Abstract
Cardiovascular diseases (CVDs) pose a significant global health threat due to their complex pathogenesis and high incidence, imposing a substantial burden on global healthcare systems. Integrins, a group of heterodimers consisting of α and β subunits that are located on the cell membrane, have emerged as key players in mediating the occurrence and progression of CVDs by regulating the physiological activities of endothelial cells, vascular smooth muscle cells, platelets, fibroblasts, cardiomyocytes, and various immune cells. The crucial role of integrins in the progression of CVDs has valuable implications for targeted therapies. In this context, the development and application of various integrin antibodies and antagonists have been explored for antiplatelet therapy and anti-inflammatory-mediated tissue damage. Additionally, the rise of nanomedicine has enhanced the specificity and bioavailability of precision therapy targeting integrins. Nevertheless, the complexity of the pathogenesis of CVDs presents tremendous challenges for monoclonal targeted treatment. This paper reviews the mechanisms of integrins in the development of atherosclerosis, cardiac fibrosis, hypertension, and arrhythmias, which may pave the way for future innovations in the diagnosis and treatment of CVDs.
1. Introduction
Cardiovascular diseases (CVDs) encompass a spectrum of disorders affecting the heart and vasculature, constituting a substantial global health burden [1]. In 2022, cardiovascular diseases were responsible for approximately 190,000 global fatalities [1]. An estimated 34% of these cardiovascular disease-related deaths occur before the age of 70 [1,2]. CVDs, which are classified according to affected sites and involve various cell types, encompass a spectrum of conditions including cardiomyopathy, arrhythmia, atherosclerosis (AS), cardiac fibrosis, heart failure, coronary artery disease, and hypertension [2,3]. Current research indicates that the interaction between cells and the extracellular matrix (ECM) plays a significantly pivotal role in the pathogenesis of CVDs [4,5,6,7]. Notably, specific focus has been directed toward dysregulated ligands that target integrins, which emphasizes their unique relevance in the context of cardioprotection [8]. Integrins, which are the principal cellular adhesion receptors for ECM constituents, comprise a family of 24 transmembrane heterodimers formed by a combination of 18 α and 8 β integrin subunits [9]. Integrins function as receptors for ECM components categorized into four distinct groups, including those containing the RGD (Arg-Gly-Asp) motif, leukocyte-specific ligands, collagen ligands, and laminin ligands (Figure 1) [10,11]. In each heterodimeric complex, there is a distinct extracellular domain, a transmembrane domain, and a concise cytoplasmic domain connected to the cytoskeleton (Figure 1) [9]. The ectodomain of the α subunit comprises four extracellular regions: a seven-bladed β-propeller, a thigh region, and two calf domains (Figure 1) [9]. The shared feature among various α subunits within their extracellular domains is the presence of seven repetitive motifs, which assemble into a seven-bladed propeller configuration on the top surface (Figure 1) [9]. Additionally, on the underside of blades 4–7, sites for binding divalent cations can be found (Figure 1) [9]. The β subunit’s ectodomain features seven domains with complex insertions: a β I domain within the hybrid domain, a plexin–semaphorin–integrin domain, four cysteine-rich epidermal growth factor modules, and a β tail domain (Figure 1) [9]. These structural components facilitate the initiation of both inside-out and outside-in signaling pathways, thereby intricately orchestrating cellular responses [12]. In the context of inside-out signaling, activated chemokines can propagate signals through G-protein-coupled receptors [11]. This signaling cascade initiates phosphorylation events within the cytoplasmic domain of the receptor subunit, thereby enhancing the affinity of integrins to their ligands [11]. This mechanism induces conformational changes in the extracellular domain of integrins, profoundly affecting critical biological processes, including cell adhesion, migration, proliferation, survival, and differentiation [11]. In the outside-in signaling cascade, ligand binding induces conformational changes in integrins situated on the plasma membrane, which leads to the separation of their heads from their tails [11]. This conformational alteration facilitates the interaction of the cytoplasmic tail domain with intracellular signaling molecules [11]. Consequently, a spectrum of cellular responses ensues, encompassing modifications in cell morphology and structure, the modulation of cell adhesion and migration, the regulation of cell proliferation and survival, and control over cellular differentiation [11].
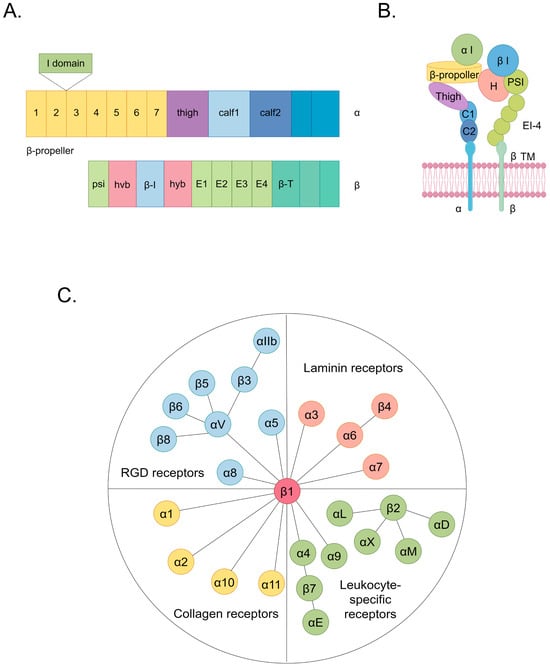
Figure 1.
The structure and classification of integrins. (A) The domain organization of integrins within primary structures. (B) The spatial arrangement of domains within the representative three-dimensional crystal structure of integrins. (C) Integrins include 24 types. They can be classified into 4 categories based on their receptor specificity: RGD receptors, laminin receptors, leukocyte-specific receptors, and collagen receptors. The same color in (A,B) indicating integrin structure corresponds to the same domain. Among them, the hvb and hyb domains in (A) correspond to the H domain in (B).
During the progression of CVDs, integrins that are present on diverse cell types establish aberrant connections between the ECM and intracellular signaling pathways. This process triggers multiple pathophysiological mechanisms characterized by dysregulation, resulting in adverse responses, including inflammation, cellular dysfunction, and aberrant proliferation, migration, adhesion, and differentiation [13]. Significantly, the interaction between ECM components and integrins that are present on diverse cell types, including cardiomyocytes, vascular endothelial cells (VECs), vascular smooth muscle cells (VSMCs), and immune cells, can potentially either facilitate or impede the pathogenesis and progression of CVDs [14,15,16,17]. As a result, the modulation of integrin-mediated signaling pathways presents a promising way of attenuating the deleterious cascades associated with these diseases, thereby mitigating the risk of CVD development and progression [18,19].
This review delineates the pathophysiological dysregulation mechanisms triggered by interactions between the ECM and integrins, elucidating their intricate roles in the progression of prevalent clinical manifestations of CVDs. Specifically, a detailed examination is provided for AS, hypertension, arrhythmias, and cardiac fibrosis [16,20,21,22]. As the intricate pathogenic mechanisms orchestrated by integrins in CVDs have become more understood, integrin-targeted therapeutics have been clinically employed for CVD treatment. These therapeutics exhibit noteworthy integrin-targeting specificity, low toxicity profiles, and robust efficacy [23]. At present, there are two integrin-targeted therapeutic modalities in clinical practice for addressing CVDs: integrin antagonists and antibodies and integrin-directed nanotherapy [23,24]. As our understanding of the contributions of integrins to the pathogenesis of CVDs advances, it is increasingly evident that therapeutic strategies targeting integrins hold considerable promise for enhancing the management of CVDs.
2. Potential Effects of Integrins in CVDs
The involvement of different cell surface integrins plays a crucial role in the development and progression of CVDs. Figure 2 demonstrates the signaling pathways of integrins and related molecules in the six specific cell types that are mainly involved in the pathogenesis of CVDs.
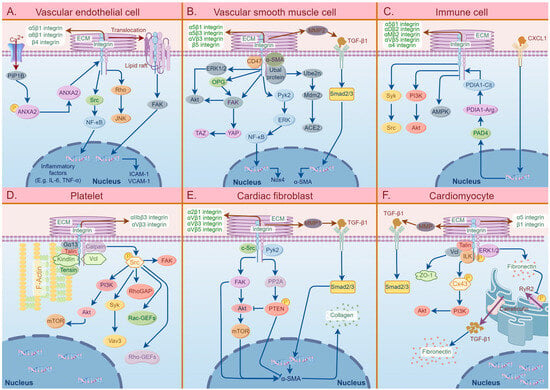
Figure 2.
A summary diagram of the effects of integrins in six specific cell types that are mainly involved in the pathogenesis of cardiovascular diseases, including vascular endothelial cells (VECs), vascular smooth muscle cells (VSMCs), immune cells, platelets, cardiac fibroblasts, and cardiomyocytes. (A) The effects of integrins in VECs involved in the pathogenesis of atherosclerosis (AS) and hypertension. The extracellular matrix (ECM)–integrin interactions mediate VEC activation, dysfunction, and oxidation, thereby inducing vascular endothelial inflammation. (B) The pathogenic roles of integrins on VSMCs in AS. The binding of integrins to ligands in ECM can activate the characteristics of VSMCs, including VSMC migration, proliferation, differentiation, and calcification, which eventually leads to vascular stiffness. (C) The effects of integrins on immune cells involved in the pathogenic process of AS. The integrins on immune cells can activate several specific signaling pathways, such as PI3K/Akt and Syk/Src, which can induce cascade reactions and further activate the characteristics of immune cells, including migration, proliferation, adhesion, and phagocytosis, thereby causing thrombus formation. (D) The specific roles of integrins in platelet in AS pathogenesis. ECM–integrin interactions mainly participate in platelet activation, migration, and secretion, which subsequently causes blood coagulation, thrombosis, and AS plaque instability. (E) The effects of integrins on cardiac fibroblasts involved in cardiac fibrosis. The binding of integrins to ECM induces fibroblast activation, migration, and trans-differentiation, which promotes collagen synthesis and deposition. (F) The pathogenic roles of integrins on cardiomyocytes in arrhythmias. The activation of cardiomyocytes induced by ECM–integrin interactions mediates myocardial damage and myocardial electrical signal dysfunction, leading to cytoskeletal remodeling. Abbreviations: ECM: extracellular matrix, Rho: Ras homolog family, JNK: C-Jun NH2-terminal kinase, Src: serum creatinine, NF-kB: nuclear factor-k-gene binding, ANXA2: annexin A2, PIP1B: Arabidopsis thaliana aquaporin gene AthH2, FAK: focal adhesion kinase, ICAM-1: intercellular cell adhesion molecule-1, VCAM-1: vascular cell adhesion molecule-1, MMP2: matrix metalloproteinase2, TGF-β1: transforming growth factor-β1, α-SMA: α-smooth muscle actin, ERK1/2: extracellular signal-regulated kinase 1/2, Akt: protein kinase B, OPG: osteoprotegerin, YAP: Yes-associated protein, TAZ: tafazzin, Pyk2: protein tyrosine kinase 2, Ube2n: ubiquitin-conjugating enzyme 2N, Mdm2: mouse double minute 2 homolog, ACE2: angiotensin-converting enzyme 2, Smad 2/3: mothers against decapentaplegic homolog 2/3, Nox4: NADPH oxidase 4, PI3K: phosphoinositide 3-kinase, AMPK: AMP-activated protein kinase, PDIA1-Cit: protein disulfide isomerase A1-citrulline, PDIA1-Arg: protein disulfide isomerase A1-arginine, PAD4: peptidylarginine deiminase 4, CXCL1: chemokine (C-X-C motif) ligand 1, Gα13: guanine nucleotide-binding protein G(13) subunit alpha, mTOR: mammalian target of rapamycin, Vav3: Vav guanine nucleotide exchange factor 3, RhoGAP: Rho GTPase-activating protein, Rac-GEFs: Rac guanine nucleotide exchange factors, c-Src: cellular serum creatinine, PP2A: protein phosphatase 2A, PTEN: phosphatase and tensin homolog, ZO-1: Zona occludens protein 1, Vcl: vinculin, ILK: integrin-linked kinase, Cx43: connexin 43, PyR2: ryanodine receptor 2.
