
Agnostic Administration of Targeted Anticancer Drugs: Looking for a Balance between Hype and Caution


Abstract
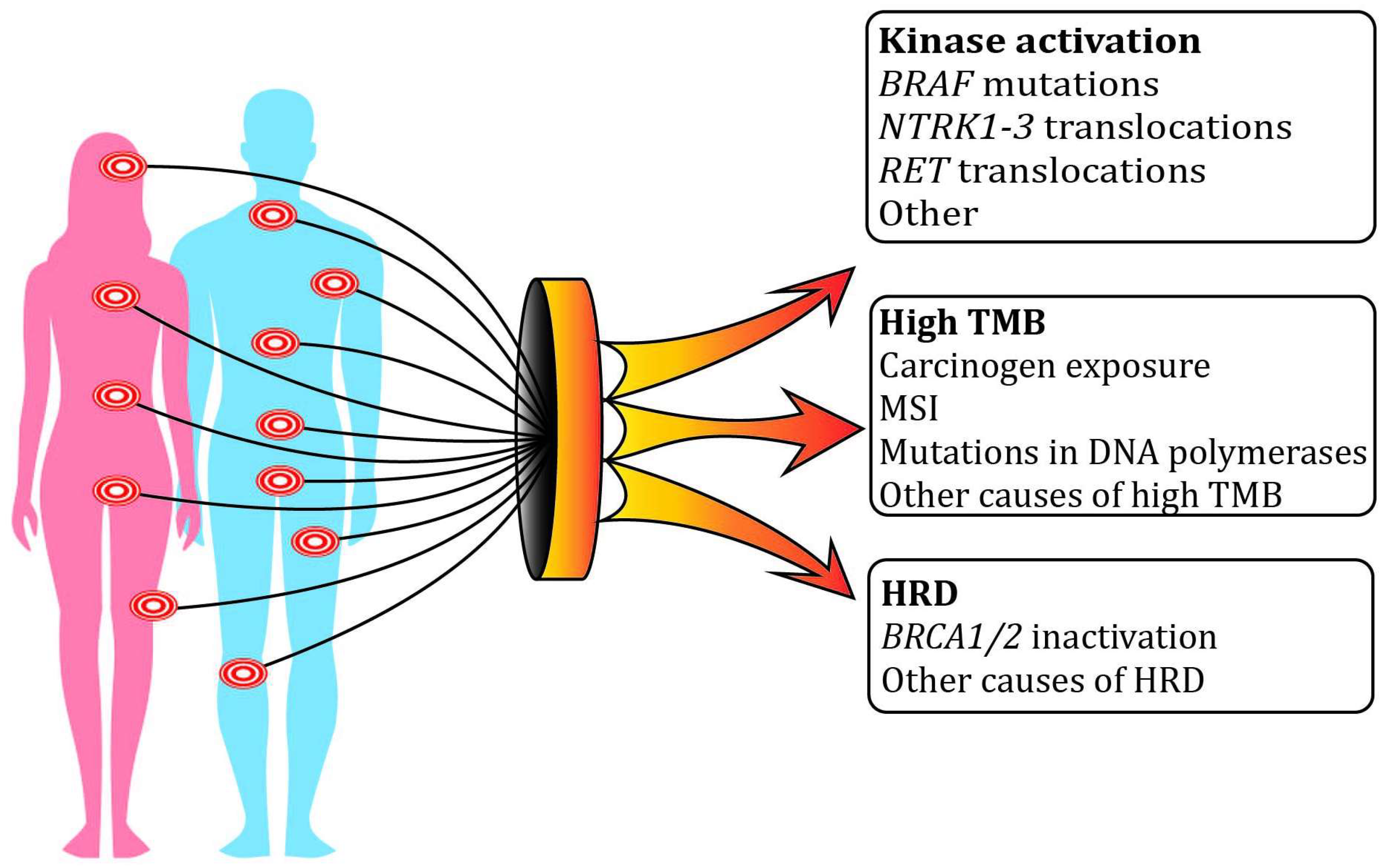
{kind=link}
{kind=link}
Share and Cite
Aleksakhina, S.N.; Ivantsov, A.O.; Imyanitov, E.N. Agnostic Administration of Targeted Anticancer Drugs: Looking for a Balance between Hype and Caution. Int. J. Mol. Sci. 2024, 25, 4094. https://doi.org/10.3390/ijms25074094
Aleksakhina SN, Ivantsov AO, Imyanitov EN. Agnostic Administration of Targeted Anticancer Drugs: Looking for a Balance between Hype and Caution. International Journal of Molecular Sciences. 2024; 25(7):4094. https://doi.org/10.3390/ijms25074094
Chicago/Turabian StyleAleksakhina, Svetlana N., Alexander O. Ivantsov, and Evgeny N. Imyanitov. 2024. "Agnostic Administration of Targeted Anticancer Drugs: Looking for a Balance between Hype and Caution" International Journal of Molecular Sciences 25, no. 7: 4094. https://doi.org/10.3390/ijms25074094
APA StyleAleksakhina, S. N., Ivantsov, A. O., & Imyanitov, E. N. (2024). Agnostic Administration of Targeted Anticancer Drugs: Looking for a Balance between Hype and Caution. International Journal of Molecular Sciences, 25(7), 4094. https://doi.org/10.3390/ijms25074094