the results of estimating their energy by the QTAIM method (Section 2.3.2). Thus, the contribution of the C···dz2(Pt) TtB interaction accounts for 45% of the total interaction energy for the dimer [3A]2, proving the important role of tetrel bonding interactions with the metal in the association of the complex 3.
Table 4.
Calculated interaction and binding energies (Eint and Eb, respectively).
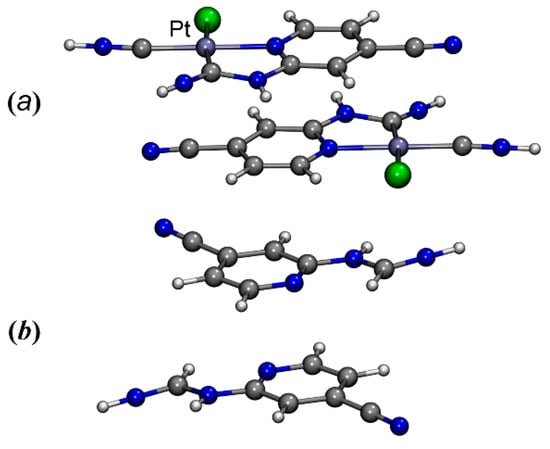
Figure 10.
Model bimolecular clusters (a) M[3A]2 and (b) M1[3A]2.
A similar calculation procedure was also conducted for the second dimer, except that the C···lp(Cl) interaction energies were calculated instead of C···Pt (according to the formula: Eint(C··· lp(Cl))SM = (Eint(M [3B]2 M) − Eint(M1[3B]2)SM)). It can be concluded that the TtB C···lp(Cl) interaction in the second dimer is the dominant one and accounts for 66% of the total interaction energy between the molecules 3B in the dimer. In comparison, in the first dimer the contribution of the TtB C···Pt interaction was 45% of the total binding energy. Thus, the results of the model system energy calculations demonstrate the significance of both platinum and chlorine as nucleophilic centers providing intermolecular interaction through the formation of TtBs with the carbon of the pyridine ligand. Both kinds of interactions play an important role and are a determining factor in the dimerization of the complexes in the considered dimer types.
Finally, we applied the GKS-EDA method [71] to M[3A]2 and M[3B]2 in order to gain further insight into the nature of the studied interaction (Table 5). In both dimers, we observe a similar composition of energy components. The GKS-EDA results indicate the predominantly dispersive character of the attractive interactions, accounting for 54% and 51% of the total attractive energy in M[3A]2 and M[3B]2, respectively. The electrostatic component makes the second largest contribution to the attractive energy, comprising 34% and 38% of the total attractive interaction in M[3A]2 and M[3B]2, respectively. The polarization component, although relatively insignificant and amounting to only 12% and 11%, however, plays a stabilizing role in the attractive interaction in M[3A]2 and M[3B]2, respectively.
Table 5.
Calculated energy interaction and Eint decomposition (all in kcal/mol).
The GKS-EDA results underscore dispersion as the predominant source of attractive interactions that stabilize the dimerization of the studied conformers of the complex 3. The comparable energetic decomposition for bimolecular model M[3A]2 and M[3B]2 further evinces the analogous interaction mechanisms for dimers connected through C···dz2(Pt) and C···lp(Cl) TtBs.
3. Conclusions
We found the Pt(II) complex 3 bearing deprotonated 3-(4-cyanopyridin-2-yl)-1,1-dimethylurea as a ligand was crystallized in form of two conformers 3A and 3B, owing to different orientation of the N,N-dimethylcarbamoyl moiety, in the structure of 3, and only one conformer 3A in 3·2MeCN. Independently on the type of conformer, molecules of 3 are associated into the “plane-to-plane” supramolecular dimers, {3A}2 or {3B}2, correspondingly. The stacking interaction between two metal square-planes, each of which consists of the 4-cyanopyridine moiety and the Pt(II)-based metallocycle, as well as hydrogen bonding, are responsible for the formation of these dimers. In its turn, the stacking interaction includes several components, namely, a π···π interaction between the heteroaromatic rings and a C···dz2(Pt) contact between the carbon atom of the substituted 4-cyanopyridyl moiety and the metal center. The contact C···dz2(Pt) in {3A}2 was attributed to metal-involving TtB, in which the metal site acts as a Lewis base (an acceptor of noncovalent interaction). This suggestion is based on structural parameters and quantum chemical calculation data for the optimized dimer [3A]2. Particularly, there is (1) the presence of a C···Pt short interatomic distance in the X-ray crystal structure; (2) the presence of a bond critical point and bond path in the QTAIM analysis; (3) the presence of a π-hole on the nitrile carbon atom and pyridine ring (as pronounced local maxima on the MEP surface); (4) the presence of significant charge transfer (more than 5 me) from the nucleophile’s orbital to the vacant π-orbital involving the π-hole of the electrophile (the carbon atom); (5) negative value of the interaction energy. A similar example of metal-involving TtB was previously described for cocrystals of a binuclear platinum(II) complex with perfluorinated aromatic compounds [55]. In both these cases, the platinum(II) centers behave as TtB acceptors, which becomes possible due to the enhanced nucleophilicity of this metal center. In contrast, the X-ray dimer {3B}2 has different geometry to the corresponding optimized dimer [3B]2: the C···Pt contact is lengthened and weakened after optimization, and the optimized dimer [3B]2 exhibits C···lp(Cl) TtB as one of the strongest noncovalent forces, which was confirmed theoretically.
The most significand finding of this work is the TtB C···dz2(Pt) in {3A}2. We believe that new example of a metal-involving TtB will significantly expand understanding the phenomenon of noncovalent interactions involving a transition metal as a Lewis base.
4. Experimental Section
4.1. Materials and Instrumentation
3-(4-Cyanopyridin-2-yl)-1,1-dimethylurea (1) [77] and cis-[PtCl2(CNXyl)2] 2 [78] were synthesized according to the literature protocols. All other reagents and solvents were purchased and were used as received in BLDPharm (Shanghai, China), Macklin (Shanghai, China).
1H, 13C, and 195Pt NMR spectra were recorded on a Bruker AVANCE III 400 spectrometer operating at room temperature at 400, 101, and 86 MHz for 1H, 13C, and 195Pt NMR spectra, respectively. All spectra were registered using CDCl3 as a solvent. The chemical shifts are given in δ-values (ppm). Multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, m = multiplet, br = broad; coupling constants, J, are reported in Hertz (Hz). High-resolution mass spectra (HRMS) were measured on Bruker Maxis HR-MS-ESI-qTOF using ESI. The most intense peak in the isotopic pattern is reported.
4.2. Synthesis of Complex 3
Triethanolamine (18 mg, 0.12 mmol) was added to a mixture of the urea 1 (19 mg, 0.10 mmol) and cis-[PtCl2(CNXyl)2] 2 (81 mg, 0.12 mmol) in CHCl3 (3 mL). The reaction mixture was stirred at RT for 24 h. After that, the reaction mixture was filtered to remove a small amount of undissolved material (triethanolamine hydrochloride) and evaporated to dryness at 45 °C in vacuo. Complex 3 was purified by reprecipitation from dichloromethane. It was dissolved in dichloromethane (0.3 mL) and diluted with MeOH (1.1 mL). The formed precipitate was collected by filtration, washed with hexane, and dried in vacuo at RT. Light yellow powder; 44% yield (30 mg). 1H NMR (400 MHz, CDCl3): δ 9.50 (dd, J = 6.3, 0.8 Hz, 1H), 7.18–7.09 (m, 3H), 6.99 (d, J = 7.6 Hz, 2H), 6.78 (d, J = 7.5 Hz, 1H), 6.64 (d, J = 7.5 Hz, 1H), 6.17 (t, J = 7.5 Hz, 1H), 3.22 (s, 3H), 3.16 (s, 3H), 2.25 (s, 6H), 2.22 (s, 3H), 2.19 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 157.1, 153.1, 150.4, 148.7, 147.7, 134.5, 129.2, 128.4, 128.0, 127.5, 127.4, 127.0, 124.9, 123.5, 115.9, 115.4, 111.5, 38.4, 36.7, 19.6, 19.3, 18.4. 195Pt NMR (86 MHz, CDCl3): δ −3801. HRMS (ESI) m/z [M–Cl]+ calculated for [C27H27ClN6OPt–Cl]+ 646.1889; found 646.1904.
4.3. Crystal Growth, Structure Solution and Refinement Details
Crystals 3 and 3·2MeCN were grown by slow evaporation of solutions of the corresponding compound in 1,2-dichloroethane (3) or acetonitrile (3·2MeCN) in air at RT. X-ray diffraction data were collected at a Rigaku XtaLAB Synergy–S diffractometers using Cu-Kα (λ = 0.154184 nm) radiation. The structures have been solved with the ShelXT [79] structure solution program using Intrinsic Phasing and refined with the ShelXL [80] refinement package incorporated in the OLEX2 program package [81] using Least Squares minimization. Supplementary crystallographic data have been deposited at Cambridge Crystallographic Data Centre: 2323847 (3) and 2323848 (3·2MeCN). They can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif (accessed on 14 February 2024).
4.4. Computational Details
Full geometry optimization of the model clusters has been carried out at the DFT level of theory using the PBE0 [82,83] functional with the atom-pairwise dispersion correction with the Becke–Johnson damping scheme (D3BJ) [84,85]. The ORCA package (version 5.0.3) was used for the calculation [86,87]. Zero-order regular approximation (ZORA) [88] was employed to account the relativistic effects. The X-ray structures (3 and 3·2MeCN) were used as initial geometries for the optimization procedure. The ZORA-def2-TZVP(–f) basis set was applied for the H, C, N, O, and Cl atoms, whereas the SARC-ZORA-TZVP basis set was used for the Pt atom [89]. The Hessian matrix was calculated analytically for the optimized structures to prove the location of correct minima (no imaginary frequencies). Combination of the “resolution of identity” and the “chain of spheres exchange” algorithms (RIJCOSX) [90] in conjunction with the auxiliary basis sets SARC/J were used [91]. Large integration grid (DEFGRID3) was used throughout the calculations.
The single point calculations based on the equilibrium geometries were performed at the PBE0-D3BJ level with the ZORA-def2-TZVP(–f) [88] (for H, C, N, O, and Cl) and the SARC-ZORA-TZVP (for Pt) basis sets [89]. This level of theory was used for the estimates of interaction energies at the super-molecular approach and for the QTAIM, ELF, IGMH, MEP, NBO, CDF, and ETS-NOCV analyses. The natural bond orbital analysis was performed using the NBO 7.0 program [70].
The QTAIM, ELF, IGMH, and MEP calculations were carried out using the Multiwfn 3.8 software [76,92,93] and results were visualized using the VMD program [94]. The generalized Kohn–Sham energy decomposition analysis (GKS-EDA) [71,95] is conducted at the PBE0-D3BJ/def2-TZVP level of theory by a local version of GAMESS-US (version 2021-R2 patch 1) [96,97]. The CDF and ETS-NOCV analyses were carried out according to the methodology described in refs. [68,98,99] using the Multiwfn 3.8 software. Basis set superposition error (BSSE) was estimated using the counterpoise method [100].
The interaction and binding energies between complex (Eint and Eb) were calculated for bimolecular clusters as
where E([3A]2), and E(3A) are total energies of the optimized structures of [3A]2 and 3A, while E{3A} are total energies of 3A in the optimized geometry of [3A]2.
Eint(3A···3A) = E([3A]2) − 2E{3A} + BSSE,
Eb(3A···3A) = E([3A]2) − 2E(3A) + BSSE,
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms25074052/s1. X–ray diffraction data; Copies of NMR and HRMS spectra of complex 3; Computational study; Cartesian coordinates for the studied molecules.
Author Contributions
Conceptualization, N.A.B. and V.P.B.; methodology, S.V.B. and E.A.K.; formal analysis, S.V.B. and E.A.K.; investigation, S.V.B., E.A.K., S.O.B. and A.V.S.; data curation, S.V.B.; writing—original draft preparation, S.V.B. and E.A.K.; writing—review and editing, N.A.B. and V.P.B.; visualization, A.V.S.; supervision, N.A.B. and V.P.B.; project administration, S.V.B.; funding acquisition, S.V.B. and E.A.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Russian Science Foundation (project 22-73-10031, synthetic work and crystallographic studies; project 23-73-01074, computational study).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
The authors are grateful to the Centre for Magnetic Resonance, Centre for X-ray Diffraction Studies, Centre for Chemical Analysis and Materials Research, Thermogravimetric and Calorimetric Research Centre (all belonging to Saint Petersburg State University).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kinzhalov, M.A.; Luzyanin, K.V. Reactivity of acyclic diaminocarbene ligands. Coord. Chem. Rev. 2019, 399, 213014. [Google Scholar] [CrossRef]
- Kinzhalov, M.A.; Grachova, E.V.; Luzyanin, K.V. Tuning the luminescence of transition metal complexes with acyclic diaminocarbene ligands. Inorg. Chem. Front. 2022, 9, 417–439. [Google Scholar] [CrossRef]
- Kinzhalov, M.A.; Luzyanin, K.V. Synthesis and Contemporary Applications of Platinum Group Metals Complexes with Acyclic Diaminocarbene Ligands (Review). Russ. J. Inorg. Chem. 2022, 67, 48–90. [Google Scholar] [CrossRef]
- Singh, C.; Prakasham, A.P.; Gangwar, M.K.; Ghosh, P. Binuclear Fused 5-membered Palladacycle and Palladium Complex of Amido-Functionalized N-heterocyclic Carbene Precatalysts for the One-Pot Tandem Hiyama Alkynylation/Cyclization Reactions. ChemistrySelect 2018, 3, 9361–9367. [Google Scholar] [CrossRef]
- Singh, C.; Prakasham, A.P.; Ghosh, P. Palladium Acyclic Diaminocarbene (ADC) Triflate Complexes as Effective Precatalysts for the Hiyama Alkynylation/Cyclization Reaction Yielding Benzofuran Compounds: Probing the Influence of the Triflate Co-Ligand in the One-Pot Tandem Reaction. ChemistrySelect 2019, 4, 329–336. [Google Scholar] [CrossRef]
- Handa, S.; Slaughter, L.M. Enantioselective Alkynylbenzaldehyde Cyclizations Catalyzed by Chiral Gold(I) Acyclic Diaminocarbene Complexes Containing Weak Au-Arene Interactions. Angew. Chem. Int. Ed. 2012, 51, 2912–2915. [Google Scholar] [CrossRef] [PubMed]
- Mikhaylov, V.; Sorokoumov, V.; Liakhov, D.; Tskhovrebov, A.; Balova, I. Polystyrene-Supported Acyclic Diaminocarbene Palladium Complexes in Sonogashira Cross-Coupling: Stability vs. Catalytic Activity. Catalysts 2018, 8, 141. [Google Scholar] [CrossRef]
- Mikhaylov, V.N.; Sorokoumov, V.N.; Korvinson, K.A.; Novikov, A.S.; Balova, I.A. Synthesis and Simple Immobilization of Palladium(II) Acyclic Diaminocarbene Complexes on Polystyrene Support as Efficient Catalysts for Sonogashira and Suzuki–Miyaura Cross-Coupling. Organometallics 2016, 35, 1684–1697. [Google Scholar] [CrossRef]
- Gee, J.C.; Fuller, B.A.; Lockett, H.-M.; Sedghi, G.; Robertson, C.M.; Luzyanin, K.V. Visible light accelerated hydrosilylation of alkynes using platinum–[acyclic diaminocarbene] photocatalysts. Chem. Commun. 2018, 54, 9450–9453. [Google Scholar] [CrossRef]
- Dobrynin, M.V.; Kasatkina, S.O.; Baykov, S.V.; Savko, P.Y.; Antonov, N.S.; Mikherdov, A.S.; Boyarskiy, V.P.; Islamova, R.M. Deprotonated diaminocarbene platinum complexes for thermoresponsive luminescent silicone materials: Both catalysts and luminophores. Dalt. Trans. 2021, 50, 14994–14999. [Google Scholar] [CrossRef]
- Deriabin, K.V.; Golovenko, E.A.; Antonov, N.S.; Baykov, S.V.; Boyarskiy, V.P.; Islamova, R.M. Platinum macrocatalyst for heterogeneous Si–O dehydrocoupling. Dalt. Trans. 2023, 52, 5854–5858. [Google Scholar] [CrossRef] [PubMed]
- Barbazanges, M.; Fensterbank, L. Chiral Acyclic Diaminocarbene Complexes: A New Opportunity for Gold Asymmetric Catalysis. ChemCatChem 2012, 4, 1065–1066. [Google Scholar] [CrossRef]
- Ruch, A.A.; Ellison, M.C.; Nguyen, J.K.; Kong, F.; Handa, S.; Nesterov, V.N.; Slaughter, L.M. Highly Sterically Encumbered Gold Acyclic Diaminocarbene Complexes: Overriding Electronic Control in Regiodivergent Gold Catalysis. Organometallics 2021, 40, 1416–1433. [Google Scholar] [CrossRef]
- Katkova, S.A.; Bunev, A.S.; Gasanov, R.E.; Khochenkov, D.A.; Kulsha, A.V.; Ivashkevich, O.A.; Serebryanskaya, T.V.; Kinzhalov, M.A. Metal-(Acyclic Diaminocarbene) Complexes Demonstrate Nanomolar Antiproliferative Activity against Triple-Negative Breast Cancer. Chem. Eur. J. 2024, e202400101. [Google Scholar] [CrossRef]
- Geyl, K.K.; Baykova, S.O.; Andoskin, P.A.; Sharoyko, V.V.; Eliseeva, A.A.; Baykov, S.V.; Semenov, K.N.; Boyarskiy, V.P. Palladium(II) and Platinum(II) Deprotonated Diaminocarbene Complexes Based on N-(2-Pyridyl)ureas with Oxadiazole Periphery. Inorganics 2022, 10, 247. [Google Scholar] [CrossRef]
- Eremina, A.A.; Kinzhalov, M.A.; Katlenok, E.A.; Smirnov, A.S.; Andrusenko, E.V.; Pidko, E.A.; Suslonov, V.V.; Luzyanin, K.V. Phosphorescent Iridium(III) Complexes with Acyclic Diaminocarbene Ligands as Chemosensors for Mercury. Inorg. Chem. 2020, 59, 2209–2222. [Google Scholar] [CrossRef] [PubMed]
- Katkova, S.A.; Kozina, D.O.; Kisel, K.S.; Sandzhieva, M.A.; Tarvanen, D.A.; Makarov, S.V.; Porsev, V.V.; Tunik, S.P.; Kinzhalov, M.A. Cyclometalated platinum(II) complexes with acyclic diaminocarbene ligands for OLED application. Dalt. Trans. 2023, 52, 4595–4605. [Google Scholar] [CrossRef]
- Mikherdov, A.S.; Kinzhalov, M.A.; Novikov, A.S.; Boyarskiy, V.P.; Boyarskaya, I.A.; Dar’in, D.V.; Starova, G.L.; Kukushkin, V.Y. Difference in Energy between Two Distinct Types of Chalcogen Bonds Drives Regioisomerization of Binuclear (Diaminocarbene)Pd II Complexes. J. Am. Chem. Soc. 2016, 138, 14129–14137. [Google Scholar] [CrossRef]
- Koshevoy, I.O.; Krause, M.; Klein, A. Non-covalent intramolecular interactions through ligand-design promoting efficient photoluminescence from transition metal complexes. Coord. Chem. Rev. 2020, 405, 213094. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Katlenok, E.A.; Zhmykhova, M.V.; Kuznetsov, M.L.; Khrustalev, V.N.; Tugashov, K.I.; Bokach, N.A.; Kukushkin, V.Y. Spodium bonding to anticrown-Hg3 boosts phosphorescence of cyclometalated-PtII complexes. Inorg. Chem. Front. 2023, 10, 493–510. [Google Scholar] [CrossRef]
- Katkova, S.A.; Antonova, E.V.; Cheranyova, A.M.; Ivanov, D.M.; Kinzhalov, M.A. Solid State Phosphorescence Enhancement of PtII-Based Emitters via Combination of π-Hole(Isocyano Group)⋅⋅⋅dz2[PtII] and I···Cl Halogen-Bonding Interactions. Inorganics 2023, 11, 403. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Carter-Fenk, K.; Herbert, J.M. Reinterpreting π-stacking. Phys. Chem. Chem. Phys. 2020, 22, 24870–24886. [Google Scholar] [CrossRef]
- Zheng, Q.; Borsley, S.; Nichol, G.S.; Duarte, F.; Cockroft, S.L. The Energetic Significance of Metallophilic Interactions. Angew. Chem. Int. Ed. 2019, 58, 12617–12623. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Resnati, G.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Krossing, I.; Legon, A.C.; Metrangolo, P.; Nicotra, F.; Rissanen, K.; Scheiner, S.; et al. Definition of the pnictogen bond (IUPAC Recommendations 2023). Pure Appl. Chem. 2024, 96, 135–145. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. Definition of the tetrel bond. CrystEngComm 2023, 25, 1411–1423. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Bokach, N.A.; Kukushkin, V.Y.; Frontera, A. Metal Centers as Nucleophiles: Oxymoron of Halogen Bond-Involving Crystal Engineering. Chem. Eur. J. 2022, 28, e202103173. [Google Scholar] [CrossRef]
- Aliyarova, I.S.; Tupikina, E.Y.; Ivanov, D.M.; Kukushkin, V.Y. Metal-Involving Halogen Bonding Including Gold(I) as a Nucleophilic Partner. The Case of Isomorphic Dichloroaurate(I)·Halomethane Cocrystals. Inorg. Chem. 2022, 61, 2558–2567. [Google Scholar] [CrossRef] [PubMed]
- Mikherdov, A.S.; Jin, M.; Ito, H. Exploring Au(I) involving halogen bonding with N-heterocyclic carbene Au(I) aryl complexes in crystalline media. Chem. Sci. 2023, 14, 4485–4494. [Google Scholar] [CrossRef] [PubMed]
- Zelenkov, L.E.; Eliseeva, A.A.; Baykov, S.V.; Suslonov, V.V.; Galmés, B.; Frontera, A.; Kukushkin, V.Y.; Ivanov, D.M.; Bokach, N.A. Electron belt-to-σ-hole switch of noncovalently bound iodine(I) atoms in dithiocarbamate metal complexes. Inorg. Chem. Front. 2021, 8, 2505–2517. [Google Scholar] [CrossRef]
- Eliseeva, A.A.; Ivanov, D.M.; Rozhkov, A.V.; Ananyev, I.V.; Frontera, A.; Kukushkin, V.Y. Bifurcated Halogen Bonding Involving Two Rhodium(I) Centers as an Integrated σ-Hole Acceptor. JACS Au 2021, 1, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Bulatova, M.; Ivanov, D.M.; Rautiainen, J.M.; Kinzhalov, M.A.; Truong, K.-N.; Lahtinen, M.; Haukka, M. Studies of Nature of Uncommon Bifurcated I–I···(I–M) Metal-Involving Noncovalent Interaction in Palladium(II) and Platinum(II) Isocyanide Cocrystals. Inorg. Chem. 2021, 60, 13200–13211. [Google Scholar] [CrossRef] [PubMed]
- Aliyarova, I.S.; Tupikina, E.Y.; Soldatova, N.S.; Ivanov, D.M.; Postnikov, P.S.; Yusubov, M.; Kukushkin, V.Y. Halogen Bonding Involving Gold Nucleophiles in Different Oxidation States. Inorg. Chem. 2022, 61, 15398–15407. [Google Scholar] [CrossRef] [PubMed]
- Rozhkov, A.V.; Katlenok, E.A.; Zhmykhova, M.V.; Ivanov, A.Y.; Kuznetsov, M.L.; Bokach, N.A.; Kukushkin, V.Y. Metal-Involving Chalcogen Bond. The Case of Platinum(II) Interaction with Se/Te-Based σ-Hole Donors. J. Am. Chem. Soc. 2021, 143, 15701–15710. [Google Scholar] [CrossRef] [PubMed]
- Burguera, S.; Gomila, R.M.; Bauzá, A.; Frontera, A. Square Planar Pt(II) Ion as Electron Donor in Pnictogen Bonding Interactions. Inorganics 2023, 11, 80. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Krykova, M.A.; Ivanov, D.M.; Novikov, A.S.; Sinelshchikova, A.A.; Volostnykh, M.V.; Konovalov, M.A.; Grigoriev, M.S.; Gorbunova, Y.G.; Kukushkin, V.Y. Reverse Arene Sandwich Structures Based upon π-Hole⋅⋅⋅[MII] (d8M=Pt, Pd) Interactions, where Positively Charged Metal Centers Play the Role of a Nucleophile. Angew. Chem. Int. Ed. 2019, 58, 4164–4168. [Google Scholar] [CrossRef]
- Baykov, S.V.; Filimonov, S.I.; Rozhkov, A.V.; Novikov, A.S.; Ananyev, I.V.; Ivanov, D.M.; Kukushkin, V.Y. Reverse Sandwich Structures from Interplay between Lone Pair−π-Hole Atom-Directed C···dz2[M] and Halogen Bond Interactions. Cryst. Growth Des. 2020, 20, 995–1008. [Google Scholar] [CrossRef]
- Zelenkov, L.E.; Eliseeva, A.A.; Baykov, S.V.; Ivanov, D.M.; Sumina, A.I.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y.; Bokach, N.A. Inorganic–Organic {dz2-MIIS4}⋯π-Hole Stacking in Reverse Sandwich Structures: The Case of Cocrystals of Group 10 Metal Dithiocarbamates with Electron-Deficient Arenes. Inorg. Chem. Front. 2022, 9, 2869–2879. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Ananyev, I.V.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y. π-Hole···dz2[PtII] Interactions with Electron-Deficient Arenes Enhance the Phosphorescence of PtII-Based Luminophores. Inorg. Chem. 2020, 59, 9308–9314. [Google Scholar] [CrossRef] [PubMed]
- Torubaev, Y.V.; Skabitsky, I.V.; Rozhkov, A.V.; Galmés, B.; Frontera, A.; Kukushkin, V.Y. Highly Polar Stacking Interactions Wrap Inorganics in Organics: Lone-Pair–π-Hole Interactions between the PdO4 Core and Electron-Deficient Arenes. Inorg. Chem. Front. 2021, 8, 4965–4975. [Google Scholar] [CrossRef]
- Seth, S.K.; Bauzá, A.; Mahmoudi, G.; Stilinović, V.; López-Torres, E.; Zaragoza, G.; Keramidas, A.D.; Frontera, A. On the importance of Pb⋯X (X = O, N, S, Br) tetrel bonding interactions in a series of tetra- and hexa-coordinated Pb(II) compounds. CrystEngComm 2018, 20, 5033–5044. [Google Scholar] [CrossRef]
- Mahmoudi, G.; Seth, S.K.; Bauzá, A.; Zubkov, F.I.; Gurbanov, A.V.; White, J.; Stilinović, V.; Doert, T.; Frontera, A. Pb⋯X (X = N, S, I) tetrel bonding interactions in Pb(II) complexes: X-ray characterization, Hirshfeld surfaces and DFT calculations. CrystEngComm 2018, 20, 2812–2821. [Google Scholar] [CrossRef]
- Eftekhari-Sis, B.; García-Santos, I.; Castiñeiras, A.; Mahmoudi, G.; Zangrando, E.; Frontera, A.; Safin, D.A. On the pivotal role of tetrel bonding in the supramolecular architectures of PbII–NCS complexes with chelating thiosemicarbazide derivatives. CrystEngComm 2024, 26, 1637–1646. [Google Scholar] [CrossRef]
- Kumar, P.; Firdoos, T.; Gomila, R.M.; Frontera, A.; Pandey, S.K. Experimental and Theoretical Study of Tetrel Bonding and Noncovalent Interactions in Hemidirected Lead(II) Phosphorodithioates: An Implication on Crystal Engineering. Cryst. Growth Des. 2023, 23, 2138–2154. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; Aliyeva, V.A.; Kuznetsov, M.L.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Tetrel Bonding in Coordination Chemistry. In Synthesis and Applications in Chemistry and Materials: Volume 11: Metal Coordination and Nanomaterials; World Scientific: Singapore, 2024; pp. 45–75. [Google Scholar]
- Bauzá, A.; Seth, S.K.; Frontera, A. Tetrel bonding interactions at work: Impact on tin and lead coordination compounds. Coord. Chem. Rev. 2019, 384, 107–125. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. The Tetrel Bond and Tetrel Halide Perovskite Semiconductors. Int. J. Mol. Sci. 2023, 24, 6659. [Google Scholar] [CrossRef]
- Servati Gargari, M.; Stilinović, V.; Bauzá, A.; Frontera, A.; McArdle, P.; Van Derveer, D.; Ng, S.W.; Mahmoudi, G. Design of Lead(II) Metal–Organic Frameworks Based on Covalent and Tetrel Bonding. Chem. Eur. J. 2015, 21, 17951–17958. [Google Scholar] [CrossRef]
- Seo, J.; Song, T.; Rasool, S.; Park, S.; Kim, J.Y. An Overview of Lead, Tin, and Mixed Tin–Lead-Based ABI3 Perovskite Solar Cells. Adv. Energy Sustain. Res. 2023, 4, 2200160. [Google Scholar] [CrossRef]
- Khadka, D.B.; Shirai, Y.; Yanagida, M.; Ryan, J.W.; Song, Z.; Barker, B.G.; Dhakal, T.P.; Miyano, K. Advancing Efficiency and Stability of Lead, Tin, and Lead/Tin Perovskite Solar Cells: Strategies and Perspectives. Sol. RRL 2023, 7, 2300535. [Google Scholar] [CrossRef]
- Katlenok, E.A.; Rozhkov, A.V.; Kuznetsov, M.L.; Suslonov, V.V.; Kukushkin, V.Y. Dichotomy of π-stacking-directing noncovalent forces in organic–inorganic planar assemblies: The case of halo-substituted benzoquinones π-stacked with a platinum(II) square-plane. Inorg. Chem. Front. 2024, 11, 1252–1265. [Google Scholar] [CrossRef]
- Katlenok, E.A.; Kuznetsov, M.L.; Cherkasov, A.V.; Kryukov, D.M.; Bokach, N.A.; Kukushkin, V.Y. Metal-involved C⋯dz2-PtII tetrel bonding as a principal component of the stacking interaction between arenes and the platinum(II) square-plane. Inorg. Chem. Front. 2023, 10, 3916–3928. [Google Scholar] [CrossRef]
- Baykova, S.O.; Baykov, S.V.; Boyarskiy, V.P. Pyridyl-Substituted Ureas and Carbamates: Synthesis and Application (A Review). Russ. J. Gen. Chem. 2024, 94, S60–S90. [Google Scholar] [CrossRef]
- Malenov, D.P.; Zarić, S.D. Stacking interactions of aromatic ligands in transition metal complexes. Coord. Chem. Rev. 2020, 419, 213338. [Google Scholar] [CrossRef]
- Chen, T.; Li, M.; Liu, J. π–π Stacking Interaction: A Nondestructive and Facile Means in Material Engineering for Bioapplications. Cryst. Growth Des. 2018, 18, 2765–2783. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii of Metals in Covalent Compounds. J. Phys. Chem. 1966, 70, 3006–3007. [Google Scholar] [CrossRef]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent radii revisited. Dalt. Trans. 2008, 2832–2838. [Google Scholar] [CrossRef]
- Alvarez, S. A cartography of the van der Waals territories. Dalt. Trans. 2013, 42, 8617. [Google Scholar] [CrossRef]
- Kumar, P.S.V.; Raghavendra, V.; Subramanian, V. Bader’s Theory of Atoms in Molecules (AIM) and its Applications to Chemical Bonding. J. Chem. Sci. 2016, 128, 1527–1536. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Lefebvre, C.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Bistoni, G.; Rampino, S.; Tarantelli, F.; Belpassi, L. Charge-displacement analysis via natural orbitals for chemical valence: Charge transfer effects in coordination chemistry. J. Chem. Phys. 2015, 142, 084112. [Google Scholar] [CrossRef] [PubMed]
- Belpassi, L.; Infante, I.; Tarantelli, F.; Visscher, L. The Chemical Bond between Au(I) and the Noble Gases. Comparative Study of NgAuF and NgAu+ (Ng = Ar, Kr, Xe) by Density Functional and Coupled Cluster Methods. J. Am. Chem. Soc. 2008, 130, 1048–1060. [Google Scholar] [CrossRef] [PubMed]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO7 2018. Available online: https://nbo6.chem.wisc.edu/index.htm (accessed on 3 April 2024).
- Tang, Z.; Song, Y.; Zhang, S.; Wang, W.; Xu, Y.; Wu, D.; Wu, W.; Su, P. XEDA, a fast and multipurpose energy decomposition analysis program. J. Comput. Chem. 2021, 42, 2341–2351. [Google Scholar] [CrossRef]
- Scheiner, S. Maximal occupation by bases of π-hole bands surrounding linear molecules. J. Comput. Chem. 2022, 43, 319–330. [Google Scholar] [CrossRef]
- Scheiner, S. Dissection of the Origin of π-Holes and the Noncovalent Bonds in Which They Engage. J. Phys. Chem. A 2021, 125, 6514–6528. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. The π-hole revisited. Phys. Chem. Chem. Phys. 2021, 23, 16458–16468. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 2022, 43, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Rassadin, V.A.; Zimin, D.P.; Raskil’dina, G.Z.; Ivanov, A.Y.; Boyarskiy, V.P.; Zlotskii, S.S.; Kukushkin, V.Y. Solvent- and halide-free synthesis of pyridine-2-yl substituted ureas through facile C–H functionalization of pyridine N-oxides. Green Chem. 2016, 18, 6630–6636. [Google Scholar] [CrossRef]
- Luzyanin, K.V.; Pombeiro, A.J.L.; Haukka, M.; Kukushkin, V.Y. Coupling between 3-Iminoisoindolin-1-ones and Complexed Isonitriles as a Metal-Mediated Route to a Novel Type of Palladium and Platinum Iminocarbene Species. Organometallics 2008, 27, 5379–5389. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef] [PubMed]
- Rolfes, J.D.; Neese, F.; Pantazis, D.A. All-electron scalar relativistic basis sets for the elements Rb–Xe. J. Comput. Chem. 2020, 41, 1842–1849. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix. J. Comput. Chem. 2003, 24, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Neese, F. All-Electron Scalar Relativistic Basis Sets for the Actinides. J. Chem. Theory Comput. 2011, 7, 677–684. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Tang, Z.; Wu, W. Generalized Kohn-Sham energy decomposition analysis and its applications. WIREs Comput. Mol. Sci. 2020, 10, e1460. [Google Scholar] [CrossRef]
- Arca, G.M.J.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Nunzi, F.; Cesario, D.; Belpassi, L.; Tarantelli, F.; Roncaratti, L.F.; Falcinelli, S.; Cappelletti, D.; Pirani, F. Insight into the halogen-bond nature of noble gas-chlorine systems by molecular beam scattering experiments, ab initio calculations and charge displacement analysis. Phys. Chem. Chem. Phys. 2019, 21, 7330–7340. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lu, T.; Chen, Q. Intermolecular interaction characteristics of the all-carboatomic ring, cyclo[18]carbon: Focusing on molecular adsorption and stacking. Carbon N. Y. 2021, 171, 514–523. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).