2.1. Atherosclerosis
AS is a prominent contributor to global mortality and is characterized as a chronic inflammatory disease with the development of fibrofatty lesions on the arterial walls of large and medium-sized arteries [25,26]. This condition predisposes individuals to a spectrum of panvascular diseases, such as ischemic heart disease, stroke, and peripheral vascular disease, leading to a subsequent reduction in life expectancy [25,26]. AS exhibits a sophisticated formation mechanism that initiates with the deposition of low-density lipoprotein (LDL) in the endothelium of blood vessels [27,28,29]. LDL is subsequently translocated to the subendothelial space, where it undergoes oxidation, resulting in the formation of oxidized LDL (ox-LDL) [28,29]. Monocytes, macrophages, and smooth muscle cells actively phagocytize ox-LDL and further assume a foam cell phenotype, while fibroblasts synthesize and release fibrous tissue into the ECM, culminating in the establishment of a fibrous cap on the plaque surface [30,31,32,33]. Upon plaque rupture, platelets aggregate at the breach site and subsequently orchestrate the formation of a fibrin meshwork, which progressively develops to a thrombus [32,34]. Extensive research has underscored the presence of integrins on VECs, VSMCs, immune cells, and platelets, which play a pivotal role in the regulation of cellular activities in AS (Table 1).

Table 1.
The interaction between integrins on specific cells and ECM components in AS.
2.1.1. Integrins That Bind to RGD Receptors
The α5, α5β1, αVβ3, αVβ5, αVβ1, α8β1, and αIIbβ3 integrins located on the cell membranes of VSMCs, VECs, immune cells, and platelets are involved in the progression of AS. The excessive activation of α5 integrins on the cell membrane of ECs triggers inflammatory activation of the ECs and contributes to the development of atherosclerosis, and this activation can be hindered by the integrins directly binding to the C-terminus of cartilage oligomeric matrix protein [14]. Demos et al. reported that the activation of the calcium ion channel Piezo1 by disturbed blood flow and subsequent conformational changes in annexin A2 induces the translocation of α5 integrins on the plasma membrane of ECs to lipid rafts (Figure 2) [35,36]. The overwhelming impact of α5 integrins further leads to the activation of the endothelial inflammatory pathway, which mediates the engulfment of ox-LDL by monocytes (foam cells) and their deposition in the vascular intima, ultimately contributing to the development of atherosclerotic plaques (Figure 2) [35,36]. In addition, it has been elucidated that the overexpression of α5β1 integrins present in the ECM can also induce VSMC calcification, which drives the trans-differentiation of VSMC into osteochondrocyte-like cells and ultimately causes the deposition of calcium salts in the arterial wall and cardiac valves [58]. Zhu et al. indicated that the overexpression of α5β1 integrins present on VSMCs and the abnormal interaction between α5β1 integrins and α-smooth muscle actin (α-SMA) on VSMCs led to vascular tone dysregulation, resulting in altered contraction function, excessive migration, and the proliferation of VSMCs, ultimately resulting in AS (Figure 2) [40]. Previous research confirmed that the upregulation of α5β1 integrins on the VSMC cell membrane mediates the transition of VSMC from a “contractile” to a “synthetic” phenotype, which significantly contributes to VSMC proliferation and migration, thus playing a crucial role in the progression of AS [41]. It has been reported that the excessive activation of α5β1 integrins that are present on ECs by ox-LDL mediates the expression of endothelial pro-inflammatory genes, such as tumor necrosis factor (TNF)-α, IL-1β, and IL-6, which results in atherosclerotic inflammation (Figure 2) [59]. One study elucidated the critical role of α5β1 integrins on macrophages in promoting cell adhesion through excessive interaction with the immune cell-specific Eph receptor A2 [50]. Budatha et al. showed that α5β1 and αVβ3 integrins located on the plasma membranes of ECs are oversensitive to inflammatory stimuli when stimulated by fibronectin [60]. This sensitivity promotes microscopic changes during atherosclerosis, such as fibronectin deposition, the activation of EC inflammation, lipid accumulation, and thickening of the arterial wall [60]. Furthermore, it has been shown that the α5β1 and αVβ3 integrins located on the plasma membrane of VSMCs can be excessively activated by changes in the ECM, inducing the cytoplasmic translocation of focal adhesion kinase (FAK) and increasing the activity of proline-rich tyrosine kinase 2 (Pyk2), which promotes the rapid proliferation of VSMCs and plaque formation (Figure 2) [61]. Moreover, studies have demonstrated that the aberrant interaction between αVβ3 integrins on platelets and fibronectin contributes significantly to thrombosis within the injured vessel wall. This interaction is pivotal for platelet adhesion and aggregation on atherosclerotic plaques, thereby highlighting its importance in the pathophysiology of vascular events associated with atherosclerosis [56,57,62]. Okamoto et al. demonstrated that vascular stiffness and shear stress upregulate α5β1 integrins on the EC membrane and connexin in the ECM; the interaction of α5β1 integrins and connexin enhances vascular inflammation and leads to atherogenesis by promoting the activation of NF-κB and the expression of pro-inflammatory genes (Figure 2) [63]. Conversely, αVβ3 integrins and β1 integrins with talin-induced binding enhance the integrity of cell–cell junctions and protect endothelial barrier function [64]. Additionally, noteworthy studies highlighted that under pathological conditions, the levels of αV integrins on the surface of endothelial microparticles, which are vesicles derived from the EC membrane, increase and exert proinflammatory effects through their interaction with monocytes to promote atherosclerosis development [44]. Notably, the existing literature has consistently documented the involvement of αVβ5 integrins that are present on macrophages, which become activated through peroxisome proliferator-activated receptor gamma (PPARγ) [45]. This activation culminates in the polarization of macrophages towards the M2 phenotype with the subsequent expression of anti-inflammatory factors, such as interleukin-10 (IL-10) [45]. This orchestrated response promotes tissue repair and fosters clot formation, illustrating the multifaceted impact of integrins in ameliorating atherosclerotic processes [45]. Recent studies have reported the role of omentin-1, a newly discovered cytokine predominantly secreted by epicardial adipose tissue and characterized by a fibrinogen-like domain, that is upregulated in AS [46]. Omentin-1 has been shown to interact with αVβ5 integrins located on the cell membrane of macrophages, eliciting the downstream activation of the Akt/AMPK signaling cascade (Figure 2) [46]. This signaling axis exerts inhibitory effects on the formation of necrotic cores and the expression of pro-inflammatory cytokines within atherosclerotic lesions, thereby fostering plaque stability [46]. Extensive research has illuminated the dual effects of osteopontin (OPN) on a spectrum of integrins, including αVβ1, αVβ3, αVβ5, and α8β1 integrins [47]. The acute elevation of OPN levels confers a protective effect by mitigating vascular calcification and promoting favorable ischemic neovascularization. Conversely, chronic elevation in OPN levels is clinically correlated with an augmented susceptibility to major adverse cardiovascular events [47]. Furthermore, it has been reported that αIIbβ3 integrins exert considerable influence on platelet functionality, with pivotal roles in activation, migration, and secretion [54,55,65,66]. Research has elucidated that the bivalent interaction between αIIbβ3 integrins presenting on platelets and fibrinogen facilitates platelet aggregation, thereby contributing to thrombus formation [65]. For example, studies have demonstrated that reelin, a glycoprotein released by platelets, exerts an overamplifying effect on the αIIbβ3 integrin-mediated outside-in signaling pathway by activating the Ras-related C3 botulinum toxin substrate and Ras homolog family member A (RhoA) [54,66]. This augmentation facilitates the cytoskeletal reorganization of platelets, thereby enhancing thrombus formation [54,66]. In contrast, the activation of αIIbβ3 integrins has been shown to be inhibited by GNE 495 and PF 06260933 (both potent and selective MAP4K4 inhibitors), the primary outcome of which is mitigating platelet aggregation and clot retraction [55]. Furthermore, Chen et al. highlighted that β1 integrins located on the plasma membrane of VECs are upregulated in response to galectin-3 stimulation, leading to the activation of the β1-RhoA-JNK signaling pathway, thereby exacerbating ox-LDL-mediated vascular endothelial injury, promoting inflammation, and facilitating foam cell deposition (Figure 2) [37]. Research has shown that β1 integrins expressed on the plasma membrane of VSMCs promote the overproduction of matrix metalloproteinases 2 (MMP) through the regulation of milk fat globule-epidermal growth factor 8 (Figure 2) [15]. Ultimately, this triggers the redundant activation of transforming growth factor-β1 (TGF-β1), which is incorporated into atherosclerotic plaques, and induces osteogenic differentiation and biomineralization in VSMCs, facilitating vascular wall calcification and sclerosis (Figure 2) [15].
2.1.2. Integrins That Bind to Laminin Receptors
The β4, α6β1, and β3 integrins that are present on ECs and VSMCs play roles in the inflammation and plaque formation of AS. Research has shown that β4 integrins on the plasma membrane of ECs activates a positive feedback signaling pathway by activating SRC and downstream NF-κB, the activation of which contributes to excessive transcriptional activity of β4 integrins; the activation of the pathway then upregulates the expression of endothelial inflammatory factors, leading to the development of AS (Figure 2) [38]. Furthermore, a similar mechanism has been reported in which the α6β1 integrins present on ECs bind to the EC-expressed matrix protein CCN1 to activate NF-κB, which forms a positive feedback loop with the CCN1 and α6β1 integrins and amplifies oxidative stress and inflammation in the arterial wall (Figure 2) [39]. Additionally, a previous study has confirmed that the β3 integrins located on the cell membrane of VSMCs can be overstimulated by thrombin and then interact with CD47 to mediate VSMC migration and proliferation (Figure 2) [41]. This process possibly leads to increased vascular wall thickness and the formation of subendothelial plaques, which promotes vascular restenosis following percutaneous transluminal angioplasty [41].
2.1.3. Integrins That Bind to Leukocyte-Specific Receptors
The β2, αMβ2, αDβ2, β1, and α4β1 integrins expressed on the cell membranes of ECs, VSMCs, and immune cells participate in AS development. Krautter et al. highlighted that the overproduction of β2 integrins with galectin-9, which is secreted by VECs, leads to the activation and differentiation of monocytes [42]. This molecular interaction consequently amplifies the advancement of atherosclerotic plaque formation [42]. Previous research has underscored the interaction between β2 integrins expressed on neutrophils and endothelial cell adhesion molecules, including E-/P-selectins and ICAM-1, particularly on human umbilical vascular endothelial cells [51]. The excessive synthesis of β2 integrins causes increased interaction that activates the Syk/Src signaling pathway, leading to a heightened calcium flux within adherent neutrophils (Figure 2) [51]. Consequently, this cascade of events enhances vascular wall stiffening and exacerbates immune responses associated with AS [51]. In addition, the upregulation of β2 integrin expression on neutrophils is facilitated by the intracellular translocation of peptidyl arginine deiminase 4 triggered by the chemokine (C-X-C motif) ligand 1 protein [53]. This phenomenon significantly contributes to the adhesive characteristics of neutrophils [53]. A previous study suggested that β2 integrins on the plasma membrane of eosinophils, activated by P-selectin glycoprotein ligand 1 and P-selectin, upregulate and bind to ICAM-1, leading to the firm adhesion of eosinophils to the vascular endothelium [52]. In addition, an investigation has demonstrated that developmental endothelial locus-1, which is a secretory protein, modulates the interaction with αMβ2 integrins presenting on leukocytes, thereby impeding αMβ2 integrins from binding to their endothelial counterpart, ICAM-1 [67]. Consequently, this impediment attenuates the adhesion and subsequent recruitment of leukocytes to inflamed sites [67]. Furthermore, Cui et al. demonstrated that the oxidative derivative resulting from the interaction of docosahexaenoic acid and 2-(w-carboxyethyl)-pyrrole acts as a ligand, facilitating binding to αDβ2 integrins situated on M1 macrophages [43]. This interaction enhances the accumulation of macrophages within the ECM of peripheral tissues, thereby contributing to the development of AS [43]. Moreover, previous investigations have shown that fatty-acid-binding protein 4 (FABP4), recognized as a member of the adipocyte fatty-acid-binding protein family, functions as an intracellular fatty acid transporter within macrophages [48,49]. Notably, its expression is upregulated by ox-LDL in monocytes and human coronary endothelial cells (HCAECs) [48,49]. Extensive research has delineated that the binding of FABP4 to α4 integrins, which are expressed on the plasma membrane of monocytes and HCAECs, facilitates the adhesion of monocytes and HCAECs [48,49]. Furthermore, cytotoxic T lymphocytes were reported to exhibit elevated expression levels of α4β1 integrins in response to various cytokines [68]. This upregulation is associated with the release of inflammatory mediators, thereby facilitating immune cell recruitment and contributing to the formation of atherosclerotic plaques [68]. A study describing the IncRNA associated with the progression and intervention of AS (RAPIA) showed that the β1 integrin/microRNA-183-5p signaling pathway in macrophages is upregulated in the presence of RAPIA expression, consequently promoting the progression of AS [69].
2.2. Cardiac Fibrosis
Cardiac fibrosis is a common terminal stage in various CVDs, such as myocardial infarction (MI), hypertension, and heart failure [22,70,71]. It refers to the increased deposition of collagen and other molecules within the ECM, resulting in changes in cardiac structure and function [22,70,71]. Research has indicated that immune cells, including monocytes, macrophages, and neutrophils, mediate inflammatory progression and ECM remodeling, while fibroblasts synthesize collagen to promote collagen deposition in the ECM [72,73,74,75]. The pathogenesis of cardiac fibrosis mainly involves the actions of fibroblasts, which cause cardiac remodeling mediated by collagen synthesis and deposition, and therefore, this section has categorized and summarized the role of cardiac cells, primarily fibroblasts, based on different integrin types. Many studies have elucidated the presence of integrins on cardiac cells, cardiac fibroblasts, and immune cells, highlighting their critical involvement in the regulation of cellular activities associated with cardiac fibrosis (Table 2).
Table 2.
The interaction between integrins on specific cells and ECM components in cardiac fibrosis.
2.2.1. αV Integrins
Numerous studies have shown that αV integrins play a pivotal role in the progression of cardiac fibrosis. They primarily participate in fibroblast proliferation, adhesion, migration, and differentiation and monocyte and macrophage migration and adhesion, thereby mediating cardiac inflammation and collagen deposition [72,73,78,83]. Moreover, αVβ5 and αVβ3 integrins on the cell membrane of human cardiac fibroblasts (CFBs), induced by mechanical stress from fibroblast contraction, demonstrate conformational changes and mediate the excessive activation and release of TGF-β1, promoting fibroblast differentiation into collagen-producing myofibroblasts in the ECM [84]. Furthermore, Olivier et al. highlighted that αV integrins facilitate the over-release of TGF-β from latent TGF-β stores by redundantly interacting with the RGD motif on latent TGF-β in the ECM [85]. Similarly, previous research indicated that the overexpression of αV integrins on the plasma membrane of CFBs bind to the RGD site of latency-associated peptide (LAP) and lead to the excessive activation and release of TGF-β1, promoting the proliferation and collagen secretion of CFBs and inflammatory responses, thereby further enhancing the development of cardiac fibrosis [76]. Furthermore, several studies have confirmed that the upregulation of αVβ3 and αVβ5 integrins on fibroblasts can significantly induce the release of TGF-β1 into the ECM and further activate the TGFβ1/Smad3 pathway through autocrine signaling to enhance the expression of α-SMA, which facilitates fibroblast trans-differentiation and the synthesis of type I collagen, ultimately resulting in cardiac fibrosis-induced heart dysfunction (Figure 2) [77,86]. In addition, Patrick et al. indicated that excessively stimulated αVβ5 integrins present on CFBs bind to FAK, an integrin-associated protein, to promote the overproduction of α-SMA and type I collagen, the result of which is associated with myofibroblast transformation and collagen deposition (Figure 2) [87]. Investigations have demonstrated that overactivated αVβ3 integrins on CFBs bind to ECM proteins containing the RGD motif to activate c-Src, Pyk2, and FAK downstream, which mediate the adhesion, spreading, proliferation, and migration of fibroblasts and induce the accumulation of interstitial fibronectin and collagen within the ECM (Figure 2) [78]. Similarly, Chen et al. demonstrated that αVβ3 and α8β1 integrins, located on the cytoplasmic membrane of VSMCs and myofibroblasts, can be upregulated by angiotensin II, ultimately facilitating collagen contraction [73]. Additionally, the findings revealed that the upregulated αVβ3 integrins located on the plasma membrane of CFBs facilitate the αVβ3 integrin-dependent excessive activation of pro-MMP2 in aortic smooth muscle cells (ASMCs), and the activated MMP2 can degrade the ECM, which results in the breakdown and remodeling of constitutive proteins, such as fibronectin and collagen [88]. In addition, αV integrins regulate the function of immune cells and cytokines. The αVβ3/FAK-Src/NF-κB signaling pathway is utilized by TN-C (an ECM protein) to accelerate macrophage migration and the synthesis of proinflammatory/profibrotic cytokines, thereby enhancing the inflammatory response and increasing the synthesis of collagen I by fibroblasts, which consequently advances fibrotic progression [73,89]. Previous research highlighted that the αVβ3-dependent activation of microfibrillar-associated protein 4 (MFAP4) facilitates monocyte migration and directly promotes the MMP9-mediated degradation of the vascular wall induced by TNF [83]. Furthermore, αVβ3 integrins located on the plasma membrane of ASMCs have been shown to stimulate the increased proliferation of CD25+ FoxP3+ Treg cells and IL-10 expression, thereby mediating the recruitment of inflammatory cells and the production of pro-inflammatory cytokines [88]. Moreover, the upregulation of platelet-derived growth factor receptor beta (PDGFRβ), which is dependent on αV integrins in mesenchymal cells, can induce cardiac fibrosis [90]. It has been shown that the αV/β5-AKT signaling pathway of cardiomyocytes in type 2 diabetes stimulates the apoptosis of cardiomyocytes and myocardial fibrosis [91]. Additionally, research has elucidated that MFAP4, located in the connective tissue of the aortic wall, binds to RGD and excessively interacts with αVβ3 integrins located on the plasma membrane of VSMCs to transmit pro-fibrotic ECM signals to VSMCs, thus promoting the phenotypic conversion, dedifferentiation, proliferation, and migration of VSMCs [83].
2.2.2. β1 Integrins
β1 integrins can produce a positive effect on the activation, proliferation, and differentiation of fibroblasts, as well as the synthesis and contraction of collagen. The overexpression of αVβ1 integrins on the cell membrane of fibroblasts has been shown to stimulate TGF-β1 expression and αVβ1-mediated FAK-Akt-mTOR and TGF-β receptor-mediated Smad2/3 signaling pathways to promote the internal transcription of α-SMA in fibroblasts, which induces the transition of fibroblasts to myofibroblasts (Figure 2) [72,92,93,94,95]. Similarly, previous research highlighted that the overactivation of β1 integrins on the fibroblast plasma membrane by ECM proteins, such as fibronectin, triggers the expression of phosphorylated FAK protein and α-SMA, ultimately promoting the migration of fibroblasts (Figure 2) [80]. β1 integrins mediate myocardial fibrosis via TGF-β signaling, with the participation of Smad2/3, a-smooth muscle actin, and collagen I (Figure 2) [79]. Previous research showed that α5β1 integrins present on the cell membrane of myofibroblasts induce the overproduction of fibronectin to mediate the further differentiation of myofibroblasts, which stimulates myofibroblasts to synthesize and secrete more collagen to enhance the deposition and reconstruction of the ECM [96]. It has been shown that α1β1 and α3β1 integrins can also promote myofibroblast differentiation and redundant collagen synthesis [96]. Leask et al. indicated that the upregulation of α11 and β1 integrins present on the cell membrane of myofibroblasts can very strongly activate downstream signaling pathways, including FAK, TGF-β-activated kinase, and reactive oxygen species by identifying high-stiffness ECM, resulting in the increased production, adhesion, and contraction of the ECM [74]. Furthermore, a recent study demonstrated that the overexpression of β1 integrins on the cytoplasmic membrane of fibroblasts activate PDGFRβ redundantly by acting on small proline-rich repeat 3 (SPRR3), which then crosstalks with PDGFRβ to regulate the Akt, FAK, ERK, and p38 signaling pathways to enhance fibroblast proliferation, collagen expression, and interstitial fibrosis [97]. Additionally, it has been reported that the overexpression of β1 integrins present on the plasma membrane of fibroblasts can identify stiffness in the ECM after MI by recognizing collagen accumulation in fibrotic tissues and triggering intracellular signaling in fibroblasts, which express vestigial-like family member 3, enhancing collagen synthesis [98]. Moreover, a previous study highlighted that the increasing association of β1 integrins and CD63 (a tissue inhibitor of metalloproteinase-1 (TIMP1) cell surface receptor) on CFBs is mediated by TIMP1 to initiate the activation and nuclear translocation of Smad2/3 and β-catenin, contributing to de novo collagen overproduction [99]. Upregulated αV integrins on the cytoplasmic membrane of cardiac PW1+ stromal cells, a type of cell that can differentiate into many cell types of the heart and is related to regeneration and repair, activate TGF-β on the surface of the cells, thus promoting the overexpression of fiber genes in cardiac PW1+ stromal cells and mediating PW1+ cells to differentiate into fibrocytes after MI [100]. In addition, it has been reported that the excessive interactions between α2β1 integrins on CFBs and type I collagen regulate the overproduction of lysyl oxidase, which catalyzes the crosslinking between collagen and elastin to form a more stable and compact network of collagen fibers, thereby increasing the persistence of fibrotic regions [101]. Furthermore, recent research has found that α7β1 integrins located on the cardiomyocyte cytoplasmic membrane are laminin receptors that facilitate laminin deposition in the cardiomyocyte lamina [102].
β1 integrins also have inhibitory effects on cardiac fibrosis. Combinations of different α subunits of β1 integrins lead to different types of integrins involved in different signaling pathways. Furthermore, α2β1 integrins located on the plasma membrane of fibroblasts recognize stiff ECM and trigger the downstream signaling pathways that induce FAK and Src kinase to inhibit collagen accumulation in human atrial fibroblasts [81]. Hong et al. indicated that α2β1 integrins on the cytoplasmic membrane of myofibroblasts regulate their downstream target PTEN through excessive crosstalk with PP2A and promote the overactivation of PETN, thus over inhibiting the activation of AKT and the expression of α-SMA, which suppresses the differentiation and proliferation of myofibroblasts (Figure 2) [82]. Additionally, α2β1 integrins present on the plasma membrane of fibroblasts can induce the upregulation of MMP1 to prevent collagen accumulation in the ECM, thereby protecting cardiomyocytes from fibrosis (Figure 2) [96]. Notably, we found that α2β1 integrins both promote and inhibit cardiac fibrosis. This was related to the different sources of cells (e.g., mice or aortic stenosis patients) and different modeling methods (e.g., mouse aortic ligation, gene editing, or fibrosis induction factor cultured cells).
2.2.3. Other Integrins
Several studies have concluded that other types of integrins can also play an important role in the development of cardiac fibrosis. Moreover, α11 integrins present on the cell membrane of fibroblasts can stimulate Smad2/3 in the promoter of α11 integrins by a TGF-β-dependent mechanism to promote the overexpression of α11 integrins, thereby mediating myofibroblast formation and extremely high type I collagen deposition [103]. Similarly, previous studies highlighted that α11, α5, β3, and α2 integrin synthesis is accelerated by the binding of TGF-β to its receptors on fibroblasts, which activates fibroblast trans-differentiation into myofibroblasts to synthesize type I and III collagens [104,105]. It has been reported that the expression of α11 integrins in patients with diabetes can be induced by the TGF-β2 autocrine signaling pathway in fibroblasts, which is triggered by glycated collagen in the cardiac stroma [106]. Previous research showed that α5 integrins on the plasma membrane of fibroblasts bind to miR-30d expressed by cardiomyocytes to inhibit fibroblast proliferation and activation, thereby inhibiting pathological left ventricle remodeling and fibrosis [107]. In addition, Liu et al. indicated that the high transcription of β2 integrins in macrophages is coordinated by myocardin-related transcription factor A and the transcription factor Sp1, contributing to the adhesion and migration of macrophages, thus promoting inflammation and ECM remodeling [75]. Upregulated αMβ2 integrins on the neutrophil plasma membrane can facilitate the increasing infiltration of neutrophils into atrial tissue, thereby facilitating the development of an inflammatory response and atrial fibrosis [73]. Furthermore, it is believed that αIIbβ3 integrins on platelets increase their expression upon the stimulation of 5-HT released from activated platelets, enhancing binding between platelets and fibronectin and the coagulation process, and they participate in inflammatory response and tissue repair, which ultimately contributes to the development of cardiac fibrosis [108]. A previous study elucidated that the upregulation of α4 integrins located on the cell membrane of type 1 T helper cells (Th1) mediate the increasing adhesion of Th1 cells in an IFN-γ-dependent manner and induce the over release of TGF-β from CFBs and the transition of CFBs to myofibroblasts [109]. It has been reported that the excessive activation of α4 integrins present on the cell membranes of monocytes and macrophages mediates the increased adhesion and infiltration of monocytes and macrophages to cardiac tissues, and monocytes can transform into macrophages that release inflammatory mediators and MMPs under inflammatory stimuli, thereby participating in the progression of cardiac inflammation and ECM remodeling [110].
2.3. Arrhythmias
Arrhythmias represent a significant health burden and are characterized by disruptions in cardiac rhythm [17,20,111,112,113,114,115,116,117]. They often stem from diverse cardiomyopathies, including valvular heart disease, arrhythmogenic cardiomyopathy (ACM), arrhythmogenic right ventricular cardiomyopathy, and cardiac aging [17,20,111,112,113,114,115,116,117]. The pathogenesis of arrhythmias involves myocardial damage, dysfunction in myocardial electrical signaling, and cytoskeletal remodeling [17,20,111,112,113,114,115,116,117]. Extensive research has underscored the pivotal role of integrins, which are prominently expressed on cardiomyocytes, in the pathogenesis of arrhythmias (Table 3).
Table 3.
The interaction between integrins on specific cells and ECM components in arrhythmias.
Integrins play a crucial role in cytoskeletal remodeling and other essential biological processes through their interaction with the ECM [20]. The β1 integrin/integrin-linked kinase (ILK) signaling pathway has been reported to exert a dual regulatory function in the modulation of cardiac aging [20]. This pathway is implicated in the colonization proximal to cardiomyocyte contacts and Z-bands, which are pivotal for the establishment and preservation of the structural integrity of the heart [20]. A slight downregulation of the β1 integrin/ILK signaling pathway has been demonstrated to attenuate age-related arrhythmia deterioration and prolong lifespan [20]. In other investigations, β1 integrins have been proposed to phosphorylate connexin-43 (Cx43), which is the predominant constituent of the gap junction channels found on cardiomyocytes (Figure 2) [111,112,113]. This phosphorylation event is believed to occur via the aberrant activation of the ILK/Akt/Cx43 signaling pathway (Figure 2) [111,112,113]. Consequently, it is suggested that this molecular mechanism attenuates cardiac remodeling and mitigates the occurrence of ischemia/reperfusion-induced ventricular arrhythmias [111,112,113]. Additionally, vinculin is localized at cell-to-cell intercalated discs and cell-to-matrix adhesion sites (Figure 2) [17]. It functions as an actin-binding protein, organizing and anchoring the actin cytoskeleton to the cardiomyocyte membrane via β1 integrin-containing focal adhesions (Figure 2) [17]. This anchoring mechanism contributes to the maintenance of a stable gap junction containing Cx43 [17]. However, vinculin is suppressed in ACM, resulting in cardiac fibrosis and arrhythmias [17]. Furthermore, contemporary investigations have highlighted that severe ventricular arrhythmias provoked by RyR2 malfunction represent notable features of arrhythmogenic right ventricular cardiomyopathy, an inherited desmosomal cardiomyopathy [114]. The absence of mutations in desmosomal genes (DSC, DSG, and DSP) leads to elevated fibronectin synthesis and secretion, promoting fibronectin binding to β1 integrins and their subsequent degradation (Figure 2) [114]. The reduction in β1 integrins present on the cell membrane of cardiomyocytes results in an upregulation of RyR2 Ser-2030 phosphorylation levels, leading to a destabilization of the RyR2 protein structure (Figure 2) [114]. Consequently, this disruption contributes to intracellular calcium dysregulation, manifesting as calcium leakage, which, in turn, triggers ventricular tachycardia (Figure 2) [114]. Furthermore, calreticulin, an endoplasmic reticulum calcium-buffering protein, has been found to play a role in mediating atrial fibrillation in valvular heart disease (Figure 2) [115]. This involvement occurs through the excessive activation of the TGF-β1/fibronectin/α5 integrin signaling pathway in fibroblasts, leading to heightened fibroblast proliferation and dysregulated ECM deposition within the cardiac interstitium (Figure 2) [115]. In the pathogenesis of arrhythmias observed in ACM that result from genetic mutations affecting the desmosomal cell–cell adhesion complex, particularly the desmoglein-2-W2A mutation, it has been reported that aberrantly activated αVβ6 integrins engage with LAP, leading to the dissociation of the TGF-β1-LAP complex [116]. Subsequently, increasing the TGF-β1 levels initiates a profibrotic downstream signaling cascade mediated through SMAD molecules [116]. The subsequent outcomes encompass fibrofatty deposition and potentially fatal arrhythmia [116].
2.4. Hypertension
As a widespread contributor to cardiovascular mortality and morbidity, hypertension entails pathological ECM–integrin interactions [21,118]. This contributes to a spectrum of biological effects, including increased blood vessel rigidity, the migration and proliferation of ASMCs, EC dysfunction, and the formation of neointima within the vascular wall [21,118]. Multiple studies have revealed the presence of integrins on ASMCs and ECs, highlighting their crucial role in regulating cellular activities linked to hypertension (Table 4).
Table 4.
The interaction between integrins on specific cells and ECM components in hypertension.
Drawing on pulmonary arterial hypertension (PAH) as an illustrative example, extensive documentation exists regarding the interaction between β3 integrins on the cell membrane of pulmonary artery smooth muscle cells (PASMCs) and MMP8, which is secreted by PASMCs (Figure 2) [119]. This interaction subsequently orchestrates the activation of the FAK/Yes-associated protein/transcriptional coactivator with the PDZ-binding motif signaling pathway, thereby facilitating PASMC proliferation and instigating vascular remodeling in the context of PAH (Figure 2) [119]. Furthermore, hypoxia has been identified as a critical factor in the pathogenesis of PAH [120]. It is implicated in the aberrant activation of the α5β3 integrin/Pyk2/ERK/NF-kB/Nox4/H2O2 signaling cascade in PASMCs, which subsequently downregulates peroxisome PPARγ expression (Figure 2) [120]. This downregulated PPARγ expression is associated with the promotion of PASMC proliferation [120]. Similarly, hypoxia can also mediate the activation of the α5β3 integrin/osteoprotegerin/FAK signaling pathway, leading to PAH (Figure 2) [61,121]. Osteoprotegerin, belonging to the tumor necrosis factor receptor superfamily and expressed in cardiac tissue under hypoxic conditions, has been investigated for its potential contribution to PAH [121]. This is postulated to occur through the aberrant activation of the αVβ3 integrin/FAK/AKT signaling pathway in PASMCs, ultimately leading to the proliferation of PASMCs (Figure 2) [121]. Furthermore, numerous studies have found that αVβ3 integrins located on the cytoplasmic membrane of PASMCs can bind to OPN, a pivotal contributor to the activated phenotype of PASMCs in hypoxic PAH [122]. Subsequently, this interaction mediates the aberrant activation of the ERK1/2 and AKT signaling pathways, culminating in the proliferation of PASMCs (Figure 2) [122]. In addition, these findings revealed that the expression of β5 integrins on the cell membrane of PASMCs is abnormally upregulated by platelet-derived growth factor beta polypeptide b (PDGF-BB) stimulation [123,124]. Elevated levels of β5 integrin aberrantly activate dual pathways [123,124]. β5 integrins engage with miR-96-5p, consequently suppressing its activity [123]. The diminished expression of miR-96-5p results in the upregulation of mTOR levels, thereby fostering a synthetic phenotype of PASMCs [123]. Alternatively, β5 integrins upregulate the expression of the Uba1 protein, activating Ube2n and Mdm2 (Figure 2) [123,124]. This cascade of events leads to the downregulation of angiotensin-converting enzyme 2 expression and the induction of a synthetic phenotype in PASMCs (Figure 2) [123,124]. Consequently, this molecular mechanism contributes to pulmonary artery remodeling and the pathogenesis of PAH [123,124]. In the context of PAH, the progression of vascular stiffness and the consequential loss of elastic properties are attributed to the involvement of the overactivation of β5 integrins [125]. β5 integrins have been demonstrated to promote barrier functions while inhibiting the endothelial-to-mesenchymal transition gene expression induced by TGF-β [125]. In addition, a plethora of studies have underscored the interaction between upregulated α5β1 integrins located on VSMCs and fibronectin, leading to VSMC adhesion [40,60,126]. Furthermore, fibronectin has been implicated in fostering inflammation by engaging α5β1 integrins on VECs, culminating in vascular remodeling and hypertension as observed in subsequent outcomes [40,60,126]. Consistently, recent research showed that the aberrant activation of αVβ5 integrin triggers the TGF-β/SMAD2/3 signaling pathway (Figure 2) [86]. This aberrant activation, in turn, promotes the upregulation of α-SMA and facilitates the differentiation of fibroblasts into contractile myofibroblasts (Figure 2) [86]. Consequently, this process contributes to the induction of ECM stiffness, which has been associated with the pathogenesis of hypertension [86]. Apart from these findings, research has revealed that pregnancy-induced hypertension represents a notable form of hypertension attributable to a deficiency in activin A [127]. This deficiency exerts its influence by suppressing the ActRII/ALK4/SMAD2/3/4 signaling pathway, subsequently upregulating β1 integrin expression on trophoblasts [127]. Consequently, this suppresses trophoblast invasion into the uterine wall tissue, ultimately culminating in vascular remodeling and the manifestation of pregnancy-induced hypertension [127].
3. Integrin-Based Therapy
The development of integrin-based therapies for CVDs is progressing slowly, with the majority of therapeutic strategies still in the research phase. Presently, two main directions for integrin therapy exist. The first involves the use of integrin antagonists or antibodies to block the binding of integrins with ligands, thereby inhibiting signaling pathway transmission and disease progression [8,19,24]. The second involves exploiting the binding mechanism between integrins and ligands to fabricate nanoparticles (NPs) for drug encapsulation, aiming to enhance targeting specificity and bioavailability, delay drug release, and mitigate drug toxicity [128,129,130,131,132]. Figure 3 and Table 5 summarize the application of integrins in these two therapeutic approaches.
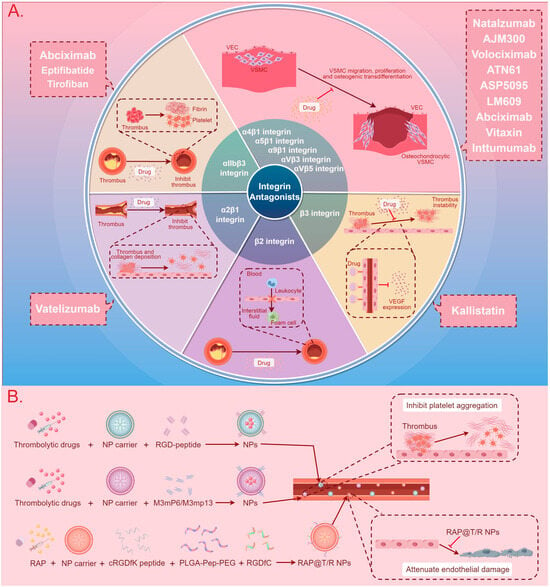
Figure 3.
A summary diagram of integrin-based cardiovascular disease treatment methods and therapeutic drugs. (A) The use of integrin antagonists to block the binding of integrins with ligands, thereby inhibiting signal pathway transmission and CVD progression. (B) The use of NPs to enhance targeting specificity and bioavailability, delay drug release, and mitigate drug toxicity in the treatment of CVDs. Abbreviations: CVD: cardiovascular disease, VEC: vascular endothelial cell, VSMC: vascular smooth muscle cell, VEGF: vascular endothelial growth factor, NP: nanoparticle, RGD-peptide: arginine–glycine–aspartate peptide, RAP: rapamycin, cRGDfK peptide: cyclic arginyl–glycyl–aspartic acid–phenylalanine–lysine peptide, PLGA-Pep-PEG: poly (lactic-co-glycolic acid)–peptide–polyethylene glycol, RGDfC: Arg-Gly-Asp-Phe-Cys, RAP@T/R NPs: RAP targeted and responsive nanoparticles.
Table 5.
Integrin-based cardiovascular disease treatment methods and therapeutic drugs.
3.1. Integrin Antagonists, Antibodies, and Inhibitors
It has been demonstrated that αIIbβ3 integrins, situated on the surface of platelets, play a crucial role in mediating platelet binding to fibrinogen, thus promoting platelet spreading, aggregation, clot retraction, and thrombus stabilization (Figure 3) [18]. Consequently, αIIbβ3 integrin antagonists are employed in anti-thrombotic therapy for CVDs [18]. Numerous studies have demonstrated that αIIbβ3 integrin antibodies, including abciximab, and αIIbβ3 integrin inhibitors, including eptifibatide (RGD-mimetics) and tirofiban (RGD-mimetics), inhibit platelet binding to fibrinogen (Figure 3) [24,133,134,135,136,137,158]. These drugs treat acute coronary syndromes, including unstable angina and MI, and reduce the risk of bleeding after percutaneous coronary intervention compared to traditional antiplatelet therapy [24,133,134,135,136,137,158]. In addition, Hind et al. indicated that α2β1 integrin antagonists can suppress thrombosis by inhibiting the adhesion of platelets to collagen and the expression of collagen and collagenase genes (Figure 3) [134]. Research has shown that kallistatin, a unique serine proteinase inhibitor, can bind to β3 integrins to inhibit NF-κB nuclear translocation and downregulate vascular endothelial growth factor expression, thus suppressing angiogenesis and the progression of atherosclerotic plaques (Figure 3) [154]. Furthermore, recent research highlighted that antibodies against β2 integrins suppress leukocyte extravasation mediated by binding between leukocytes and ICAM-1 to reduce inflammatory tissue damage, thus treating AS development and plaque progression, MI, reperfusion injury, valvular stenosis, and cardiomyopathy (Figure 3) [8]. Additionally, it has been reported that antibodies against α2β1 integrins (vatelizumab) inhibit the transition of VSMCs from a “contractile” to a “synthetic” phenotype, which is known as SMC phenotypic modulation, and promote the aberrant proliferation, differentiation, and migration of VSMCs mediated by α2β1 integrins, thereby suppressing the formation of atherosclerotic plaques [142,159,160]. Similarly, the antibodies and antagonists of α4β1, α5β1, α9β1, αVβ3, and αVβ5 also restrain the chemotaxis of VSMCs and neointimal hyperplasia to limit the expansion of plaques and platelet aggregation (Figure 3) [141,142,144,145,146,147,150,151,152,153].
3.2. Nanotherapy
Nanomedicine has the potential to overcome certain obstacles faced by traditional medicine. Some of its advantages include high stability, high drug-carrying capacity, and various drug delivery modalities [155,161,162]. Overall, nanodrugs can be used in many ways to complement current therapeutic regimens, and the further exploration of NPs in CVDs will allow nanomedicine to reduce the global burden of CVDs. Several studies have reported the use of RGD-peptide, which specifically binds to the αIIbβ3 integrin ligand, as a surface molecule on nanocarriers for loading thrombolytic drugs. This enables targeted drug delivery to sites of platelet aggregation, thereby reducing the bleeding and toxicity risks associated with systemic administration (Figure 3) [19,155]. The selective inhibition of outside-in signals shows potential for inducing robust antithrombotic effects while mitigating the risk of bleeding complications [23]. The use of nanocarriers containing the Gα13-binding ExE motif within the integrin β3 cytoplasmic domain (M3mP6 or M3mp13) selectively targets the intracellular and extracellular signaling pathways of αIIbβ3 integrin, which effectively prevents secondary thrombus expansion mediated by outside-in signaling, subsequent vascular occlusion, and intravascular thrombosis, while allowing for primary platelet adhesion and aggregation, and was thus applied in the treatment of acute coronary syndrome and thrombotic cardiovascular events (Figure 3) [23,129,156]. Similarly, Linsey et al. demonstrated that ανβ3 integrins located on the surface of ECs are targeted by NP-wrapping cRGDfK peptides to deliver drug-containing NPs to atherosclerotic plaques (Figure 3) [130]. In addition, αVβ3 integrin-targeted and cathepsin k (CTSK)-responsive NPs are employed for the localized controlled release of the anti-inflammatory drug rapamycin (Figure 3) [157]. Rapamycin-targeted and responsive NPs (RAP@T/R NPs), containing the targeting polymer PLGA-PEG-c and the CTSK-sensitive polymer PLGA-Pep-PEG, accelerate rapamycin release in response to CTSK stimulation, leading to the significant inhibition of inflammatory macrophage uptake of ox-LDL and cytokine release, thereby suppressing systemic and local inflammation (Figure 3) [157]. Additionally, it is reported that α4β1 integrin-modified macrophage membrane NPs promote their accumulation in atherosclerotic plaques by binding to VCAM-1 on ECs [128].
4. Discussion
The current understanding of the mechanisms underlying CVDs revealed by research remains incomplete. Thus, a more in-depth exploration of the molecular mechanisms and associations between integrins and other CVDs that are not explicitly mentioned in this paper is required. Looking forward, CVDs increasingly stand as one of the most serious public health challenges, posing a significant threat to global health [163,164]. Abundant research indicates that integrins, as ubiquitous transmembrane heterodimeric proteins, are widely distributed on the cell membranes of VSMCs, ECs, fibroblasts, platelets, immune cells, and cardiomyocytes, and they play a crucial role in mediating cell signaling and physiological functions within the cardiovascular system [9]. Through integration with various cellular processes, integrins become indispensable in regulating cell functions such as inflammation, proliferation, migration, adhesion, and differentiation. The dysregulation of integrin signaling and conduction can lead to a variety of pathophysiological mechanisms, ultimately contributing to the initiation and progression of CVDs [165]. In the context of AS, integrins can orchestrate plaque formation by promoting EC oxidative stress and inflammation activation, recruiting and promoting the proliferation of VSMCs, inducing calcification, and allowing for the infiltration of immune cells and platelets [15,35,44,62]. Moreover, in the progression of cardiac fibrosis, integrins play a pivotal role in modulating inflammation driven by immune cells, the remodeling of the ECM, and the synthesis of collagen, thus exerting a significant impact on fibrosis development [75,85,108]. Additionally, in the realm of arrhythmias, integrins are implicated in the regulation of myocardial injury, dysfunction in cardiac electrical signaling, and the reconstruction of the cellular cytoskeleton, establishing a close association with the occurrence of rhythm disorders [20]. Furthermore, integrins contribute to the cellular and molecular mechanisms underlying the development of hypertension, participating in the regulation of increased vascular stiffness, VSMC migration and proliferation, EC dysfunction, and the formation of neointima within the vascular wall [21,118]. Notably, the long-term loading of Na+ in high-sodium-salt-diet-sensitive hypertension induces the destruction and reduction in glycocalyx (a protein covering the VEC membrane as a sensor of blood flow shear stress), which affects the excitation–secretion coupling of VECs, the excitation–contraction coupling of VSMCs, and the cellular homeostasis of Ca2+, Na+, and reactive oxygen species, ultimately leading to endothelial damage [166]. Salt-sensitive hypertension and the remodeling of human vascular smooth muscle cells induced by high sodium levels permanently reshape the sensitivity of the cells to short-term increases in circulating sodium levels following normalization at the systemic level [167]. Given the complexity of the mechanisms, unraveling the complex network of molecular interactions and pathways associated with CVDs holds tremendous potential to deepen our understanding of these complex diseases. Through an in-depth investigation into the relevant mechanisms, researchers can reveal the potential role of integrins in other types of CVDs, expanding our current knowledge base. Additionally, investigating the complex interplay between integrins and various types of CVDs beyond those explicitly mentioned in this review may lead to the discovery of new therapeutic targets and treatment strategies. Researchers can identify shared pathways and mechanisms by studying the intricate interactions between integrins and different types of CVDs, providing targets for developing innovative therapies and interventions. Therefore, due to the incomplete understanding of the potential mechanisms underlying CVDs, a more comprehensive exploration of molecular complexity and unexplored associations between integrins and other CVDs is urgently needed. Conducting such scientific investigations could potentially pave the way for breakthroughs in the diagnosis, prevention, and treatment of CVDs, ultimately improving the overall health outcomes of future patients.
A broad potential for integrin-related therapeutic approaches becomes evident given the impact of integrins on CVDs. Current research primarily concentrates on the treatment of AS, employing two main modalities—antibodies or antagonists and nanotherapy [8,129]. Integrin-targeting antibodies and antagonists, which include agents countering platelet and VSMC proliferation and migration, can inhibit integrin-mediated pro-plaque effects, thereby impeding AS progression [18,159]. Antiplatelet therapy continues to be the standard treatment for thrombotic diseases in the heart, peripheral arteries, and cerebral arteries with atherosclerotic thrombosis [135]. Based on the molecular mechanisms of thrombosis, three αIIbβ3 antagonists are currently used clinically, including eptifibatide and tirofiban from snake venom disintegrins and abciximab [135]. While various oral integrin αIIbβ3 antagonists have undergone testing, clinical trials have been halted due to adverse effects, such as high mortality rates or the lack of improvement in existing treatments [135]. Future research directions may involve structure–function analyses based on exogenous disintegrins, aiding in identifying platelet antagonists that maintain hemostasis while minimizing adverse effects [135]. Additionally, anti-VSMC antibodies and antagonists, such as vatelizumab and AJM300, can achieve therapeutic effects by inhibiting the integrin-mediated transition of VSMCs from a contractile phenotype to a synthetic phenotype, thereby suppressing VSMC proliferation, migration, and differentiation, ultimately blockading plaque formation [142,159,160]. Furthermore, α7β1 and α8β1 integrins prevent VSMC proliferation and migration, thus inspiring the development of integrin agonists, although currently, no relevant integrin-targeted drugs have been reported [168,169,170,171]. In addition, nanotherapy, a treatment method that uses NPs to deliver drugs, offers a unique approach. Unlike integrin antibodies and antagonists, nanotherapy not only possesses a targeting effect but also enhances targeting specificity and bioavailability, delays drug release, and mitigates drug toxicity [131,132]. Several studies have demonstrated that nanotherapy can be applied in the treatment of acute coronary syndromes and thrombotic cardiovascular events [23,129,156]. However, anti-atherosclerotic nanotherapies have undergone preclinical testing but have not yet been applied in clinical settings [172].
Current integrin-based therapeutic approaches are single-target integrin therapies. Future research should focus on developing multi-target therapies addressing diverse signaling pathways, which may yield superior therapeutic outcomes. Moreover, integrin-targeted drugs for other CVDs are promising and require further development. Additionally, the predominant treatment paradigm for contemporary CVDs continues to center on post-onset management and correction, exemplified by interventions such as thrombolysis, vasodilators, percutaneous coronary intervention, and coronary artery bypass grafting [173,174]. However, these approaches fail to address the underlying etiology and are therefore categorized as palliative treatments. Whether future efforts should concentrate on preventive treatments is a potential avenue for exploration, as indicated by our summary of integrin-related targeted therapies. Chinese scholars have proposed the concept of panvascular medicine, which provides a new perspective for the management of CVDs [174]. Panvascular diseases, characterized by vascular lesions and predominantly AS in 95% of cases, primarily affect vital organs, including the heart, brain, kidneys, limbs, and major arteries [174]. Panvascular medicine considers the systemic vasculature as a unified entity, explores the mechanisms underlying the development and progression of CVDs from a holistic perspective that encompasses both structural and functional aspects, and provides unified management of diseases [174]. The management of CVDs encompasses four aspects: prevention, diagnosis, treatment, and prognosis [174]. This includes targeting risk factors for vascular diseases, identifying more precise biomarkers, developing diagnostic tools or techniques with higher sensitivity and specificity (computed tomography angiography, magnetic resonance angiography, and ultrasound), developing more potent and compliant medications, strengthening patient follow-up, and organizing and summarizing data to further guide research and clinical practice [174]. Building upon this foundational concept, exploring the intricate relationship between integrins and CVDs could help catalyze advancements in early diagnostic processes and therapeutic interventions. Consequently, integrins could be strategically leveraged as pivotal biomarkers or therapeutic targets, thereby facilitating the early identification and management of cardiovascular conditions.
Author Contributions
Conceptualization, Z.X. and J.C.; investigation, J.C., Q.Z. and S.Z.; writing—original draft preparation, S.Z., Q.Z. and Y.L.; writing—review and editing, S.Z., Q.Z., J.C., Z.X. and Y.L.; figure design, and editing, S.Z., Q.Z., J.L. and Z.L.; visualization, S.Z. and Q.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by funding from the National Natural Science Foundation of China (41966006).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Servier Home for Researchers. Parts of the figures were drawn by using pictures from Servier Home for Researchers.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mensah, G.A.; Fuster, V.; Roth, G.A. A Heart-Healthy and Stroke-Free World: Using Data to Inform Global Action. J. Am. Coll. Cardiol. 2023, 82, 2343–2349. [Google Scholar] [CrossRef]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report from the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Liu, J.; Wang, M.; Zhang, X.; Zhou, M. Epidemiology of cardiovascular disease in China: Current features and implications. Nat. Rev. Cardiol. 2018, 16, 203–212. [Google Scholar] [CrossRef]
- Ferro, F.; Spelat, R.; Pandit, A.; Martin-Ventura, J.L.; Rabinovich, G.A.; Contessotto, P. Glycosylation of blood cells during the onset and progression of atherosclerosis and myocardial infarction. Trends Mol. Med. 2024, 30, 178–196. [Google Scholar] [CrossRef]
- Wulf-Johansson, H.; Lock Johansson, S.; Schlosser, A.; Trommelholt Holm, A.; Rasmussen, L.M.; Mickley, H.; Diederichsen, A.C.; Munkholm, H.; Poulsen, T.S.; Tornøe, I.; et al. Localization of microfibrillar-associated protein 4 (MFAP4) in human tissues: Clinical evaluation of serum MFAP4 and its association with various cardiovascular conditions. PLoS ONE 2013, 8, e82243. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.H.; Mouton, A.J.; DeLeon-Pennell, K.Y.; Genovese, F.; Karsdal, M.; Lindsey, M.L. Understanding cardiac extracellular matrix remodeling to develop biomarkers of myocardial infarction outcomes. Matrix Biol. 2019, 75–76, 43–57. [Google Scholar] [CrossRef]
- Toba, H.; Lindsey, M.L. Extracellular matrix roles in cardiorenal fibrosis: Potential therapeutic targets for CVD and CKD in the elderly. Pharmacol. Ther. 2019, 193, 99–120. [Google Scholar] [CrossRef] [PubMed]
- Haverslag, R.; Pasterkamp, G.; Hoefer, I.E. Targeting adhesion molecules in cardiovascular disorders. Cardiovasc. Hematol. Disord. Drug Targets 2008, 8, 252–260. [Google Scholar] [CrossRef]
- Pang, X.; He, X.; Qiu, Z.; Zhang, H.; Xie, R.; Liu, Z.; Gu, Y.; Zhao, N.; Xiang, Q.; Cui, Y. Targeting integrin pathways: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 1. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Seetharaman, S.; Etienne-Manneville, S. Integrin diversity brings specificity in mechanotransduction. Biol. Cell 2018, 110, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Samarel, A.M. Costameres, focal adhesions, and cardiomyocyte mechanotransduction. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2291–H2301. [Google Scholar] [CrossRef]
- Lv, H.; Wang, H.; Quan, M.; Zhang, C.; Fu, Y.; Zhang, L.; Lin, C.; Liu, X.; Yi, X.; Chen, J.; et al. Cartilage oligomeric matrix protein fine-tunes disturbed flow-induced endothelial activation and atherogenesis. Matrix Biol. J. Int. Soc. Matrix Biol. 2021, 95, 32–51. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.Y.; Chu, P.H.; Chen, S.C.; Lee, T.H. MFG-E8 promotes osteogenic transdifferentiation of smooth muscle cells and vascular calcification by regulating TGF-β1 signaling. Commun. Biol. 2022, 5, 364. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.; Li, X.; Ramos-Echazabal, G.; Faridi, H.; Zigmond, Z.M.; Santos Falcon, N.; Hernandez, D.R.; Shehadeh, S.A.; Velazquez, O.C.; Gupta, V.; et al. A Genetic Model of Constitutively Active Integrin CD11b/CD18. J. Immunol. 2020, 205, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Zemljic-Harpf, A.E.; Godoy, J.C.; Platoshyn, O.; Asfaw, E.K.; Busija, A.R.; Domenighetti, A.A.; Ross, R.S. Vinculin directly binds zonula occludens-1 and is essential for stabilizing connexin-43-containing gap junctions in cardiac myocytes. J. Cell Sci. 2014, 127, 1104–1116. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, X.; Shi, X.; Zhu, M.; Wang, J.; Huang, S.; Huang, X.; Wang, H.; Li, L.; Deng, H.; et al. Platelet integrin αIIbβ3: Signal transduction, regulation, and its therapeutic targeting. J. Hematol. Oncol. 2019, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Arosio, D.; Casagrande, C.; Manzoni, L. Integrin-mediated drug delivery in cancer and cardiovascular diseases with peptide-functionalized nanoparticles. Curr. Med. Chem. 2012, 19, 3128–3151. [Google Scholar] [CrossRef]
- Nishimura, M.; Kumsta, C.; Kaushik, G.; Diop, S.B.; Ding, Y.; Bisharat-Kernizan, J.; Catan, H.; Cammarato, A.; Ross, R.S.; Engler, A.J.; et al. A dual role for integrin-linked kinase and β1-integrin in modulating cardiac aging. Aging Cell 2014, 13, 431–440. [Google Scholar] [CrossRef]
- Xu, J.; Shi, G.P. Vascular wall extracellular matrix proteins and vascular diseases. Biochim. Biophys. Acta 2014, 1842, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Estevez, B.; Shen, B.; Du, X. Targeting integrin and integrin signaling in treating thrombosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.J.; Byzova, T.V.; Qin, J.; Topol, E.J.; Plow, E.F. Integrin alphaIIbbeta3 and its antagonism. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Khatana, C.; Saini, N.K.; Chakrabarti, S.; Saini, V.; Sharma, A.; Saini, R.V.; Saini, A.K. Mechanistic Insights into the Oxidized Low-Density Lipoprotein-Induced Atherosclerosis. Oxid. Med. Cell Longev. 2020, 2020, 5245308. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Gisterå, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Guo, X.; Xia, Y.; Mao, L. An update on the phenotypic switching of vascular smooth muscle cells in the pathogenesis of atherosclerosis. Cell Mol. Life Sci. 2021, 79, 6. [Google Scholar] [CrossRef] [PubMed]
- Falk, E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, C7–C12. [Google Scholar] [CrossRef] [PubMed]
- Demos, C.; Williams, D.; Jo, H. Disturbed Flow Induces Atherosclerosis by Annexin A2-Mediated Integrin Activation. Circ. Res. 2020, 127, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhou, T.; Chen, Z.; Yan, M.; Li, B.; Lv, H.; Wang, C.; Xiang, S.; Shi, L.; Zhu, Y.; et al. Coupling of Integrin α5 to Annexin A2 by Flow Drives Endothelial Activation. Circ. Res. 2020, 127, 1074–1090. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lin, J.; Hu, T.; Ren, Z.; Li, L.; Hameed, I.; Zhang, X.; Men, C.; Guo, Y.; Xu, D.; et al. Galectin-3 exacerbates ox-LDL-mediated endothelial injury by inducing inflammation via integrin β1-RhoA-JNK signaling activation. J. Cell Physiol. 2019, 234, 10990–11000. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Chen, S.; Luo, S.; Chen, A.; Wang, L.; Tang, H.; Wang, F.; Wang, Z.; Gao, X.; Zuo, G.; et al. Shear-Induced ITGB4 Promotes Endothelial Cell Inflammation and Atherosclerosis. Oxid. Med. Cell Longev. 2022, 2022, 5842677. [Google Scholar] [CrossRef]
- Hsu, P.L.; Chen, J.S.; Wang, C.Y.; Wu, H.L.; Mo, F.E. Shear-Induced CCN1 Promotes Atheroprone Endothelial Phenotypes and Atherosclerosis. Circulation 2019, 139, 2877–2891. [Google Scholar] [CrossRef]
- Zhu, Y.; Qu, J.; He, L.; Zhang, F.; Zhou, Z.; Yang, S.; Zhou, Y. Calcium in Vascular Smooth Muscle Cell Elasticity and Adhesion: Novel Insights Into the Mechanism of Action. Front. Physiol. 2019, 10, 852. [Google Scholar] [CrossRef]
- Govatati, S.; Pichavaram, P.; Kumar, R.; Rao, G.N. Blockade of CD47 function attenuates restenosis by promoting smooth muscle cell efferocytosis and inhibiting their migration and proliferation. J. Biol. Chem. 2023, 299, 104594. [Google Scholar] [CrossRef] [PubMed]
- Krautter, F.; Hussain, M.T.; Zhi, Z.; Lezama, D.R.; Manning, J.E.; Brown, E.; Marigliano, N.; Raucci, F.; Recio, C.; Chimen, M.; et al. Galectin-9: A novel promoter of atherosclerosis progression. Atherosclerosis 2022, 363, 57–68. [Google Scholar] [CrossRef]
- Cui, K.; Podolnikova, N.P.; Bailey, W.; Szmuc, E.; Podrez, E.A.; Byzova, T.V.; Yakubenko, V.P. Inhibition of integrin α(D)β(2)-mediated macrophage adhesion to end product of docosahexaenoic acid (DHA) oxidation prevents macrophage accumulation during inflammation. J. Biol. Chem. 2019, 294, 14370–14382. [Google Scholar] [CrossRef] [PubMed]
- Favretto, G.; Cunha, R.S.D.; Dalboni, M.A.; Oliveira, R.B.; Barreto, F.C.; Massy, Z.A.; Stinghen, A.E.M. Endothelial Microparticles in Uremia: Biomarkers and Potential Therapeutic Targets. Toxins 2019, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, X.; Han, J.; Peng, W.; Fang, Z.; Zhou, Y.; Xu, X.; Lin, J.; Xiao, F.; Zhao, L.; et al. Convallatoxin Promotes M2 Macrophage Polarization to Attenuate Atherosclerosis through PPARγ-Integrin α(v)β(5) Signaling Pathway. Drug Des. Devel Ther. 2021, 15, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Sun, Y.; Yang, S.; Yu, M.; Pan, L.; Yang, J.; Yang, J.; Shao, Q.; Liu, J.; Liu, Y.; et al. Omentin-1 Modulates Macrophage Function via Integrin Receptors αvβ3 and αvβ5 and Reverses Plaque Vulnerability in Animal Models of Atherosclerosis. Front. Cardiovasc. Med. 2021, 8, 757926. [Google Scholar] [CrossRef] [PubMed]
- Lok, Z.S.Y.; Lyle, A.N. Osteopontin in Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Chang, T.T.; Chang, C.C.; Chen, J.W. Fatty-Acid-Binding Protein 4 as a Novel Contributor to Mononuclear Cell Activation and Endothelial Cell Dysfunction in Atherosclerosis. Int. J. Mol. Sci. 2020, 21, 9245. [Google Scholar] [CrossRef] [PubMed]
- Marchini, T.; Mitre, L.S.; Wolf, D. Inflammatory Cell Recruitment in Cardiovascular Disease. Front. Cell Dev. Biol. 2021, 9, 635527. [Google Scholar] [CrossRef]
- Finney, A.C.; Scott, M.L.; Reeves, K.A.; Wang, D.; Alfaidi, M.; Schwartz, J.C.; Chitmon, C.M.; Acosta, C.H.; Murphy, J.M.; Alexander, J.S.; et al. EphA2 signaling within integrin adhesions regulates fibrillar adhesion elongation and fibronectin deposition. Matrix Biol. 2021, 103–104, 1–21. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, D.; Lü, S.; Zhang, Y.; Long, M. Mechanical features of endothelium regulate cell adhesive molecule-induced calcium response in neutrophils. APL Bioeng. 2019, 3, 016104. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.; Novotny, J.; Salbeck, D.; Zellner, K.R.; Nicolai, L.; Pekayvaz, K.; Kilani, B.; Stockhausen, S.; Bürgener, N.; Kupka, D.; et al. Eosinophil-platelet interactions promote atherosclerosis and stabilize thrombosis with eosinophil extracellular traps. Blood 2019, 134, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, J.; Osaka, M.; Deushi, M.; Hosoya, S.; Ishigami, A.; Maehara, T.; Yoshida, M. CXCL1-Triggered PAD4 Cytoplasmic Translocation Enhances Neutrophil Adhesion through Citrullination of PDIA1. J. Atheroscler. Thromb. 2022, 29, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Krueger, I.; Gremer, L.; Mangels, L.; Klier, M.; Jurk, K.; Willbold, D.; Bock, H.H.; Elvers, M. Reelin Amplifies Glycoprotein VI Activation and AlphaIIb Beta3 Integrin Outside-In Signaling via PLC Gamma 2 and Rho GTPases. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2391–2403. [Google Scholar] [CrossRef] [PubMed]
- Nam, G.S.; Kim, S.; Kwon, Y.S.; Kim, M.K.; Nam, K.S. A new function for MAP4K4 inhibitors during platelet aggregation and platelet-mediated clot retraction. Biochem. Pharmacol. 2021, 188, 114519. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, W.S.; Vesey, A.T.; Vickers, A.; Neale, A.; Moles, C.; Connell, M.; Joshi, N.V.; Lucatelli, C.; Fletcher, A.M.; Spratt, J.C.; et al. In vivo alpha-V beta-3 integrin expression in human aortic atherosclerosis. Heart 2019, 105, 1868–1875. [Google Scholar] [CrossRef]
- Sheikhvatan, M.; Boroumand, M.A.; Behmanesh, M.; Ziaee, S.; Cheraghee, S. Integrin Beta-3 Gene Polymorphism and Risk for Myocardial Infarction in Premature Coronary Disease. Iran. J. Biotechnol. 2019, 17, e1921. [Google Scholar] [CrossRef]
- Ngai, D.; Lino, M.; Bendeck, M.P. Cell-Matrix Interactions and Matricrine Signaling in the Pathogenesis of Vascular Calcification. Front. Cardiovasc. Med. 2018, 5, 174. [Google Scholar] [CrossRef]
- Al-Yafeai, Z.; Yurdagul, A., Jr.; Peretik, J.M.; Alfaidi, M.; Murphy, P.A.; Orr, A.W. Endothelial FN (Fibronectin) Deposition by α5β1 Integrins Drives Atherogenic Inflammation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2601–2614. [Google Scholar] [CrossRef]
- Budatha, M.; Zhang, J.; Schwartz, M.A. Fibronectin-Mediated Inflammatory Signaling Through Integrin α5 in Vascular Remodeling. J. Am. Heart Assoc. 2021, 10, e021160. [Google Scholar] [CrossRef]
- Murphy, J.M.; Jeong, K.; Lim, S.S. FAK Family Kinases in Vascular Diseases. Int. J. Mol. Sci. 2020, 21, 3630. [Google Scholar] [CrossRef] [PubMed]
- Dietz, M.; Kamani, C.H.; Deshayes, E.; Dunet, V.; Mitsakis, P.; Coukos, G.; Nicod Lalonde, M.; Schaefer, N.; Prior, J.O. Imaging angiogenesis in atherosclerosis in large arteries with (68)Ga-NODAGA-RGD PET/CT: Relationship with clinical atherosclerotic cardiovascular disease. EJNMMI Res. 2021, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Park, E.J.; Kawamoto, E.; Usuda, H.; Wada, K.; Taguchi, A.; Shimaoka, M. Endothelial connexin-integrin crosstalk in vascular inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166168. [Google Scholar] [CrossRef] [PubMed]
- Aman, J.; Margadant, C. Integrin-Dependent Cell-Matrix Adhesion in Endothelial Health and Disease. Circ. Res. 2023, 132, 355–378. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, S.; Nesbitt, W.S.; Rooney, M.; Jackson, S.P. Bidirectional integrin alphaIIbbeta3 signalling regulating platelet adhesion under flow: Contribution of protein kinase C. Biochem. J. 2003, 372, 163–172. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Larivière, M.; Lorenzato, C.S.; Adumeau, L.; Bonnet, S.; Hémadou, A.; Jacobin-Valat, M.J.; Noubhani, A.; Santarelli, X.; Minder, L.; Di Primo, C.; et al. Multimodal molecular imaging of atherosclerosis: Nanoparticles functionalized with scFv fragments of an anti-αIIbβ3 antibody. Nanomedicine 2019, 22, 102082. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zheng, Z.; Li, C.; Wan, J.; Wang, M. Developmental endothelial locus-1 in cardiovascular and metabolic diseases: A promising biomarker and therapeutic target. Front. Immunol. 2022, 13, 1053175. [Google Scholar] [CrossRef]
- Potashnikova, D.M.; Saidova, A.A.; Tvorogova, A.V.; Anisimova, A.S.; Botsina, A.Y.; Vasilieva, E.Y.; Margolis, L.B. CTLs From Patients With Atherosclerosis Show Elevated Adhesiveness and Distinct Integrin Expression Patterns on 2D Substrates. Front. Med. 2022, 9, 891916. [Google Scholar] [CrossRef]
- Sun, C.; Fu, Y.; Gu, X.; Xi, X.; Peng, X.; Wang, C.; Sun, Q.; Wang, X.; Qian, F.; Qin, Z.; et al. Macrophage-Enriched lncRNA RAPIA: A Novel Therapeutic Target for Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1464–1478. [Google Scholar] [CrossRef]
- Weber, K.T.; Brilla, C.G. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 1991, 83, 1849–1865. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Al-Hasani, J.; Sens-Albert, C.; Ghosh, S.; Trogisch, F.A.; Nahar, T.; Friede, P.A.P.; Reil, J.C.; Hecker, M. Zyxin protects from hypertension-induced cardiac dysfunction. Cell Mol. Life Sci. 2022, 79, 93. [Google Scholar] [CrossRef]
- Chen, C.; Li, R.; Ross, R.S.; Manso, A.M. Integrins and integrin-related proteins in cardiac fibrosis. J. Mol. Cell Cardiol. 2016, 93, 162–174. [Google Scholar] [CrossRef]
- Leask, A. A sticky wicket: Overexpression of integrin alpha 11 is sufficient for cardiac fibrosis. Acta Physiol. 2018, 222, e13025. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, Q.; Kong, M.; Mao, L.; Yang, Y.; Xu, Y. Myocardin-related transcription factor A regulates integrin beta 2 transcription to promote macrophage infiltration and cardiac hypertrophy in mice. Cardiovasc. Res. 2022, 118, 844–858. [Google Scholar] [CrossRef]
- van Putten, S.; Shafieyan, Y.; Hinz, B. Mechanical control of cardiac myofibroblasts. J. Mol. Cell Cardiol. 2016, 93, 133–142. [Google Scholar] [CrossRef]
- Sui, S.; Hou, Y. Dual integrin αvβ3 and αvβ5 blockade attenuates cardiac dysfunction by reducing fibrosis in a rat model of doxorubicin-induced cardiomyopathy. Scand. Cardiovasc. J. 2021, 55, 287–296. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Quinones, L.; Kasiganesan, H.; Zhang, Y.; Pleasant, D.L.; Sundararaj, K.P.; Zile, M.R.; Bradshaw, A.D.; Kuppuswamy, D. β3 integrin in cardiac fibroblast is critical for extracellular matrix accumulation during pressure overload hypertrophy in mouse. PLoS ONE 2012, 7, e45076. [Google Scholar] [CrossRef]
- Sun, M.; Ishii, R.; Okumura, K.; Krauszman, A.; Breitling, S.; Gomez, O.; Hinek, A.; Boo, S.; Hinz, B.; Connelly, K.A.; et al. Experimental Right Ventricular Hypertension Induces Regional β1-Integrin-Mediated Transduction of Hypertrophic and Profibrotic Right and Left Ventricular Signaling. J. Am. Heart Assoc. 2018, 7, e13025. [Google Scholar] [CrossRef]
- Valiente-Alandi, I.; Potter, S.J.; Salvador, A.M.; Schafer, A.E.; Schips, T.; Carrillo-Salinas, F.; Gibson, A.M.; Nieman, M.L.; Perkins, C.; Sargent, M.A.; et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation 2018, 138, 1236–1252. [Google Scholar] [CrossRef]
- Gałdyszyńska, M.; Zwoliński, R.; Piera, L.; Szymański, J.; Jaszewski, R.; Drobnik, J. Stiff substrates inhibit collagen accumulation via integrin α2β1, FAK and Src kinases in human atrial fibroblast and myofibroblast cultures derived from patients with aortal stenosis. Biomed. Pharmacother. 2023, 159, 114289. [Google Scholar] [CrossRef]
- Hong, J.; Chu, M.; Qian, L.; Wang, J.; Guo, Y.; Xu, D. Fibrillar Type I Collagen Enhances the Differentiation and Proliferation of Myofibroblasts by Lowering α2β1 Integrin Expression in Cardiac Fibrosis. Biomed. Res. Int. 2017, 2017, 1790808. [Google Scholar] [CrossRef]
- Kanaan, R.; Medlej-Hashim, M.; Jounblat, R.; Pilecki, B.; Sorensen, G.L. Microfibrillar-associated protein 4 in health and disease. Matrix Biol. J. Int. Soc. Matrix Biol. 2022, 111, 1–25. [Google Scholar] [CrossRef]
- Sarrazy, V.; Koehler, A.; Chow, M.L.; Zimina, E.; Li, C.X.; Kato, H.; Caldarone, C.A.; Hinz, B. Integrins αvβ5 and αvβ3 promote latent TGF-β1 activation by human cardiac fibroblast contraction. Cardiovasc. Res. 2014, 102, 407–417. [Google Scholar] [CrossRef]
- Schussler, O.; Chachques, J.C.; Alifano, M.; Lecarpentier, Y. Key Roles of RGD-Recognizing Integrins During Cardiac Development, on Cardiac Cells, and After Myocardial Infarction. J. Cardiovasc. Transl. Res. 2022, 15, 179–203. [Google Scholar] [CrossRef]
- Perrucci, G.L.; Barbagallo, V.A.; Corlianò, M.; Tosi, D.; Santoro, R.; Nigro, P.; Poggio, P.; Bulfamante, G.; Lombardi, F.; Pompilio, G. Integrin ανβ5 in vitro inhibition limits pro-fibrotic response in cardiac fibroblasts of spontaneously hypertensive rats. J. Transl. Med. 2018, 16, 352. [Google Scholar] [CrossRef]
- Meagher, P.B.; Lee, X.A.; Lee, J.; Visram, A.; Friedberg, M.K.; Connelly, K.A. Cardiac Fibrosis: Key Role of Integrins in Cardiac Homeostasis and Remodeling. Cells 2021, 10, 770. [Google Scholar] [CrossRef]
- Failer, T.; Amponsah-Offeh, M.; Neuwirth, A.; Kourtzelis, I.; Subramanian, P.; Mirtschink, P.; Peitzsch, M.; Matschke, K.; Tugtekin, S.M.; Kajikawa, T.; et al. Developmental endothelial locus-1 protects from hypertension-induced cardiovascular remodeling via immunomodulation. J. Clin. Investig. 2022, 132, e126155. [Google Scholar] [CrossRef]
- Shimojo, N.; Hashizume, R.; Kanayama, K.; Hara, M.; Suzuki, Y.; Nishioka, T.; Hiroe, M.; Yoshida, T.; Imanaka-Yoshida, K. Tenascin-C may accelerate cardiac fibrosis by activating macrophages via the integrin αVβ3/nuclear factor-κB/interleukin-6 axis. Hypertension 2015, 66, 757–766. [Google Scholar] [CrossRef]
- Murray, I.R.; Gonzalez, Z.N.; Baily, J.; Dobie, R.; Wallace, R.J.; Mackinnon, A.C.; Smith, J.R.; Greenhalgh, S.N.; Thompson, A.I.; Conroy, K.P.; et al. αv integrins on mesenchymal cells regulate skeletal and cardiac muscle fibrosis. Nat. Commun. 2017, 8, 1118. [Google Scholar] [CrossRef]
- Lin, C.; Guo, Y.; Xia, Y.; Li, C.; Xu, X.; Qi, T.; Zhang, F.; Fan, M.; Hu, G.; Zhao, H.; et al. FNDC5/Irisin attenuates diabetic cardiomyopathy in a type 2 diabetes mouse model by activation of integrin αV/β5-AKT signaling and reduction of oxidative/nitrosative stress. J. Mol. Cell Cardiol. 2021, 160, 27–41. [Google Scholar] [CrossRef]
- Nakamura, Y.; Kita, S.; Tanaka, Y.; Fukuda, S.; Obata, Y.; Okita, T.; Kawachi, Y.; Tsugawa-Shimizu, Y.; Fujishima, Y.; Nishizawa, H.; et al. A disintegrin and metalloproteinase 12 prevents heart failure by regulating cardiac hypertrophy and fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H238–H251. [Google Scholar] [CrossRef]
- Fan, D.; Takawale, A.; Shen, M.; Samokhvalov, V.; Basu, R.; Patel, V.; Wang, X.; Fernandez-Patron, C.; Seubert, J.M.; Oudit, G.Y.; et al. A Disintegrin and Metalloprotease-17 Regulates Pressure Overload-Induced Myocardial Hypertrophy and Dysfunction Through Proteolytic Processing of Integrin β1. Hypertension 2016, 68, 937–948. [Google Scholar] [CrossRef]
- Angelini, A.; Trial, J.; Ortiz-Urbina, J.; Cieslik, K.A. Mechanosensing dysregulation in the fibroblast: A hallmark of the aging heart. Ageing Res. Rev. 2020, 63, 101150. [Google Scholar] [CrossRef]
- Niu, L.; Cheng, B.; Huang, G.; Nan, K.; Han, S.; Ren, H.; Liu, N.; Li, Y.; Genin, G.M.; Xu, F. A positive mechanobiological feedback loop controls bistable switching of cardiac fibroblast phenotype. Cell Discov. 2022, 8, 84. [Google Scholar] [CrossRef]
- Schroer, A.K.; Merryman, W.D. Mechanobiology of myofibroblast adhesion in fibrotic cardiac disease. J. Cell Sci. 2015, 128, 1865–1875. [Google Scholar] [CrossRef]
- Saraswati, S.; Lietman, C.D.; Li, B.; Mathew, S.; Zent, R.; Young, P.P. Small proline-rich repeat 3 is a novel coordinator of PDGFRβ and integrin β1 crosstalk to augment proliferation and matrix synthesis by cardiac fibroblasts. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 7885–7904. [Google Scholar] [CrossRef]
- Horii, Y.; Matsuda, S.; Toyota, C.; Morinaga, T.; Nakaya, T.; Tsuchiya, S.; Ohmuraya, M.; Hironaka, T.; Yoshiki, R.; Kasai, K.; et al. VGLL3 is a mechanosensitive protein that promotes cardiac fibrosis through liquid-liquid phase separation. Nat. Commun. 2023, 14, 550. [Google Scholar] [CrossRef]
- Takawale, A.; Zhang, P.; Patel, V.B.; Wang, X.; Oudit, G.; Kassiri, Z. Tissue Inhibitor of Matrix Metalloproteinase-1 Promotes Myocardial Fibrosis by Mediating CD63-Integrin β1 Interaction. Hypertension 2017, 69, 1092–1103. [Google Scholar] [CrossRef]
- Bouvet, M.; Claude, O.; Roux, M.; Skelly, D.; Masurkar, N.; Mougenot, N.; Nadaud, S.; Blanc, C.; Delacroix, C.; Chardonnet, S.; et al. Anti-integrin α(v) therapy improves cardiac fibrosis after myocardial infarction by blunting cardiac PW1(+) stromal cells. Sci. Rep. 2020, 10, 11404. [Google Scholar] [CrossRef]
- Gao, A.E.; Sullivan, K.E.; Black, L.D., 3rd. Lysyl oxidase expression in cardiac fibroblasts is regulated by α2β1 integrin interactions with the cellular microenvironment. Biochem. Biophys. Res. Commun. 2016, 475, 70–75. [Google Scholar] [CrossRef]
- Tannous, C.; Deloux, R.; Karoui, A.; Mougenot, N.; Burkin, D.; Blanc, J.; Coletti, D.; Lavery, G.; Li, Z.; Mericskay, M. NMRK2 Gene Is Upregulated in Dilated Cardiomyopathy and Required for Cardiac Function and NAD Levels during Aging. Int. J. Mol. Sci. 2021, 22, 3524. [Google Scholar] [CrossRef]
- Civitarese, R.A.; Talior-Volodarsky, I.; Desjardins, J.F.; Kabir, G.; Switzer, J.; Mitchell, M.; Kapus, A.; McCulloch, C.A.; Gullberg, D.; Connelly, K.A. The α11 integrin mediates fibroblast-extracellular matrix-cardiomyocyte interactions in health and disease. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H96–H106. [Google Scholar] [CrossRef]
- Hanna, A.; Humeres, C.; Frangogiannis, N.G. The role of Smad signaling cascades in cardiac fibrosis. Cell Signal 2021, 77, 109826. [Google Scholar] [CrossRef]
- Romaine, A.; Sørensen, I.W.; Zeltz, C.; Lu, N.; Erusappan, P.M.; Melleby, A.O.; Zhang, L.; Bendiksen, B.; Robinson, E.L.; Aronsen, J.M.; et al. Overexpression of integrin α11 induces cardiac fibrosis in mice. Acta Physiol. 2018, 222, e12932. [Google Scholar] [CrossRef]
- Talior-Volodarsky, I.; Arora, P.D.; Wang, Y.; Zeltz, C.; Connelly, K.A.; Gullberg, D.; McCulloch, C.A. Glycated Collagen Induces α11 Integrin Expression Through TGF-β2 and Smad3. J. Cell. Physiol. 2015, 230, 327–336. [Google Scholar] [CrossRef]
- Li, J.; Salvador, A.M.; Li, G.; Valkov, N.; Ziegler, O.; Yeri, A.; Yang Xiao, C.; Meechoovet, B.; Alsop, E.; Rodosthenous, R.S.; et al. Mir-30d Regulates Cardiac Remodeling by Intracellular and Paracrine Signaling. Circ. Res. 2021, 128, e1–e23. [Google Scholar] [CrossRef]
- Rouzaud-Laborde, C.; Delmas, C.; Pizzinat, N.; Tortosa, F.; Garcia, C.; Mialet-Perez, J.; Payrastre, B.; Sié, P.; Spreux-Varoquaux, O.; Sallerin, B.; et al. Platelet activation and arterial peripheral serotonin turnover in cardiac remodeling associated to aortic stenosis. Am. J. Hematol. 2015, 90, 15–19. [Google Scholar] [CrossRef]
- Nevers, T.; Salvador, A.M.; Velazquez, F.; Ngwenyama, N.; Carrillo-Salinas, F.J.; Aronovitz, M.; Blanton, R.M.; Alcaide, P. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J. Exp. Med. 2017, 214, 3311–3329. [Google Scholar] [CrossRef]
- Walker, J.A.; Beck, G.A.; Campbell, J.H.; Miller, A.D.; Burdo, T.H.; Williams, K.C. Anti-α4 Integrin Antibody Blocks Monocyte/Macrophage Traffic to the Heart and Decreases Cardiac Pathology in a SIV Infection Model of AIDS. J. Am. Heart Assoc. 2015, 4, e001932. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.; Yang, D.; Jiang, X.; Wang, W.E.; Yue, R.; Fang, Y. Integrin-Linked Kinase Activation Prevents Ventricular Arrhythmias Induced by Ischemia/Reperfusion Via Inhibition of Connexin 43 Remodeling. J. Cardiovasc. Transl. Res. 2021, 14, 610–618. [Google Scholar] [CrossRef]
- Quang, K.L.; Maguy, A.; Qi, X.Y.; Naud, P.; Xiong, F.; Tadevosyan, A.; Shi, Y.F.; Chartier, D.; Tardif, J.C.; Dobrev, D.; et al. Loss of cardiomyocyte integrin-linked kinase produces an arrhythmogenic cardiomyopathy in mice. Circ. Arrhythm. Electrophysiol. 2015, 8, 921–932. [Google Scholar] [CrossRef]
- Brodehl, A.; Rezazadeh, S.; Williams, T.; Munsie, N.M.; Liedtke, D.; Oh, T.; Ferrier, R.; Shen, Y.; Jones, S.J.M.; Stiegler, A.L.; et al. Mutations in ILK, encoding integrin-linked kinase, are associated with arrhythmogenic cardiomyopathy. Transl. Res. 2019, 208, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, C.; Shi, L.; Chen, X.; Cui, C.; Huang, J.; Chen, B.; Hall, D.D.; Pan, Z.; Lu, M.; et al. Integrin β1D Deficiency-Mediated RyR2 Dysfunction Contributes to Catecholamine-Sensitive Ventricular Tachycardia in Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1477–1493. [Google Scholar] [CrossRef]
- Zhao, F.; Zhang, S.; Shao, Y.; Wu, Y.; Qin, J.; Chen, Y.; Chen, L.; Gu, H.; Wang, X.; Huang, C.; et al. Calreticulin overexpression correlates with integrin-α5 and transforming growth factor-β1 expression in the atria of patients with rheumatic valvular disease and atrial fibrillation. Int. J. Cardiol. 2013, 168, 2177–2185. [Google Scholar] [CrossRef]
- Schinner, C.; Xu, L.; Franz, H.; Zimmermann, A.; Wanuske, M.T.; Rathod, M.; Hanns, P.; Geier, F.; Pelczar, P.; Liang, Y.; et al. Defective Desmosomal Adhesion Causes Arrhythmogenic Cardiomyopathy by Involving an Integrin-αVβ6/TGF-β Signaling Cascade. Circulation 2022, 146, 1610–1626. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, D.J.; Schmeckpeper, J.; Knollmann, B.C. Animal Models to Study Cardiac Arrhythmias. Circ. Res. 2022, 130, 1926–1964. [Google Scholar] [CrossRef]
- Kamble, R.N.; Gaikwad, S.; Maske, A.; Patil, S.S. Fabrication of electrospun nanofibres of BCS II drug for enhanced dissolution and permeation across skin. J. Adv. Res. 2016, 7, 483–489. [Google Scholar] [CrossRef]
- Dieffenbach, P.B.; Mallarino Haeger, C.; Rehman, R.; Corcoran, A.M.; Coronata, A.M.F.; Vellarikkal, S.K.; Chrobak, I.; Waxman, A.B.; Vitali, S.H.; Sholl, L.M.; et al. A Novel Protective Role for Matrix Metalloproteinase-8 in the Pulmonary Vasculature. Am. J. Respir. Crit. Care Med. 2021, 204, 1433–1451. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Yu, P.B. Periostin: A Novel Integrator of Hypoxic Signaling in Pulmonary Hypertension. Circ. Res. 2020, 127, 1153–1155. [Google Scholar] [CrossRef]
- Jia, D.; Zhu, Q.; Liu, H.; Zuo, C.; He, Y.; Chen, G.; Lu, A. Osteoprotegerin Disruption Attenuates HySu-Induced Pulmonary Hypertension Through Integrin αvβ3/FAK/AKT Pathway Suppression. Circ. Cardiovasc. Genet. 2017, 10, e001591. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Liu, X.; Teng, X.; Gu, H.; Yuan, W.; Meng, J.; Li, J.; Zheng, Z.; Wei, Y.; Hu, S. Osteopontin plays important roles in pulmonary arterial hypertension induced by systemic-to-pulmonary shunt. FASEB J. 2019, 33, 7236–7251. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Martini, A.G.; Brown, R.I.; Liang, X.; Medrano, S.; Goto, S.; Narita, I.; Arend, L.J.; Sequeira-Lopez, M.L.S.; Gomez, R.A. Inhibition of the renin-angiotensin system causes concentric hypertrophy of renal arterioles in mice and humans. JCI Insight 2021, 6, e154337. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Zhu, H.; Wang, S.; Li, Y.; Yan, C.; Wang, J.; Ying, K. CircItgb5 promotes synthetic phenotype of pulmonary artery smooth muscle cells via interacting with miR-96-5p and Uba1 in monocrotaline-induced pulmonary arterial hypertension. Respir. Res. 2023, 24, 165. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, N.; Link, P.A.; Farkas, D.; Harmon, B.; Hudson, J.; Bogamuwa, S.; Piper, B.; Authelet, K.; