Abstract
Cellular survival hinges on a delicate balance between accumulating damages and repair mechanisms. In this intricate equilibrium, oxidants, currently considered physiological molecules, can compromise vital cellular components, ultimately triggering cell death. On the other hand, cells possess countermeasures, such as autophagy, which degrades and recycles damaged molecules and organelles, restoring homeostasis. Lysosomes and their enzymatic arsenal, including cathepsins, play critical roles in this balance, influencing the cell’s fate toward either apoptosis and other mechanisms of regulated cell death or autophagy. However, the interplay between reactive oxygen species (ROS) and cathepsins in these life-or-death pathways transcends a simple cause-and-effect relationship. These elements directly and indirectly influence each other’s activities, creating a complex web of interactions. This review delves into the inner workings of regulated cell death and autophagy, highlighting the pivotal role of ROS and cathepsins in these pathways and their intricate interplay.
1. Introduction
While the intricate interplay between modulation of biochemical pathways and oxidative stress may not always be characterized by a straightforward cause-and-effect relationship, it undoubtedly represents a crucial juncture in cellular homeostasis and pathology. Many cellular processes are accompanied by the generation of reactive metabolites. Such metabolites include oxidants, which are generally defined as reactive oxygen (ROS), nitrogen (RNS), chlorine (RCS), and sulfur species (RSS). Oxidants can be interconverted into each other spontaneously or via catalytic aid [1,2]; for example, H2O2 (the most abundant physiological oxidant) can be converted into hydroxyl radical ·OH (the most potent physiological oxidant) [3] by interaction with appropriate electron donors, such as Fe2+, or into HOCl via myeloperoxidase activity in the phagosomes [4,5,6]. Major oxidants include hydroxyl radical (·OH), peroxynitrite (ONOO−), hypohalous acids (HOCl, HOBr, and HOSCN−), superoxide anion radical (O2·−), peroxides (ROOH) and peroxide radical (ROO·), reactive carbonyls (RC(O)·), and others [7]. Oxidant action depends on their activation energy, reaction kinetics, site of generation, and concentrations [2,8,9]. Oxidants have traditionally been viewed as harmful byproducts of cellular metabolic processes, such as the leakage of superoxide anion radicals (O2·−) from the mitochondrial electron transport chain into the cytosol [10] or during lipid metabolism via lipoxygenase activity, converting unsaturated fatty acids into oxidized products. However, more recently, it was shown that oxidant generation is tightly controlled [11] and has physiological functions, including cellular and intercellular signaling, fighting infections, and fine-tuned protein function regulation [12,13,14,15,16]. The proper regulation of these functions is determined by the cellular location of the enzymes that generate the ROS. In this scenario, the superoxide anion radical is “deliberately” generated together with H2O2 by NADPH-oxidizing NOX enzymes outside the mitochondria and by NADH-dependent enzymes inside the mitochondria [17]. H2O2 is recognized as a physiological second messenger, orchestrating many processes [7]. Due to its relatively low reactivity, H2O2 can travel long distances until it encounters an H2O2-sensitive site or is enzymatically scavenged [14,18]. Oxidative post-translational modifications in proteins can change the physical–chemical properties of amino acid residues, potentially leading to gain or loss of function and conformational changes [19,20,21]. Regulation of cellular and physiological processes is achieved by direct oxidation of sensitive sites or via targeted enzymatic oxidation of Cys and Met residues with thioredoxins, peroxiredoxins, and other enzymes, acting as redox switches [2,22,23]. In 2020, the visionary review of Lalmanach et al. [24] highlighted the key role of reactive oxygen species in regulating the activity of a particular class of proteases, referred to as cathepsins. Lysosomal cathepsins, a family of proteolytic enzymes mainly residing within the acidic lysosomal compartment, play a pivotal role in the regulation of the cellular processes, including (but not limited to) protein degradation, antigen presentation, and tissue remodeling [25,26], both in physiologic and pathologic conditions. Cathepsins are classified as a function of the presence of specific amino acids in their catalytic site in cysteine, serine, or aspartic proteases, or as a function of the substrate cleavage site in endo-, exo-, and endo/exopeptidase (Table 1).

Table 1.
Cathepsin activity, expression, and gene name. Table implemented from [27].
The reciprocal regulation of ROS and cathepsins can potentially occur every time oxidative stress rises to a particular level of intensity, but the connection between these molecules is particularly evident in the cellular processes of controlled cell death (i.e., apoptosis, necroptosis, ferroptosis, pyroptosis, and NETosis) and autophagy, where ROS play a key role as inducers, while cathepsins represent the effectors of these phenomena. The interdependence of lysosomal cathepsins, oxidative stress, cell death, and autophagy underscores the multifaceted nature of cellular responses to environmental cues and stressors. Understanding the molecular intricacies of these interactions holds promise for unraveling novel therapeutic targets for conditions associated with dysregulated cell death and disrupted cellular quality control mechanisms. In this review, we describe the interactions between ROS and cathepsins, highlighting their reciprocal influence, in particular in apoptosis and autophagy. However, before describing the dynamics of this interplay in detail, some definitions of oxidative stress need to be reported.
2. Oxidative Stress General Definition and Methods of Investigation
Oxidative stress can be defined as the result of the disproportion between generated oxidants and antioxidants, which modulates or disrupts the redox signaling, leading to structural and functional molecular damages [39]. Currently, oxidative stress is classified into oxidative eustress and oxidative distress (Figure 1).
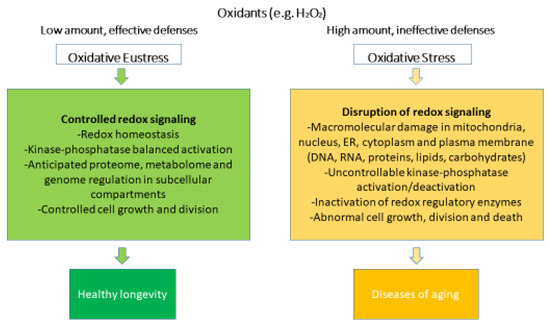
Figure 1.
Oxidative stress and oxidative eustress within redox biology. Rearranged from [40].
Oxidative eustress occurs after physical exercise [41] in response to small stressors, such as mild pharmacological interventions [42], and it is usually considered a physiological and endogenous process [43,44], also known as “mitohormesis” [44]. In oxidative eustress, low (nanomolar for H2O2) concentrations of oxidants interacting with their specific targets maintain the physiological signaling [45].
On the other hand, in oxidative distress, when concentrations of oxidants rise substantially (micromolar and millimolar for H2O2) [4], antioxidant systems are overwhelmed [46], leading to excessive oxidation of biomolecules. Reactive metabolites that are not scavenged by antioxidants may promiscuously react with proteins, DNA, and lipids. Oxidized molecules may show altered or loss of functions depending on the location of the oxidation site [47,48,49]. Oxidative distress is a hallmark of many pathologies, such as ischemia-reperfusion, sepsis, and aging [50].
To cope with the deleterious consequences of oxidative distress, several antioxidant and repairing systems have evolved [51,52,53,54,55,56,57]. Many of them rely on glutathione as a reducing equivalent. Glutathione is a short peptide used as a regenerative source for several antioxidant enzymes, such as peroxiredoxins, thioredoxins, and methionine sulfoxide reductases. Glutathione can also act alone, reducing oxidized Cys residues in a process known as glutathionilation [57]. Once oxidized, glutathione is reduced enzymatically by glutathione reductases, mostly in a NADPH-dependent manner [58]. Hydrogen sulfide H2S and persulfides (RSSH) may act as reductants, maintaining proper redox homeostasis and signaling [59,60,61]. Other small molecules, such as vitamins C and E, tocopherol, taurine, or protein-bound Met and Tyr, can directly scavenge the oxidants [52,53,55,62]. Monocytes secrete catalase, an enzyme that shields extracellular enzymes from oxidative damage [63]. Additionally, macrophages, when added to chemical and biological insults, can release cytotoxic ROS [64], and they can survive thanks to antioxidant enzymes, such as catalase [65].
There are several approaches to study oxidative stress in living (single) cells, dynamically and non-invasively, that implement more traditional tests based on chromatography and mass spectrometry that, on the other hand, require a relatively larger number of cells and their lysis. Small molecules, such as dichlorodihydrofluorescein diacetate, Amplex Red, or luminol, are convenient due to their ease of use and compatibility with primary cells, since they do not require any transfection or genetic manipulation. However, due to the absence of specificity and the potential generation of false-positive results, ROS identification with these dyes generally necessitates the use of orthogonal methods [66,67,68]. Recent genetically encoded redox indicators and sensors allow unambiguous detection of oxidants and oxidative stress effects with subcellular resolution. These molecular tools include sensors to measure markers of oxidative stress, such as a reduced/oxidized glutathione ratio [69], NADP+/NADPH [70], NAD+/NADH [71], redox potential [72], and Met oxidation [73,74,75], as well as sensors specific for oxidants, such as H2O2 [76], HOCl [77], and ONOO− [78], and numerous sensors based on enzymes involved in oxidant-assisted signaling.
Both cathepsins and ROS can mutually influence each other’s activity and effects. For example, oxidative stress can influence the subcellular localization of cathepsins. Under normal conditions, cathepsins are predominantly localized within lysosomes, performing their proteolytic functions. However, oxidative stress can disrupt the integrity of lysosomes, causing their rupture and leading to the release of cathepsins into the cytosol, where they can contribute to pathological processes, such as apoptosis and inflammation [79].
On the other hand, the proteolytic activity of cathepsins can target the enzymes responsible for ROS generation [24]. More information about this phenomenon is provided in the text below.
3. Cathepsin and ROS in Apoptosis
Apoptosis is a vital cellular process, observed in all cell types, that involves distinct intrinsic and extrinsic pathways that converge on the activation of biochemical cascades, leading to cell death. Depending on the pathway, specific signals trigger mitochondrial membrane damage, resulting in the release of cytochrome C and ultimately leading to the activation of effector caspases. Effector caspases, along with mitochondrial proteins, translocate to the nucleus, where they carry out essential processes, such as nuclear protein cleavage, DNA fragmentation, and chromatin condensation. Apoptosis serves as a key mechanism in tissue maintenance, organ development, and immune system balance, ensuring cellular health [80]. However, apoptosis dysregulation can have significant implications for diseases, such as cancer, neurodegenerative disorders, and autoimmune conditions [81,82]. By targeting specific regulators and components of apoptotic pathways, it is possible to modulate cell death, offering potential therapeutic strategies for various disorders [83,84].
Emerging evidence suggests that cathepsin dysregulation can have significant implications in different cellular processes, including apoptosis [85]. In fact, while cathepsins may not play a predominant role in this process, they do contribute to apoptosis regulation by influencing some of its key steps. Cathepsins can act as regulators of apoptosis by affecting the activity of different enzymes. In the cytoplasm, cathepsins can directly cleave specific apoptotic regulators, such as pro-apoptotic and anti-apoptotic proteins. For example, research has shown that neutrophils isolated from cathepsin D-deficient mice undergo spontaneous apoptosis at later times compared to normal cells [86]. During this phenomenon, cathepsin D directly cleaves caspase-8, as demonstrated in both cellular and pure recombinant protein studies. Cathepsin D-mediated cleavage of caspase-8 produces an enzymatically active fragment, also known as initiator caspase, which further activates caspase-3 [86]. Cathepsin-mediated apoptosis can be induced by lysosomotropic agents, such as the 2-amino acid compound Leu-Leu-OMe, inducing the release of these enzymes in the cytoplasm. Upon this treatment, Bid cleavage and degradation of anti-apoptotic proteins, such as Bcl-2, Bcl-xL, Mcl-1, and XIAP, were detected in various cell lines [83]. Studies have shown that lysosomal proteases activate Bid protein in a time-dependent manner [87,88]. In this context, cathepsin B- and L-mediated activation of Bid and degradation of Mcl-1 were observed during Type-1-fimbriated E. coli-induced neutrophil apoptosis [42].
Oxidative stress plays a significant role not only in inducing, but also in regulating apoptosis [89,90]. As a trigger, ROS can induce various DNA and protein modifications, gene expression modulation, and increase mitochondrial membrane permeability [25]. If the damage inflicted by ROS becomes irreparable or overwhelms the cellular repair mechanisms, pro-apoptotic signaling pathways are activated [79,91]. For instance, mitochondrial membrane integrity disruption results in mitochondrial dysfunction and release of pro-apoptotic factors (i.e., cytochrome C). This, in turn, triggers the activation of caspases, culminating in apoptotic cell death [92]. ROS were particularly investigated as apoptotic inducers upon treatment with xenobiotics, such as cadmium, in osteosarcoma cells [93] or methacrylate monomers [94], which can induce apoptosis in dental pulp cells. In all these cases, antioxidant mechanisms or molecules were efficient in protecting the cells from apoptosis.
Interplay between ROS and Cathepsins in Apoptosis
Oxidative stress can influence the subcellular localization of cathepsins by disrupting the integrity of lysosomes and causing their rupture, leading to the release of cathepsins into the cytosol, where they can contribute to apoptosis [87]. As a result of this phenomenon, recent studies have indicated that cathepsins can also translocate in the nucleus [95,96,97]. Interestingly, their activity within the nucleus appears to increase during apoptosis, even though more investigations are necessary to unveil the mechanisms underlying these phenomena [98]. Despite the role of ROS as a lysosome-permeabilizing agent, treatment of murine hepatoma cells with inhibitors of cathepsins L, B, and D did not prevent Bid activation after treatment with N-aspartyl chlorin e6 (NPe6) photosensitizer nanoparticles and subsequent irradiation [87]. In contrast to that described above, these data suggest that other lysosomal proteases might be involved in Bid activation under these specific conditions. On the other hand, a separate investigation observed changes in the expression and activity of cathepsins B and D in the rat pheochromocytoma cell line PC12 when treated with H2O2. Notably, H2O2 exposure increased cathepsin D activity, while cathepsin B activity remained unaffected [99]. During H2O2 and nitric oxide-induced apoptosis, cytoplasmic acidification is a well-established phenomenon. As cathepsins are known to activate at acidic pH within lysosomes, it was hypothesized that these proteases might also become active in the acidified cytoplasm during apoptosis. However, this hypothesis remains untested and requires further investigation [100].
The member of the Bcl-2 family Bax and its polyubiquitinated intermediate were found to be cleaved by cathepsin S during paclitaxel- or hydrogen-peroxide-induced apoptosis in renal cancer cells [101]. The activation of Bid and cleavage of anti-apoptotic proteins can result in mitochondrial outer membrane permeabilization and subsequent apoptotic stages, followed by caspase-3 activation. In this scenario, ROS, especially highly reactive hydroxyl radicals, can directly modify cathepsins’ activity. Specifically, Cys25, constituting the catalytic triad in the active center of cysteine cathepsins, can undergo post-translational modifications under the influence of ROS [24]. This modification can alter the active site of cysteine cathepsins and result in the inactivation of these enzymes [102].
However, moderate levels of oxidative stress can, in fact, activate pro-survival and pro-apoptotic signaling pathways simultaneously, allowing the cells to respond or to adapt to environmental conditions [103,104]. The precise outcome depends on factors such as the extent and duration of oxidative stress, the cellular antioxidant defense system, and the specific context and cell type involved. Cellular defense systems rely on antioxidant enzymes and molecules counteracting the effects of oxidative stress [25]. The disruption of molecular defense expression or function plays a crucial role in determining the fate of the cells, either promoting their survival or triggering apoptosis [105]. In this context, cathepsins have been implicated in regulating oxidative stress by controlling the expression and activity of antioxidant enzymes. A study investigating left ventricular (LV) dysfunction induced by overexpression of cathepsin A in cardiomyocytes revealed a reduction in the activity of the extracellular antioxidant enzyme, superoxide dismutase (EC-SOD), which catalyzes the dismutation of superoxide radicals to H2O2 and oxygen in the extracellular space.
The decrease in EC-SOD in LV tissue in mice resulted in the accumulation of superoxide radicals that induced elevated expression of CTGF, TNF-α, IL-6, IL-10, and IL-2, and generated a high amount of apoptotic cells [106,107]. TNF-α is a cytokine produced by natural killer cells and cytotoxic T lymphocytes that induces various inflammatory and immune responses [108]. Furthermore, it was discovered that the inhibition of cathepsin B by Z-FA.FMK effectively blocked TNF-α/D-galactosamine-induced oxidative damage in the mouse brain. Injection of Z-FA.FMK resulted in decreased levels of lipid peroxidation and increased levels of glutathione. Additionally, the activity of catalase, superoxide dismutase, paraoxonase 1, and glutathione peroxidase increased compared with the TNF-α/D-galactosamine-treated group [109]. Cathepsin activity can also increase ROS production, via mitochondrial dysfunction. Conversely, in S. cervi parasites, inhibiting cathepsin D with E-64 caused a substantial reduction in glutathione levels, as well as glutathione reductase and glutathione-S-transferase activity, accompanied by an increase in NADPH oxidase activity.
This resulted in an elevation of ROS, lipid, and protein peroxidation in the E-64-treated parasites [110]. Additionally, the inhibition of cathepsin K can disrupt the degradation of regulatory-associated protein of mammalian target of rapamycin (Raptor), resulting in heightened mitochondrial ROS levels [111]. To summarize, cathepsins have been found to regulate oxidative stress through their influence on antioxidant enzymes. In various systems, inhibiting specific cathepsins has shown promising results in mitigating oxidative damage and improving antioxidant defense mechanisms.
The relationship between ROS and cathepsins is bidirectional: ROS can promote cathepsin release from lysosomes and affect the activity of cathepsin inhibitors. These interactions significantly impact the apoptotic pathway, ultimately influencing cell survival and death. Understanding the interplay between ROS and cathepsins in apoptosis can provide insights into the molecular mechanisms underlying cell fate determination and may have implications for developing therapeutic strategies targeting these pathways in various diseases.
A schematic of the interplay between ROS and cathepsins during apoptosis is shown in Figure 2.
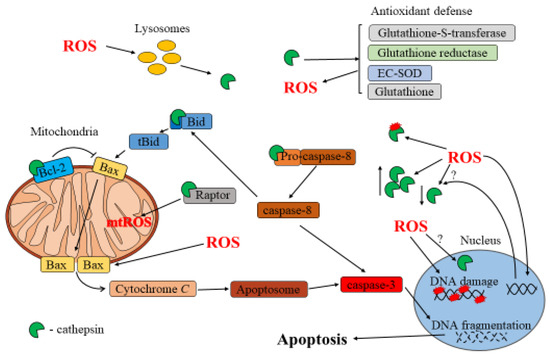
Figure 2.
Interplay between ROS and cathepsins in apoptosis.
4. Other Types of Regulated Cell Death
The interplay between ROS and cathepsins was also highlighted in other forms of regulated cell death, including necroptosis, ferroptosis, pyroptosis, and NETosis.
Necroptosis is a form of programmed cell death resembling necrosis [112]. It was mainly observed during physiologic development or viral infection. Necroptosis pathways are usually associated with biochemical stimuli, including activation of death receptors (TNFR1 and Fas), toll-like receptors (TLR4), and others. When the necroptotic pathway is activated, receptor-interacting serine/threonine kinase 1 (RIPK1) associates with RIPK3 into a complex [113], which activates the mixed-lineage kinase domain-like pseudo-kinase (MLKL). This chain of events leads to post-translational modifications, resulting in the formation of a complex known as the necrosome (RIPK1–RIPK3–MLKL) [114]. The necrosome complex affects cell membrane continuity, eventually resulting in its permeabilization and cellular death.
Ferroptosis is a type of iron-dependent regulated cell death that is characterized by lipid peroxidation, leading to damage of the cell membrane [115]. The cell regulates the amount of iron through a system of transport, which involves transferrin and its receptor TFR for import and ferroportin for export. Cellular iron is transported as a complex with ferritin in its inactive form, Fe3+, predominantly localized in the cytoplasm, mitochondria, and nucleus [116]. The disruption of these iron transport systems and the release of iron from ferritin lead to the accumulation of intracellular iron, triggering ferroptosis. As a result of this process, lipid peroxidation occurs by iron-dependent enzymatic (lipoxygenases) and non-enzymatic (Fenton reactions) processes. These phenomena induce a significant oxidation of polyunsaturated fatty acids (PUFAs) in the cell membrane and organelles [117]. The glutathione peroxidase 4 (GPX4) complex plays a critical role in ferroptosis: its inactivation leads to the accumulation of PUFA-OOH [118]. Glutathione depletion, synthesized with the participation of system xc− (SLC3A2 and SLC7A11) and used in GPX4 complex reactions, can trigger ferroptosis. Blockade of system xc− triggers activation of voltage-dependent anion channel 2 (VDAC2) and VDAC3 on the outer mitochondrial membrane, leading to increased production of ROS [119]. The interplay between iron accumulation, glutathione depletion, ROS production, and increased lipid peroxidation ultimately drives ferroptotic cell death.
Pyroptosis is a type of regulated cell death observed during viral or bacterial infections, tissue damage, or metabolic disturbances, involving the activation of inflammasomes and consequent activation of pro-inflammatory caspases (such as caspase-1, 4, 5, and 11) [120]. These enzymes cleave interleukin-1 family members (i.e., pro-IL-1β and pro-IL-18) into their mature forms and gasdermin D (GSDMD) into two products: GSDMD-N and GSDMD-C. GSDMD-N can translocate to the inner layer of the cell membrane, binding cell phospholipids and affecting the continuity of this structure by generating pores. These pores allow the release of IL-1β and IL-18 from the cell, initiating an immune response. The damage to the cell membrane eventually leads to its rupture, which is a hallmark of pyroptosis.
Finally, NETosis is a specific form of cell death characteristic of neutrophils and other leukocytes (i.e., eosinophils, mast cells, and macrophages) [121,122,123] and is associated with release of neutrophil extracellular trap (NET) from the cell. Pathogens or external stimuli can trigger NADPH oxidase activation, inducing ROS production [124,125]. The azurophilic granules contain antimicrobial peptides, neutrophil elastase (NE), cathepsin G, and myeloperoxidase (MPO), which are released into the cell cytoplasm in response to ROS. NE moves into the nucleus, causing cleavage of nuclear proteins. Additionally, peptidyl arginine deiminase 4 (PAD4) leads to DNA de-condensation through histone citrullination. The decondensed chromatin, along with histones and proteases, is released into the cytoplasm and, subsequently, into the extracellular matrix, resulting in the formation of a neutrophil extracellular trap.
In all these kinds of cellular death, the interplay between ROS and cathepsin is very similar (Figure 3).
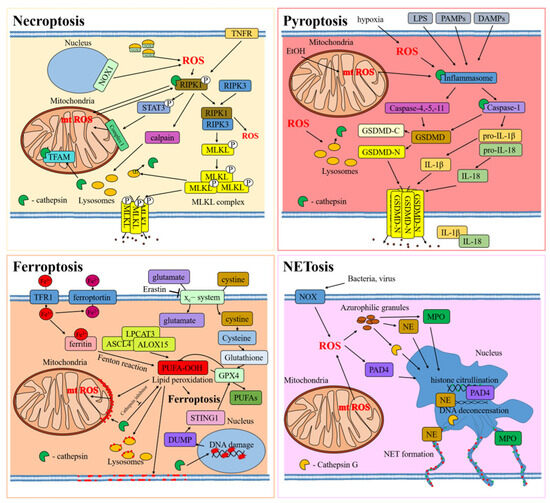
Figure 3.
Interplay between ROS and cathepsins in types of regulated cell death. The cellular proteins are released from the cell through protein pores in the cell membrane, which are indicated by black dots. Red dots indicate lipid peroxidation in cell membranes.
ROS is a constant characteristic of necroptosis, ferroptosis, pyroptosis, and NETosis. In necroptosis, ROS can be produced through the activation of several pathways, including mitochondrial damage, that further increase ROS production [126]. For example, the transcriptional factor STAT3 is phosphorylated by the RIPK1 kinase, causing its translocation into the mitochondria, binding with respiratory chain complex I and increasing ROS levels [127]. The surge in mitochondrial ROS can, in turn, generate post-translational modifications in RIPK1, favoring the formation of the necrosome [128,129]. This phenomenon can further increase ROS levels [130], as observed in macrophages upon TNF-alpha activation [131]. Furthermore, in certain instances, ROS accumulation is directly linked to NADPH oxidase 1 activity, which can favor RIPK1 activation [132].
During ferroptosis, ROS and oxidation products can drastically increase, as observed during Fenton reactions, where H2O2 is converted to a hydroxyl radical (·OH). On the other hand, the enzymatic peroxidation of PUFAs, including phosphatidylethanolamine, occurs through a series of enzymatic reactions [133,134]. In these processes, iron ions play a key role as catalyzers. In this enzymatic balance, the GPX4 can mitigate ferroptosis activation and accumulation of oxidized PUFA. In this context, it was shown that erastin, a major activator of ferroptosis, can affect GPX4 activity by inhibiting glutathione formation, with consequent cell death by ferroptosis [119]. Similar to necroptosis, during ferroptosis, mitochondrial membrane undergo lipid oxidation, resulting in mitochondrial membrane damage [135,136] and a further increase in ROS production.
Additionally, pyroptosis is associated with a significant increase in ROS levels that can eventually affect mitochondrial biology, fueling oxidative chain reactions. In cases of bacterial infection, cells exhibit increased production of ROS, resulting in lysosomal membrane permeabilization.
In the process of NETosis, ROS are a key element in the formation of extracellular traps [137]. These ROS are essential in initiating the sequences that result in the release of NET. Studies have shown that the development of NET during fungal infection relies on NADPH oxidase, which generates ROS [138]. Additionally, ROS are responsible for triggering the release of azurophilic granules [139].
The oxidative stress generated during these kinds of cell death does not impact only mitochondrial biology but can also damage other organelles, including the lysosomes. An increase of ROS-mediated lysosome permeability was observed in necroptosis [140], ferroptosis [136,141], and pyroptosis during bacterial infection (Table 2) [142,143]. Cathepsin B was shown to play an important role in all these processes. In necroptosis, the necroptotic p-MLKL complex can be activated under ROS and is sequestered into lysosomes. This prompts the release of cathepsin B and further activates the pyroptotic cell death pathway [144]. In other works, together with cathepsin B, the release of cathepsins L and D was also shown [127,140,145,146,147,148,149]. The resultant release of lysosomal proteinases, including cathepsins, can further lead to the permeabilization of the mitochondrial membrane, increase of ROS, and ultimately, cell death [149]. In a different study, it was shown that the degradation of mitochondrial transcriptional factor A (TFAM) by lysosomal cathepsin B results in increased intracellular ROS levels that can eventually activate necroptosis [147], while in macrophages treated with LPS/zVAD, cathepsins B and L directly cleaved and activated RIPK1 [150].
During erastin-induced ferroptosis in PANC1 and MIAPaCa2 cells, lysosomes are destroyed and lysosomal proteases are released [141], and the activity and expression of cathepsins L and B were also found to be increased in glutamate-induced HT22 cells [136]. After being released into the cytoplasm of the cells, cathepsin B is transported to the nucleus, where it mediates DNA damage and releases nuclear DAMP into the cytoplasm. This, in turn, activates the STING1 pathway, leading to autophagy-dependent ferroptosis. The process results in the degradation of the antioxidant protein GPX4 and further ferroptosis [141]. In another study, inhibition of cathepsin B with CA-074-me reduced lipid oxidation, mitochondrial dysfunction, and ferroptotic cell death in spinal cord cells after spinal cord injury [151].
Following release from lysosomes during pyroptosis, which can be induced by ROS activity, cathepsin B activates formation of inflammasomes, such as NLRP3 [142,143,144]. Cathepsin B is necessary for inflammasome activation through its interaction with NLRP3 [152,153]. Experiments on mice that were fed either a special diet or acid showed an increase in the level of reactive oxygen species. This led to the release of cathepsin B, which activated the NLRP3 inflammasome [154].
Finally, during NETosis, cathepsin G, released from azurophilic granules, contributes to NET formation. Additionally, during this process, cathepsin G induces activation of cytokines IL-1α and IL-36 in PMA-treated neutrophils [155,156].
In conclusion, in these pathways, a vicious cycle between ROS and cathepsins occurs, eventually fueling cell death pathways.
Table 2.
The relationship between cathepsin and reactive oxygen species in the pathways of various forms of cell death.
Table 2.
The relationship between cathepsin and reactive oxygen species in the pathways of various forms of cell death.
| Cell Death | Cell Death Inducer | Cathepsin Assessment | ROS Assessment | Cells and Tissues | Trigger of the Interplay | Ref. |
|---|---|---|---|---|---|---|
| Necroptosis | Ischemic condition | Cathepsin release from lysosome undergoes lysosome membrane permeabilization. | Potential increase | Ischemic flaps | Cathepsins | [145] |
| Necroptosis | LPS + zVAD | CtsB and CtsL cleave RIPK1 protein. Cathepsin inhibition induces cell death. | Potential increase | Macrophages | Cathepsins | [150] |
| Necroptosis | Acute pancreatitis | Degrades TFAM. | Increase | Pancreatic acinar cells | Cathepsins | [147] |
| Necroptosis | TNF | Cathepsin L activation. | Increase | Mouse fibrosarcoma cells | ROS | [148] |
| Necroptosis | Tag7-Hsp70 | Cathepsin B and D leakage from lysosomes. | Increase | Mouse fibroblast | Cathepsins | [149] |
| Necroptosis | FasL | Cathepsin B and D leakage from lysosomes. | Increase | Lymphoblast | Cathepsins | [127] |
| Necroptosis | Sodium sulfite | Cathepsin B and D leakage from lysosomes. | Increase | Mouse liver cells | ROS | [144] |
| Ferroptosis | Spinal cord injury | Increased Cathepsin B expression. CtsB inhibition decreases lipid peroxidation and mitochondrial disfunction. | Lipid peroxidation increase | Spinal cord | Cathepsins | [151] |
| Ferroptosis | Erastin | CtsB leakage from lysosomes. CtsB induces DNA damage. | Degradation of antioxidant protein GPX4 | Pancreatic carcinoma cell | Cathepsins | [141] |
| Ferroptosis | Glutamate | CtsB is released from lysosomes, increases expression and activity, and cleaves H3. | Lipid peroxidation increase | Mouse hippocampal neuronal cell line | Cathepsins | [136] |
| Pyroptosis | T. gondii infection | CtsB release from lysosomes and its activation. | Increase | Human placental trophoblast, amniotic cells | Unknown | [142] |
| Pyroptosis | High-fat diet, palmitic acid | CtsB release from lysosomes and further NLRP3 activation. | Increase | C57BL/6J mice and AML12 cells | ROS | [154] |
| Pyroptosis | B. cereus strain, H2 | Lysosomal damage and cathepsin release. | Increase | Macrophages | Unknown | [143] |
| Pyroptosis | All-trans retinal | Lysosomal damage and cathepsin release. | Increase | Spontaneously arising retinal pigment epithelia cells | Unknown | [153] |
| Pyroptosis | Sodium sulfite | Cathepsin release and NLRP3 activation. | Increase | Mouse liver cells | ROS | [144] |
| NETosis | PMA | Cathepsin contributes to NET formation. | Increase | Neutrophils | ROS | [139,156] |
LPS: lipopolysaccharides; RIPK1: receptor-interacting serine/threonine kinase 1; CtsB: cathepsin B; CtsL: cathepsin L; FasL: Fas ligand; TFAM: mitochondrial transcription factor A; Hsp70: heat shock protein 70; ROS: reactive oxygen species; TNF: tumor necrosis factor; GPX4: glutathione peroxidase 4; NLRP3: NLR family pyrin domain containing 3; NET: neutrophil extracellular trap.
5. Cathepsin and ROS in Autophagy
Autophagy is the cellular process responsible for degrading and recycling cellular components through the formation of autophagosomes, playing a crucial role in maintaining cellular homeostasis and typical for eukaryotic cells [157,158].
This process can be stimulated by different kinds of cellular stress, such as starvation, hypoxia, oxidative stress, DNA damage, and intracellular pathogens [159,160,161], even though a certain degree of genetic correlation between autophagy and senescence was demonstrated [162]. Cellular senescence is another pathway of stress response [163,164,165], and this correlation has been the subject of numerous studies [166,167], demonstrating the activation of similar signaling pathways between these processes [168,169,170]. Recent studies have revealed a complex interplay between autophagy and oxidative stress, where autophagy serves as a key mechanism to mitigate cellular damage by selectively removing damaged organelles and protein aggregates generated under oxidative stress conditions [171]. In response to lipid and protein oxidation, the redox-regulated protease ATG4 cleaves Atg8/microtubule-associated protein light chain 3 (LC3), inducing its binding to phosphatidylethanolamine (PE) and its localization on the autophagosomal membrane (Figure 4).
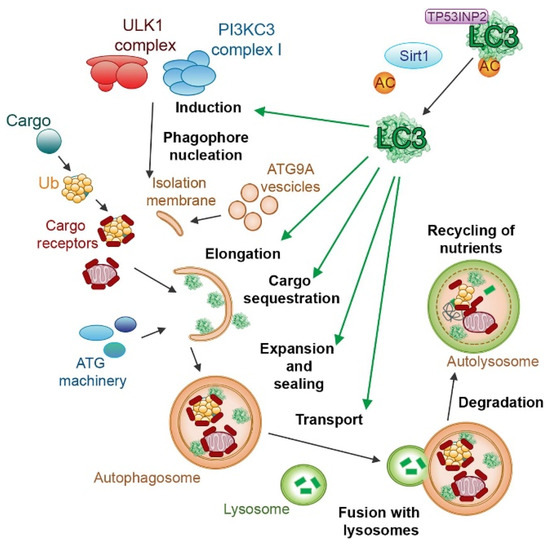
Figure 4.
The role of LC3 proteins in selective autophagy. The figure illustrates the involvement of the LC3 subfamily of ATG8 proteins in different steps of selective autophagy. Reproduced with permission from [172].
ATG5, ATG7, and other modifiers also play a role in the maturation of autophagosomes and in the formation of autolysosomes.
Autophagy regulation is orchestrated by a complex interplay between three main signaling pathways. The class I phosphatidylinositol 3-kinase (PI3K) pathway is activated by growth factors, promoting cell growth, while the class III PI3K pathway responds to the number of amino acids within the cell. When amino acids are scarce, this pathway triggers autophagy to recycle cellular components for survival. Similarly, the LKB1/AMPK pathway primarily reacts to cellular ATP levels. When ATP is low, it activates autophagy to generate energy by breaking down cellular components. mTOR kinase acts as a central checkpoint for autophagy, functioning as its main repressor in these pathways when the cell has sufficient resources. Another key player in autophagy is the extracellular signal-regulated kinase (ERK), which can induce cytoplasm vacuolization [173] and induction of LC3, beclin-1, and p53 phosphorylation [174], all of which are involved in regulating this process.
Autophagy can be induced as a protective response against oxidative stress, promoting cell survival through the removal of dysfunctional mitochondria, which are major sources of ROS generation [175,176,177]. Autophagy dysregulation was linked to various diseases, including neurodegenerative disorders and cancer, emphasizing the need to elucidate the molecular mechanisms driving this process for effective therapeutic strategies and understanding pathologic mechanisms. Lysosomes play a pivotal role in autophagy, serving as the final destination for the degradation of sequestered cytoplasmic components delivered through autophagosomes [178,179]. In this scenario, cathepsins are the key mediators of the autophagic flux by cleaving and breaking down the cargo within lysosomes, ensuring the efficient degradation of proteins, organelles, and other cellular structures [180,181]. Stringent regulation of cathepsin expression and activity ensures efficient lysosomal degradation, preventing uncontrolled proteolysis and promoting controlled autophagic cargo breakdown. Cathepsins also contribute to autophagy modulation by participating in the regulation of autophagosome–lysosome fusion, a crucial factor that influences the efficiency of this process [182,183]. For example, a mutation in the lysosomal factor Saposin C, that favors the activity of acid β-gluocosidase, can induce aberrant autophagy by inducing accumulation of autophagosomes and decrease cathepsin B and D expression and activity [182]. Furthermore, studies have shown that inhibiting cathepsin S can directly induce autophagy. This activation occurs through the phosphorylation of the epidermal growth factor receptor (EGFR), leading to the activation of the ERK/MAPK signaling pathway, which is known to regulate autophagy [184].
Interplay of ROS and Cathepsins in Autophagy
Extensive research has investigated alterations in the proteolytic activity during autophagy, but the underlying mechanisms orchestrating the autophagic processes in response to oxidative stress remain elusive. On the other hand, substantial attention has been devoted to the intricate relationship between autophagy and apoptosis [185,186]. While autophagy typically exerts a protective and anti-apoptotic function, it can trigger programmed cell death under conditions of extreme external stress [187], and oxidative stress and cathepsins cover a key role in this fine balance. It was demonstrated that cathepsin D can enhance the survival of HeLa cells under oxidative conditions, challenging the established link between apoptosis and autophagy. The authors hypothesized that elevated cathepsin D expression activates autophagy, and this phenomenon was substantiated by increased efficiency in autophagic vacuole formation and the autophagy marker LC3-II (Figure 5).
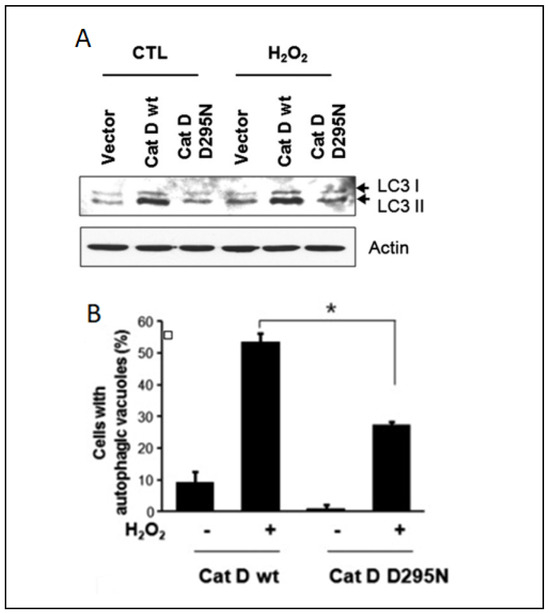
Figure 5.
Correlation between cathepsin D expression and autophagy in HeLa cells: (A) Western blotting analysis of HeLa cells expressing wildtype cathepsin D and cathepsin D D295N after treatment with 1 mM of H2O2 for 24 h. (B) Percentage of HeLa cells with autophagy vacuoles after H2O2 treatment. Data were obtained from confocal images of cells transfected with GFP-LC3 plasmid. +/− means HeLa cells with or without H2O2 treatment, * indicates the significance. Reproduced with permission from [188].
Conversely to the studies reported in the apoptosis section, these findings indicate that cathepsin D can act as an anti-apoptotic mediator by inducing autophagy during cellular oxidative stress. A similar anti-apoptotic effect of cathepsin D was also observed in colorectal cancer cells [189]. Another investigation revealing a connection between oxidative stress and cathepsin L and B activity indicated these proteases as pro-autophagic and pro-apoptotic enzymes, respectively [190]. It was observed that oxidative stress induced by auranofin-mediated inhibition of thioredoxin reductase led to a significant increase in cathepsin B activity, while the protein levels of this enzyme remained relatively unchanged. Conversely, cathepsin L exhibited an opposite pattern, with a substantial increase in protein levels not accompanied by a corresponding change in activity. The authors demonstrated, via the thiol-trapping method, that the oxidative stress disrupts cathepsin L processing, impairing its pro-autophagic function. However, no discernible impact of oxidative stress on cathepsin B was identified. The authors proposed the following mechanism to account for these observations: protective autophagy prevents oxidative stress by inhibiting cathepsin L processing, while apoptosis is induced by an increased lysosomal membrane permeability that favors cathepsin B release into the cytoplasm, which can eventually induce the activation of pro-apoptotic enzymes. Inhibition of cathepsin B under these conditions suppresses apoptosis, enhancing cell viability. The interplay of this mechanism is shown in Figure 6.
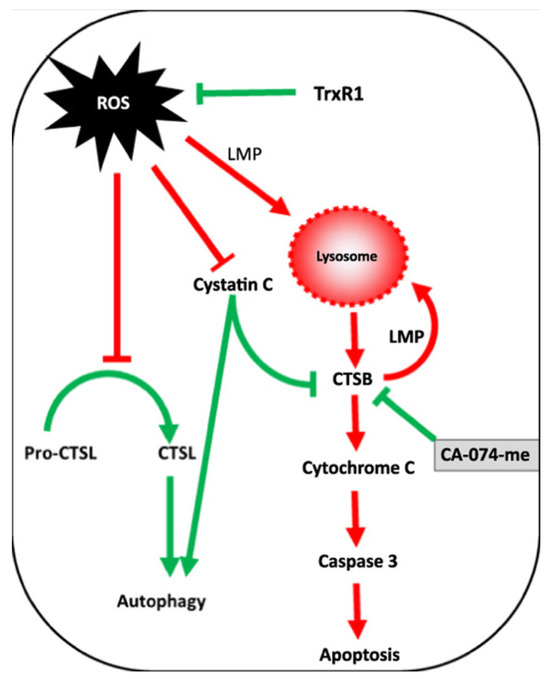
Figure 6.
Correlation between ROS, cathepsin B (CTSB), and cathepsin L (CTSL) in regulating autophagy and apoptosis as a result of thioredoxin reductase inhibition. Reproduced with permission from [190].
A recent study has shed light on the interplay between cathepsin S and autophagy. This study demonstrated that the autophagic process is accompanied by an increase in ROS; in fact, by inhibiting the expression of ATG-related proteins via gene silencing or pharmacological agents, ROS levels decreased. On the other hand, the inhibition of cathepsin S induced ROS production and autophagy, as well as DNA damage. The authors indicated that the enzyme xanthine oxidase is at the basis of the working mechanism regulating the balance between autophagy and cathepsin S activity. However, while xanthine oxidase is a key player in ROS generation during autophagy, direct proof of the interaction between this enzyme and cathepsin S was not reported. Additionally, the relationship between cathepsin E and oxidative stress during autophagy was analyzed in mice macrophages [191]. In this context, cathepsin E-deficient murine macrophages displayed an aberrant autophagic behavior, characterized by heightened levels of autophagy markers, such as LC3 and phosphorylated p62, which can cause autophagy [192]. Cathepsin E deficiency also induced perturbations in signaling pathways associated with autophagy, impacting mTOR and ERK signaling. Furthermore, cathepsin E deficiency hindered the fusion of autophagosomes with lysosomes via inhibition of LC3 transport to the vesicular compartment. The macrophages exhibited an increase in ROS levels accompanied by the activation of oxidized peroxiredoxin-6 and a concomitant reduction of glutathione. Hence, it can be postulated that cathepsin E can exert a substantial influence on oxidative stress, operating through a NADPH oxidase-independent pathway. The aforementioned studies collectively underscore a robust correlation between oxidative stress, autophagy, and lysosomal cathepsins. Targeting cathepsins could thus be contemplated as a viable strategy for the manipulation of autophagy, early ROS generation, and cell death, potentially offering therapeutic avenues for a spectrum of diseases.
6. Conclusions
ROS can have a destructive effect on cellular structures and initiate free radical oxidation of nucleic acids, lipids, and proteins, which underlie the pathogenesis of many diseases. These phenomena usually activate proteolytic processes that can determine cell life or death in the contexts of autophagy and cell death, respectively. Cathepsins can regulate these phenomena as effectors or indirectly, favoring the development of these processes. In this review, we highlighted different mechanisms in which cathepsins regulate apoptosis and other kinds of cell death and autophagy induced by ROS. However, the interplay between ROS and cathepsins is significantly more complex than a simple cause–effect relationship since they can directly and reciprocally affect each other’s function and activity. For this reason, further investigations are needed to understand the fine regulation of the proteolytic machinery during oxidative stress and the contribution of cathepsin in degrading antioxidant enzymes. These studies might reveal new targets for the treatment of different diseases since ROS is a common element in many pathological conditions. For this reason, parallel efforts should be performed in developing more sensors to precisely understand the oxidant origin, conversion, and effects on different molecules and, in particular, on cathepsins.
Author Contributions
Conceptualization, M.V.V. and A.S.F.; writing—original draft preparation, M.V.V., A.S.F., E.P.K., N.A.K. and A.P.; writing—review and editing, M.V.V., A.S.F. and A.P.; visualization, A.S.F. and E.P.K.; project administration, A.P. and A.A.Z.J.; funding acquisition, A.P. and A.A.Z.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation (grant number 21-75-30020).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ferrer-Sueta, G.; Campolo, N.; Trujillo, M.; Bartesaghi, S.; Carballal, S.N.; Romero, N.; Alvarez, B.; Radi, R. Biochemistry of peroxynitrite and protein tyrosine nitration. Chem. Rev. 2018, 118, 1338–1408. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.L.; Davies, M.J. Detection, identification, and quantification of oxidative protein modifications. J. Biol. Chem. 2019, 294, 19683–19708. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J. Protein oxidation and peroxidation. Biochem. J. 2016, 473, 805–825. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase: A key regulator of neutrophil oxidant production. Redox Rep. 1997, 3, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Armstrong, D.A.; Huie, R.E.; Koppenol, W.H.; Lymar, S.V.; Merényi, G.; Neta, P.; Ruscic, B.; Stanbury, D.M.; Steenken, S.; Wardman, P. Standard electrode potentials involving radicals in aqueous solution: Inorganic radicals (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1139–1150. [Google Scholar] [CrossRef]
- Kaludercic, N.; Deshwal, S.; Di Lisa, F. Reactive oxygen species and redox compartmentalization. Front. Physiol. 2014, 5, 285. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Cremers, C.M.; Jakob, U. Oxidant sensing by reversible disulfide bond formation. J. Biol. Chem. 2013, 288, 26489–26496. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.J.; Han, Z.Q.; Li, Z.Y. Modulating protein activity and cellular function by methionine residue oxidation. Amino Acids 2012, 43, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Jacobs, K.; Haucke, E.; Santos, A.N.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Nordzieke, D.E.; Medraño-Fernandez, I. The plasma membrane: A platform for intra-and intercellular redox signaling. Antioxidants 2018, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Hider, R. Iron and redox cycling. Do’s and don’ts. Free. Radic. Biol. Med. 2019, 133, 3–10. [Google Scholar] [CrossRef]
- Erickson, J.R.; Mei-ling, A.J.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef]
- Kassmann, M.; Hansel, A.; Leipold, E.; Birkenbeil, J.; Lu, S.-Q.; Hoshi, T.; Heinemann, S.H. Oxidation of multiple methionine residues impairs rapid sodium channel inactivation. Pflug. Arch.-Eur. J. Physiol. 2008, 456, 1085–1095. [Google Scholar] [CrossRef][Green Version]
- Lewis, A.K.; Dunleavy, K.M.; Senkow, T.L.; Her, C.; Horn, B.T.; Jersett, M.A.; Mahling, R.; McCarthy, M.R.; Perell, G.T.; Valley, C.C. Oxidation increases the strength of the methionine-aromatic interaction. Nat. Chem. Biol. 2016, 12, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Hung, R.-J.; Pak, C.W.; Terman, J.R. Direct redox regulation of F-actin assembly and disassembly by Mical. Science 2011, 334, 1710–1713. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.; Mokhosoev, I.; Feldman, N.; Lutsenko, S. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modifications. Free. Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef] [PubMed]
- Lalmanach, G.; Saidi, A.; Bigot, P.; Chazeirat, T.; Lecaille, F.; Wartenberg, M. Regulation of the proteolytic activity of cysteine cathepsins by oxidants. Int. J. Mol. Sci. 2020, 21, 1944. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Petushkova, A.I.; Zamyatnin, A.A., Jr. Redox-mediated post-translational modifications of proteolytic enzymes and their role in protease functioning. Biomolecules 2020, 10, 650. [Google Scholar] [CrossRef] [PubMed]
- Vidak, E.; Javoršek, U.; Vizovišek, M.; Turk, B. Cysteine cathepsins and their extracellular roles: Shaping the microenvironment. Cells 2019, 8, 264. [Google Scholar] [CrossRef] [PubMed]
- Reich, M.; Spindler, K.-D.; Burret, M.; Kalbacher, H.; Boehm, B.O.; Burster, T. Cathepsin A is expressed in primary human antigen-presenting cells. Immunol. Lett. 2010, 128, 143–147. [Google Scholar] [CrossRef]
- Siming, G.; Honglin, Z.; Xiaoxia, Z.; Hui, L. Cathepsin G and its role in inflammation and autoimmune diseases. Arch. Rheumatol. 2018, 33, 498. [Google Scholar]
- Kolesova, E.P.; Egorova, V.S.; Syrocheva, A.O.; Frolova, A.S.; Kostyushev, D.; Kostyusheva, A.; Brezgin, S.; Trushina, D.B.; Fatkhutdinova, L.; Zyuzin, M. Proteolytic Resistance Determines Albumin Nanoparticle Drug Delivery Properties and Increases Cathepsin B, D, and G Expression. Int. J. Mol. Sci. 2023, 24, 10245. [Google Scholar] [CrossRef]
- Drake, F.H.; Dodds, R.A.; James, I.E.; Connor, J.R.; Debouck, C.; Richardson, S.; Lee-Rykaczewski, E.; Coleman, L.; Rieman, D.; Barthlow, R. Cathepsin K, but Not Cathepsins B, L, or S, is abundantly expressed in human osteoclasts (∗). J. Biol. Chem. 1996, 271, 12511–12516. [Google Scholar] [CrossRef] [PubMed]
- Riese, R.J.; Mitchell, R.N.; Villadangos, J.A.; Shi, G.-P.; Palmer, J.T.; Karp, E.R.; De Sanctis, G.T.; Ploegh, H.L.; Chapman, H.A. Cathepsin S activity regulates antigen presentation and immunity. J. Clin. Investig. 1998, 101, 2351–2363. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.Y.; Brissette, W.H.; Lira, P.D.; Griffiths, R.J.; Petrushova, N.; Stock, J.; McNeish, J.D.; Eastman, S.E.; Howard, E.D.; Clarke, S.R. Impaired invariant chain degradation and antigen presentation and diminished collagen-induced arthritis in cathepsin S null mice. Immunity 1999, 10, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Brömme, D.; Li, Z.; Barnes, M.; Mehler, E. Human cathepsin V functional expression, tissue distribution, electrostatic surface potential, enzymatic characterization, and chromosomal localization. Biochemistry 1999, 38, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.-P.; Villadangos, J.A.; Dranoff, G.; Small, C.; Gu, L.; Haley, K.J.; Riese, R.; Ploegh, H.L.; Chapman, H.A. Cathepsin S required for normal MHC class II peptide loading and germinal center development. Immunity 1999, 10, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Wex, T.; Bühling, F.; Wex, H.; Günther, D.; Malfertheiner, P.; Weber, E.; Brömme, D. Human cathepsin W, a cysteine protease predominantly expressed in NK cells, is mainly localized in the endoplasmic reticulum. J. Immunol. 2001, 167, 2172–2178. [Google Scholar] [CrossRef] [PubMed]
- Fusek, M.; Mares, M.; Vetvicka, V. Cathepsin D. In Handbook of Proteolytic Enzymes; Elsevier: Amsterdam, The Netherlands, 2013; pp. 54–63. [Google Scholar]
- Pontious, C.; Kaul, S.; Hong, M.; Hart, P.A.; Krishna, S.G.; Lara, L.F.; Conwell, D.L.; Cruz-Monserrate, Z. Cathepsin E expression and activity: Role in the detection and treatment of pancreatic cancer. Pancreatology 2019, 19, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.-M.; Jones, D.P. Redox theory of aging: Implications for health and disease. Clin. Sci. 2017, 131, 1669–1688. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef]
- Blomgran, R.; Zheng, L.; Stendahl, O. Cathepsin-cleaved Bid promotes apoptosis in human neutrophils via oxidative stress-induced lysosomal membrane permeabilization. J. Leucoc. Biol. 2007, 81, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Bazopoulou, D.; Knoefler, D.; Zheng, Y.; Ulrich, K.; Oleson, B.J.; Xie, L.; Kim, M.; Kaufmann, A.; Lee, Y.-T.; Dou, Y. Developmental ROS individualizes organismal stress resistance and lifespan. Nature 2019, 576, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose-Response 2014, 12, 288–341. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative eustress: On constant alert for redox homeostasis. Redox Biol. 2021, 41, 101867. [Google Scholar] [CrossRef] [PubMed]
- Degrossoli, A.; Müller, A.; Xie, K.; Schneider, J.F.; Bader, V.; Winklhofer, K.F.; Meyer, A.J.; Leichert, L.I. Neutrophil-generated HOCl leads to non-specific thiol oxidation in phagocytized bacteria. Elife 2018, 7, e32288. [Google Scholar] [CrossRef] [PubMed]
- Bettinger, J.Q.; Welle, K.A.; Hryhorenko, J.R.; Ghaemmaghami, S. Quantitative analysis of in vivo methionine oxidation of the human proteome. J. Proteome Res. 2019, 19, 624–633. [Google Scholar] [CrossRef]
- Gęgotek, A.; Skrzydlewska, E. Biological effect of protein modifications by lipid peroxidation products. Chem. Phys. Lipids 2019, 221, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J. A quantitative tissue-specific landscape of protein redox regulation during aging. Cell 2020, 180, 968–983.e924. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Gray, H.B.; Winkler, J.R. Hole hopping through tyrosine/tryptophan chains protects proteins from oxidative damage. Proc. Natl. Acad. Sci. USA 2015, 112, 10920–10925. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Levine, R.L. Methionine in proteins defends against oxidative stress. FASEB J. 2009, 23, 464. [Google Scholar] [CrossRef] [PubMed]
- Moosmann, B.; Schindeldecker, M.; Hajieva, P. Cysteine, glutathione and a new genetic code: Biochemical adaptations of the primordial cells that spread into open water and survived biospheric oxygenation. Biol. Chem. 2020, 401, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Polizzi, N.F.; Migliore, A.; Therien, M.J.; Beratan, D.N. Defusing redox bombs? Proc. Natl. Acad. Sci. USA 2015, 112, 10821–10822. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, K.; Jakob, U. The role of thiols in antioxidant systems. Free. Radic. Biol. Med. 2019, 140, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta-Gen. Subj. 2008, 1780, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef]
- Ono, K.; Akaike, T.; Sawa, T.; Kumagai, Y.; Wink, D.A.; Tantillo, D.J.; Hobbs, A.J.; Nagy, P.; Xian, M.; Lin, J. Redox chemistry and chemical biology of H2S, hydropersulfides, and derived species: Implications of their possible biological activity and utility. Free. Radic. Biol. Med. 2014, 77, 82–94. [Google Scholar] [CrossRef]
- Wu, Z.; Khodade, V.S.; Chauvin, J.-P.R.; Rodriguez, D.; Toscano, J.P.; Pratt, D.A. Hydropersulfides inhibit lipid peroxidation and protect cells from ferroptosis. J. Am. Chem. Soc. 2022, 144, 15825–15837. [Google Scholar] [CrossRef]
- Levine, R.L.; Berlett, B.S.; Moskovitz, J.; Mosoni, L.; Stadtman, E.R. Methionine residues may protect proteins from critical oxidative damage. Mech. Ageing Dev. 1999, 107, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Hervé-Grépinet, V.; Veillard, F.; Godat, E.; Heuzé-Vourc’h, N.; Lecaille, F.; Lalmanach, G. Extracellular catalase activity protects cysteine cathepsins from inactivation by hydrogen peroxide. FEBS Lett. 2008, 582, 1307–1312. [Google Scholar] [CrossRef] [PubMed]
- Albina, J.E.; Cui, S.; Mateo, R.B.; Reichner, J.S. Nitric oxide-mediated apoptosis in murine peritoneal macrophages. J. Immunol. 1993, 150, 5080–5085. [Google Scholar] [CrossRef]
- Sibille, Y.; Reynolds, H.Y. Macrophages and Polymorphonuclear neutrophils in lung defense and Injury1-2. Am. Rev. Respir. Dis. 1990, 141, 471–501. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Lambeth, J.D.; Kalyanaraman, B. On the use of L-012, a luminol-based chemiluminescent probe, for detecting superoxide and identifying inhibitors of NADPH oxidase: A reevaluation. Free. Radic. Biol. Med. 2013, 65, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Wang, Z.; Zhou, J.; Zhu, M.; Liu, J.; James, T.D. Recent progress in the development of fluorescent probes for imaging pathological oxidative stress. Chem. Soc. Rev. 2023, 52, 3873–3926. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Augusto, O.; Brigelius-Flohe, R.; Dennery, P.A.; Kalyanaraman, B.; Ischiropoulos, H.; Mann, G.E.; Radi, R.; Roberts, L.J., II; Vina, J. Even free radicals should follow some rules: A guide to free radical research terminology and methodology. Free Radic. Biol. Med. 2015, 78, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Gutscher, M.; Pauleau, A.-L.; Marty, L.; Brach, T.; Wabnitz, G.H.; Samstag, Y.; Meyer, A.J.; Dick, T.P. Real-time imaging of the intracellular glutathione redox potential. Nat. Methods 2008, 5, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Zhao, Y.; Chu, H.; Wang, A.; Zhu, J.; Chen, X.; Zou, Y.; Shi, M.; Liu, R.; Su, N. Genetically encoded fluorescent sensors reveal dynamic regulation of NADPH metabolism. Nat. Methods 2017, 14, 720–728. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, Q.; Cheng, F.; Su, N.; Wang, A.; Zou, Y.; Hu, H.; Chen, X.; Zhou, H.-M.; Huang, X. SoNar, a highly responsive NAD+/NADH sensor, allows high-throughput metabolic screening of anti-tumor agents. Cell Metab. 2015, 21, 777–789. [Google Scholar] [CrossRef]
- Hanson, G.T.; Aggeler, R.; Oglesbee, D.; Cannon, M.; Capaldi, R.A.; Tsien, R.Y.; Remington, S.J. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 2004, 279, 13044–13053. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W.; Roh, Y.J.; Kim, S.; Lee, H.M.; Kim, M.; Shin, D.; Park, J.H.; Cho, Y.; Park, H.H.; Ok, Y.S. Development of a novel fluorescent biosensor for dynamic monitoring of metabolic methionine redox status in cells and tissues. Biosens. Bioelectron. 2021, 178, 113031. [Google Scholar] [CrossRef]
- Kuldyushev, N.; Schönherr, R.; Coburger, I.; Ahmed, M.; Hussein, R.A.; Wiesel, E.; Godbole, A.; Pfirrmann, T.; Hoshi, T.; Heinemann, S.H. A GFP-based ratiometric sensor for cellular methionine oxidation. Talanta 2022, 243, 123332. [Google Scholar] [CrossRef]
- Tarrago, L.; Péterfi, Z.; Lee, B.C.; Michel, T.; Gladyshev, V.N. Monitoring methionine sulfoxide with stereospecific mechanism-based fluorescent sensors. Nat. Chem. Biol. 2015, 11, 332–338. [Google Scholar] [CrossRef]
- Smolyarova, D.D.; Podgorny, O.V.; Bilan, D.S.; Belousov, V.V. A guide to genetically encoded tools for the study of H2O2. FEBS J. 2022, 289, 5382–5395. [Google Scholar] [CrossRef]
- Kostyuk, A.I.; Tossounian, M.-A.; Panova, A.S.; Thauvin, M.; Raevskii, R.I.; Ezeriņa, D.; Wahni, K.; Van Molle, I.; Sergeeva, A.D.; Vertommen, D. Hypocrates is a genetically encoded fluorescent biosensor for (pseudo) hypohalous acids and their derivatives. Nat. Commun. 2022, 13, 171. [Google Scholar] [CrossRef]
- Chen, Z.-J.; Ren, W.; Wright, Q.E.; Ai, H.-W. Genetically encoded fluorescent probe for the selective detection of peroxynitrite. J. Am. Chem. Soc. 2013, 135, 14940–14943. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.-N.; Sim, S.-P.; Khoo, A.S. Potential role of oxidative stress-induced apoptosis in mediating chromosomal rearrangements in nasopharyngeal carcinoma. Cell Biosci. 2016, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef]
- Maniati, E.; Potter, P.; Rogers, N.; Morley, B.J. Control of apoptosis in autoimmunity. J. Pathol. J. Pathol. Soc. Great Br. Irel. 2008, 214, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Droga-Mazovec, G.; Bojic, L.; Petelin, A.; Ivanova, S.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008, 283, 19140–19150. [Google Scholar] [CrossRef] [PubMed]
- Broecker-Preuss, M.; Becher-Boveleth, N.; Müller, S.; Mann, K. The BH3 mimetic drug ABT-737 induces apoptosis and acts synergistically with chemotherapeutic drugs in thyroid carcinoma cells. Cancer Cell Int. 2016, 16, 27. [Google Scholar] [CrossRef] [PubMed]
- Chwieralski, C.; Welte, T.; Bühling, F. Cathepsin-regulated apoptosis. Apoptosis 2006, 11, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Conus, S.; Perozzo, R.; Reinheckel, T.; Peters, C.; Scapozza, L.; Yousefi, S.; Simon, H.-U. Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J. Exp. Med. 2008, 205, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Reiners, J., Jr.; Caruso, J.; Mathieu, P.; Chelladurai, B.; Yin, X.; Kessel, D. Release of cytochrome c and activation of pro-caspase-9 following lysosomal photodamage involves Bid cleavage. Cell Death Differ. 2002, 9, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Stoka, V.; Turk, B.; Schendel, S.L.; Kim, T.-H.; Cirman, T.; Snipas, S.J.; Ellerby, L.M.; Bredesen, D.; Freeze, H.; Abrahamson, M. Lysosomal protease pathways to apoptosis: Cleavage of Bid, not pro-caspases, is the most likely route. J. Biol. Chem. 2001, 276, 3149–3157. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Matés, J.M.; Segura, J.A.; Alonso, F.J.; Márquez, J. Oxidative stress in apoptosis and cancer: An update. Arch. Toxicol. 2012, 86, 1649–1665. [Google Scholar] [CrossRef]
- Simon, H.-U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.-H.; Li, W.-X.; Sun, M.-Y.; Zhang, S.-B.; Fan, C.-X.; Wu, Q.; Zhu, W.; Xu, X. Cadmium induced apoptosis in MG63 cells by increasing ROS, activation of p38 MAPK and inhibition of ERK 1/2 pathways. Cell. Physiol. Biochem. 2015, 36, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Jin, X.; Yu, Q.; Zhang, X.; Zheng, B.; Wang, K.; Sun, X.; Chen, Y.; Ren, X.; Ma, J. The vicious circle between mitochondrial oxidative stress and dynamic abnormality mediates triethylene glycol dimethacrylate-induced preodontoblast apoptosis. Free. Radic. Biol. Med. 2019, 134, 644–656. [Google Scholar]
- Al-Hashimi, A.; Venugopalan, V.; Sereesongsaeng, N.; Tedelind, S.; Pinzaru, A.M.; Hein, Z.; Springer, S.; Weber, E.; Führer, D.; Scott, C.J. Significance of nuclear cathepsin V in normal thyroid epithelial and carcinoma cells. Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 118846. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Liu, Y.; Xie, Z.; Qing, H.; Lei, P.; Ni, J. Nucleus distribution of cathepsin B in senescent microglia promotes brain aging through degradation of sirtuins. Neurobiol. Aging 2020, 96, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Frolova, A.S.; Tikhomirova, N.K.; Kireev, I.I.; Zernii, E.Y.; Parodi, A.; Ivanov, K.I.; Zamyatnin, A.A., Jr. Expression, Intracellular Localization, and Maturation of Cysteine Cathepsins in Renal Embryonic and Cancer Cell Lines. Biochemistry 2023, 88, 1034–1044. [Google Scholar] [CrossRef]
- Frolova, A.S.; Chepikova, O.E.; Deviataikina, A.S.; Solonkina, A.D.; Zamyatnin, A.A., Jr. New Perspectives on the Role of Nuclear Proteases in Cell Death Pathways. Biology 2023, 12, 797. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Mason, C.W.; Goodman, C.B.; Holder, M.S.; Kirksey, O.W.; Womble, T.A.; Severs, W.B.; Palm, D.E. Hydrogen peroxide induces lysosomal protease alterations in PC12 cells. Neurochem. Res. 2007, 32, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Lagadic-Gossmann, D.; Huc, L.; Lecureur, V. Alterations of intracellular pH homeostasis in apoptosis: Origins and roles. Cell Death Differ. 2004, 11, 953–961. [Google Scholar] [CrossRef]
- Soond, S.M.; Savvateeva, L.V.; Makarov, V.A.; Gorokhovets, N.V.; Townsend, P.A.; Zamyatnin, A.A., Jr. Cathepsin S cleaves BAX as a novel and therapeutically important regulatory mechanism for apoptosis. Pharmaceutics 2021, 13, 339. [Google Scholar] [CrossRef]
- Wartenberg, M.; Andrault, P.-M.; Saidi, A.; Bigot, P.; Nadal-Desbarats, L.; Lecaille, F.; Lalmanach, G. Oxidation of cathepsin S by major chemicals of cigarette smoke. Free. Radic. Biol. Med. 2020, 150, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Kaminskyy, V.O.; Zhivotovsky, B. Free radicals in cross talk between autophagy and apoptosis. Antioxid. Redox Signal. 2014, 21, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Shiau, J.-P.; Chuang, Y.-T.; Tang, J.-Y.; Yang, K.-H.; Chang, F.-R.; Hou, M.-F.; Yen, C.-Y.; Chang, H.-W. The impact of oxidative stress and AKT pathway on cancer cell functions and its application to natural products. Antioxidants 2022, 11, 1845. [Google Scholar] [CrossRef] [PubMed]
- Kahl, R.; Kampkötter, A.; Wätjen, W.; Chovolou, Y. Antioxidant enzymes and apoptosis. Drug Metab. Rev. 2004, 36, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Hohl, M.; Mayr, M.; Lang, L.; Nickel, A.G.; Barallobre-Barreiro, J.; Yin, X.; Speer, T.; Selejan, S.-R.; Goettsch, C.; Erb, K. Cathepsin A contributes to left ventricular remodeling by degrading extracellular superoxide dismutase in mice. J. Biol. Chem. 2020, 295, 12605–12617. [Google Scholar] [CrossRef] [PubMed]
- Jurisic, V.; Srdic-Rajic, T.; Konjevic, G.; Bogdanovic, G.; Colic, M. TNF-α induced apoptosis is accompanied with rapid CD30 and slower CD45 shedding from K-562 cells. J. Membr. Biol. 2011, 239, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Jurisic, V.; Bumbasirevic, V.; Konjevic, G.; Djuricic, B.; Spuzic, I. TNF-α induces changes in LDH isotype profile following triggering of apoptosis in PBL of non-Hodgkin’s lymphomas. Ann. Hematol. 2004, 83, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Oztay, F.; Gezginci-Oktayoglu, S.; Bayrak, B.B.; Yanardag, R.; Bolkent, S. Cathepsin B inhibition improves lung injury associated to D-galactosamine/tumor necrosis factor-alpha-induced liver injury in mice. Mol. Cell. Biochem. 2010, 333, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Wadhawan, M.; Singh, N.; Rathaur, S. Inhibition of cathepsin B by E-64 induces oxidative stress and apoptosis in filarial parasite. PLoS ONE 2014, 9, e93161. [Google Scholar] [CrossRef]
- Seo, S.U.; Woo, S.M.; Kim, M.W.; Lee, H.-S.; Kim, S.H.; Kang, S.C.; Lee, E.-W.; Min, K.-J.; Kwon, T.K. Cathepsin K inhibition-induced mitochondrial ROS enhances sensitivity of cancer cells to anti-cancer drugs through USP27x-mediated Bim protein stabilization. Redox Biol. 2020, 30, 101422. [Google Scholar] [CrossRef]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 199. [Google Scholar] [CrossRef] [PubMed]
- Wegner, K.W.; Saleh, D.; Degterev, A. Complex pathologic roles of RIPK1 and RIPK3: Moving beyond necroptosis. Trends Pharmacol. Sci. 2017, 38, 202–225. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kos, R.; Garssen, J.; Redegeld, F. Molecular insights into the mechanism of necroptosis: The necrosome as a potential therapeutic target. Cells 2019, 8, 1486. [Google Scholar] [CrossRef]
- Dixon, S.J.; Olzmann, J.A. The cell biology of ferroptosis. Nat. Rev. Mol. Cell Biol. 2024, 1–19. [Google Scholar] [CrossRef]
- Mackenzie, E.L.; Iwasaki, K.; Tsuji, Y. Intracellular iron transport and storage: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 997–1030. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Liu, Y.; Wan, Y.; Jiang, Y.; Zhang, L.; Cheng, W. GPX4: The hub of lipid oxidation, ferroptosis, disease and treatment. Biochim. Biophys. Acta-Rev. Cancer 2023, 1878, 188890. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The role of erastin in ferroptosis and its prospects in cancer therapy. OncoTargets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Morshed, M.; Hlushchuk, R.; Simon, D.; Walls, A.F.; Obata-Ninomiya, K.; Karasuyama, H.; Djonov, V.; Eggel, A.; Kaufmann, T.; Simon, H.-U. NADPH oxidase–independent formation of extracellular DNA traps by basophils. J. Immunol. 2014, 192, 5314–5323. [Google Scholar] [CrossRef]
- Granger, V.; Faille, D.; Marani, V.; Noël, B.; Gallais, Y.; Szely, N.; Flament, H.; Pallardy, M.; Chollet-Martin, S.; de Chaisemartin, L. Human blood monocytes are able to form extracellular traps. J. Leukoc. Biol. 2017, 102, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes-Costa, A.B.; Nascimento, M.T.; Wardini, A.B.; Pinto-da-Silva, L.H.; Saraiva, E.M. ETosis: A microbicidal mechanism beyond cell death. J. Parasitol. Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Azzouz, D.; Khan, M.A.; Palaniyar, N. ROS induces NETosis by oxidizing DNA and initiating DNA repair. Cell Death Discov. 2021, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, X.; Nie, Y.; Zhan, F.; Zhu, B. Oxidative stress and ROS-mediated cellular events in RSV infection: Potential protective roles of antioxidants. Virol. J. 2023, 20, 224. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-K.; Chang, W.-T.; Lin, I.-L.; Chen, Y.-F.; Padalwar, N.B.; Cheng, K.-C.; Teng, Y.-N.; Wang, C.-H.; Chiu, C.-C. The role of necroptosis in ROS-mediated cancer therapies and its promising applications. Cancers 2020, 12, 2185. [Google Scholar] [CrossRef] [PubMed]
- Sharapova, T.N.; Romanova, E.A.; Sashchenko, L.P.; Yashin, D.V. FasL on the surface of Tag7 (PGRP-S)-activated lymphocytes induces necroptosis in HLA-negative tumor cells with the involvement of lysosomes and mitochondria. Biochimie 2018, 152, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.-Q.; Chen, X.; Cai, Q.; Yang, Z.-H.; Huang, D.; Wu, R. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [PubMed]
- Deragon, M.A.; McCaig, W.D.; Patel, P.S.; Haluska, R.J.; Hodges, A.L.; Sosunov, S.A.; Murphy, M.P.; Ten, V.S.; LaRocca, T.J. Mitochondrial ROS prime the hyperglycemic shift from apoptosis to necroptosis. Cell Death Discov. 2020, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- Barati, M.; Javidi, M.A.; Darvishi, B.; Shariatpanahi, S.P.; Moosavi, Z.S.M.; Ghadirian, R.; Khani, T.; Sanati, H.; Simaee, H.; Barough, M.S. Necroptosis triggered by ROS accumulation and Ca2+ overload, partly explains the inflammatory responses and anti-cancer effects associated with 1 Hz, 100 mT ELF-MF in vivo. Free. Radic. Biol. Med. 2021, 169, 84–98. [Google Scholar] [CrossRef]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3–mediated pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Morgan, M.J.; Choksi, S.; Liu, Z.-G. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell 2007, 26, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target. Ther. 2020, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.S.; Ruiz, J.; Watts, J.L. Polyunsaturated fatty acids drive lipid peroxidation during ferroptosis. Cells 2023, 12, 804. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Nagakannan, P.; Islam, M.I.; Conrad, M.; Eftekharpour, E. Cathepsin B is an executioner of ferroptosis. Biochim. Biophys. Acta-Mol. Cell Res. 2021, 1868, 118928. [Google Scholar] [CrossRef] [PubMed]
- Stoiber, W.; Obermayer, A.; Steinbacher, P.; Krautgartner, W.-D. The role of reactive oxygen species (ROS) in the formation of extracellular traps (ETs) in humans. Biomolecules 2015, 5, 702–723. [Google Scholar] [CrossRef] [PubMed]
- Röhm, M.; Grimm, M.J.; D’Auria, A.C.; Almyroudis, N.G.; Segal, B.H.; Urban, C.F. NADPH oxidase promotes neutrophil extracellular trap formation in pulmonary aspergillosis. Infect. Immun. 2014, 82, 1766–1777. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef]
- Berghe, T.V.; Vanlangenakker, N.; Parthoens, E.; Deckers, W.; Devos, M.; Festjens, N.; Guerin, C.; Brunk, U.; Declercq, W.; Vandenabeele, P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010, 17, 922–930. [Google Scholar] [CrossRef]
- Kuang, F.; Liu, J.; Li, C.; Kang, R.; Tang, D. Cathepsin B is a mediator of organelle-specific initiation of ferroptosis. Biochem. Biophys. Res. Commun. 2020, 533, 1464–1469. [Google Scholar] [CrossRef]
- Quan, J.-H.; Gao, F.F.; Ma, T.-Z.; Ye, W.; Gao, X.; Deng, M.-Z.; Yin, L.-L.; Choi, I.-W.; Yuk, J.-M.; Cha, G.-H. Toxoplasma gondii Induces Pyroptosis in Human Placental Trophoblast and Amniotic Cells by Inducing ROS Production and Activation of Cathepsin B and NLRP1/NLRP3/NLRC4/AIM2 Inflammasome. Am. J. Pathol. 2023, 193, 2047–2065. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jiang, S.; Zhang, J.; Guan, X.-L.; Sun, B.-G.; Sun, L. A virulent Bacillus cereus strain from deep-sea cold seep induces pyroptosis in a manner that involves NLRP3 inflammasome, JNK pathway, and lysosomal rupture. Virulence 2021, 12, 1362–1376. [Google Scholar] [CrossRef]
- Liu, M.; Lu, J.; Hu, J.; Chen, Y.; Deng, X.; Wang, J.; Zhang, S.; Guo, J.; Li, W.; Guan, S. Sodium sulfite triggered hepatic apoptosis, necroptosis, and pyroptosis by inducing mitochondrial damage in mice and AML-12 cells. J. Hazard. Mater. 2024, 467, 133719. [Google Scholar] [CrossRef] [PubMed]
- Lou, J.; Wang, X.; Zhang, H.; Yu, G.; Ding, J.; Zhu, X.; Li, Y.; Wu, Y.; Xu, H.; Xu, H. Inhibition of PLA2G4E/cPLA2 promotes survival of random skin flaps by alleviating Lysosomal membrane permeabilization-Induced necroptosis. Autophagy 2022, 18, 1841–1863. [Google Scholar] [CrossRef]
- Talukdar, R.; Sareen, A.; Zhu, H.; Yuan, Z.; Dixit, A.; Cheema, H.; George, J.; Barlass, U.; Sah, R.; Garg, S.K. Release of cathepsin B in cytosol causes cell death in acute pancreatitis. Gastroenterology 2016, 151, 747–758.e745. [Google Scholar] [CrossRef]
- Wu, Z.; Han, X.; Bao, J.; Li, B.; Shen, J.; Song, P.; Peng, Q.; Wang, X.; Hu, G. Dopamine D2 receptor signaling attenuates acinar cell necroptosis in acute pancreatitis through the cathepsin B/TFAM/ROS pathway. Oxidative Med. Cell. Longev. 2022, 2022, 4499219. [Google Scholar] [CrossRef]
- Pacheco, F.J.; Almaguel, F.G.; Evans, W.; Rios-Colon, L.; Filippov, V.; Leoh, L.S.; Rook-Arena, E.; Mediavilla-Varela, M.; De Leon, M.; Casiano, C.A. Docosahexanoic acid antagonizes TNF-α-induced necroptosis by attenuating oxidative stress, ceramide production, lysosomal dysfunction, and autophagic features. Inflamm. Res. 2014, 63, 859–871. [Google Scholar] [CrossRef]
- Yashin, D.V.; Romanova, E.A.; Ivanova, O.K.; Sashchenko, L.P. The Tag7–Hsp70 cytotoxic complex induces tumor cell necroptosis via permeabilisation of lysosomes and mitochondria. Biochimie 2016, 123, 32–36. [Google Scholar] [CrossRef] [PubMed]
- McComb, S.; Shutinoski, B.; Thurston, S.; Cessford, E.; Kumar, K.; Sad, S. Cathepsins limit macrophage necroptosis through cleavage of Rip1 kinase. J. Immunol. 2014, 192, 5671–5678. [Google Scholar] [CrossRef]
- Xu, J.; Ding, Y.; Sheng, X.; Shi, C.; Yuan, F.; Liu, Y.; Xie, Y.; Lu, H.; Jiang, L.; Hu, J. Identification of Cathepsin B as a Pharmacological Target for Ferroptosis after Spinal Cord Injury Via Combined Transcriptome Analysis. SSRN Electron. J. 2022, 15, 421–443. [Google Scholar] [CrossRef]
- Chevriaux, A.; Pilot, T.; Derangère, V.; Simonin, H.; Martine, P.; Chalmin, F.; Ghiringhelli, F.; Rébé, C. Cathepsin B is required for NLRP3 inflammasome activation in macrophages, through NLRP3 interaction. Front. Cell Dev. Biol. 2020, 8, 167. [Google Scholar] [CrossRef]
- Liao, Y.; Zhang, H.; He, D.; Wang, Y.; Cai, B.; Chen, J.; Ma, J.; Liu, Z.; Wu, Y. Retinal pigment epithelium cell death is associated with NLRP3 inflammasome activation by all-trans retinal. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3034–3045. [Google Scholar] [CrossRef]
- Meng, Z.; Gao, M.; Wang, C.; Guan, S.; Zhang, D.; Lu, J. Apigenin alleviated high-fat-diet-induced hepatic pyroptosis by mitophagy-ROS-CTSB-NLRP3 pathway in mice and AML12 cells. J. Agric. Food Chem. 2023, 71, 7032–7045. [Google Scholar] [CrossRef]
- Burster, T.; Macmillan, H.; Hou, T.; Boehm, B.O.; Mellins, E.D. Cathepsin G: Roles in antigen presentation and beyond. Mol. Immunol. 2010, 47, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Clancy, D.M.; Henry, C.M.; Sullivan, G.P.; Martin, S.J. Neutrophil extracellular traps can serve as platforms for processing and activation of IL-1 family cytokines. FEBS J. 2017, 284, 1712–1725. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Tang, J.; Bassham, D.C. Autophagy during drought: Function, regulation, and potential application. Plant J. 2022, 109, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Vanzo, R.; Bartkova, J.; Merchut-Maya, J.M.; Hall, A.; Bouchal, J.; Dyrskjøt, L.; Frankel, L.B.; Gorgoulis, V.; Maya-Mendoza, A.; Jäättelä, M. Autophagy role(s) in response to oncogenes and DNA replication stress. Cell Death Differ. 2020, 27, 1134–1153. [Google Scholar] [CrossRef]
- Ryter, S.W.; Choi, A.M. Autophagy: An integral component of the mammalian stress response. J. Biochem. Pharmacol. Res. 2013, 1, 176. [Google Scholar]
- Kang, C.; Elledge, S.J. How autophagy both activates and inhibits cellular senescence. Autophagy 2016, 12, 898–899. [Google Scholar] [CrossRef]
- Martyshkina, Y.S.; Tereshchenko, V.P.; Bogdanova, D.A.; Rybtsov, S.A. Reliable Hallmarks and Biomarkers of Senescent Lymphocytes. Int. J. Mol. Sci. 2023, 24, 15653. [Google Scholar] [CrossRef]
- Ishikawa, F. Cellular senescence as a stress response. Cornea 2006, 25, S3–S6. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Weinberg, R.A. When cells get stressed: An integrative view of cellular senescence. J. Clin. Investig. 2004, 113, 8–13. [Google Scholar] [CrossRef]
- Cassidy, L.D.; Narita, M. Autophagy at the intersection of aging, senescence, and cancer. Mol. Oncol. 2022, 16, 3259–3275. [Google Scholar] [CrossRef]
- Rastaldo, R.; Vitale, E.; Giachino, C. Dual role of autophagy in regulation of mesenchymal stem cell senescence. Front. Cell Dev. Biol. 2020, 8, 276. [Google Scholar] [CrossRef]
- Cayo, A.; Segovia, R.; Venturini, W.; Moore-Carrasco, R.; Valenzuela, C.; Brown, N. mTOR activity and autophagy in senescent cells, a complex partnership. Int. J. Mol. Sci. 2021, 22, 8149. [Google Scholar] [CrossRef]
- Sui, X.; Han, W.; Pan, H. p53-induced autophagy and senescence. Oncotarget 2015, 6, 11723. [Google Scholar] [CrossRef]
- Tan, P.; Wang, Y.-J.; Li, S.; Wang, Y.; He, J.-Y.; Chen, Y.-Y.; Deng, H.-Q.; Huang, W.; Zhan, J.-K.; Liu, Y.-S. The PI3K/Akt/mTOR pathway regulates the replicative senescence of human VSMCs. Mol. Cell. Biochem. 2016, 422, 1–10. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Sora, V.; Kumar, M.; Maiani, E.; Lambrughi, M.; Tiberti, M.; Papaleo, E. Structure and dynamics in the ATG8 family from experimental to computational techniques. Front. Cell Dev. Biol. 2020, 8, 420. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death-apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Cheng, Y.; Qiu, F.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. ERK and JNK mediate TNFα-induced p53 activation in apoptotic and autophagic L929 cell death. Biochem. Biophys. Res. Commun. 2008, 376, 483–488. [Google Scholar] [CrossRef]
- Su, T.; Li, X.; Yang, M.; Shao, Q.; Zhao, Y.; Ma, C.; Wang, P. Autophagy: An intracellular degradation pathway regulating plant survival and stress response. Front. Plant Sci. 2020, 11, 164. [Google Scholar] [CrossRef]
- Ornatowski, W.; Lu, Q.; Yegambaram, M.; Garcia, A.E.; Zemskov, E.A.; Maltepe, E.; Fineman, J.R.; Wang, T.; Black, S.M. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 2020, 36, 101679. [Google Scholar] [CrossRef]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay between NLRP3 inflammasome and autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef]
- Kroemer, G.; Jäättelä, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897. [Google Scholar] [CrossRef]
- Li, S.-S.; Zhang, M.; Wang, J.-H.; Yang, F.; Kang, B.; Xu, J.-J.; Chen, H.-Y. Monitoring the changes of pH in lysosomes during autophagy and apoptosis by plasmon enhanced Raman imaging. Anal. Chem. 2019, 91, 8398–8405. [Google Scholar] [CrossRef]
- Uchiyama, Y. Autophagic cell death and its execution by lysosomal cathepsins. Arch. Histol. Cytol. 2001, 64, 233–246. [Google Scholar] [CrossRef]
- Weiss-Sadan, T.; Maimoun, D.; Oelschlagel, D.; Kaschani, F.; Misiak, D.; Gaikwad, H.; Ben-Nun, Y.; Merquiol, E.; Anaki, A.; Tsvirkun, D. Cathepsins drive anti-inflammatory activity by regulating autophagy and mitochondrial dynamics in macrophage foam cells. Cell. Physiol. Biochem. 2019, 53, 550–572. [Google Scholar]
- Tatti, M.; Motta, M.; Di Bartolomeo, S.; Cianfanelli, V.; Salvioli, R. Cathepsin-mediated regulation of autophagy in saposin C deficiency. Autophagy 2013, 9, 241–243. [Google Scholar] [CrossRef]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 2016, 32, 22–37. [Google Scholar] [CrossRef]
- Chen, K.-L.; Chang, W.-S.W.; Cheung, C.H.A.; Lin, C.-C.; Huang, C.-C.; Yang, Y.-N.; Kuo, C.-P.; Kuo, C.-C.; Chang, Y.-H.; Liu, K.-J. Targeting cathepsin S induces tumor cell autophagy via the EGFR–ERK signaling pathway. Cancer Lett. 2012, 317, 89–98. [Google Scholar] [CrossRef]
- Ravegnini, G.; Sammarini, G.; Nannini, M.; Pantaleo, M.A.; Biasco, G.; Hrelia, P.; Angelini, S. Gastrointestinal stromal tumors (GIST): Facing cell death between autophagy and apoptosis. Autophagy 2017, 13, 452–463. [Google Scholar] [CrossRef]
- Gao, L.; Loveless, J.; Shay, C.; Teng, Y. Targeting ROS-mediated crosstalk between autophagy and apoptosis in cancer. Rev. New Drug Targets Age-Relat. Disord. 2020, 1260, 1–12. [Google Scholar]
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12, 1528–1534. [Google Scholar] [CrossRef]
- Hah, Y.-S.; Noh, H.S.; Ha, J.H.; Ahn, J.S.; Hahm, J.R.; Cho, H.Y.; Kim, D.R. Cathepsin D inhibits oxidative stress-induced cell death via activation of autophagy in cancer cells. Cancer Lett. 2012, 323, 208–214. [Google Scholar] [CrossRef]
- Oliveira, C.S.; Pereira, H.; Alves, S.; Castro, L.; Baltazar, F.; Chaves, S.; Preto, A.; Côrte-Real, M. Cathepsin D protects colorectal cancer cells from acetate-induced apoptosis through autophagy-independent degradation of damaged mitochondria. Cell Death Dis. 2015, 6, e1788. [Google Scholar] [CrossRef]
- Nagakannan, P.; Eftekharpour, E. Differential redox sensitivity of cathepsin B and L holds the key to autophagy-apoptosis interplay after Thioredoxin reductase inhibition in nutritionally stressed SH-SY5Y cells. Free. Radic. Biol. Med. 2017, 108, 819–831. [Google Scholar] [CrossRef]
- Tsukuba, T.; Yanagawa, M.; Kadowaki, T.; Takii, R.; Okamoto, Y.; Sakai, E.; Okamoto, K.; Yamamoto, K. Cathepsin E deficiency impairs autophagic proteolysis in macrophages. PLoS ONE 2013, 8, e82415. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).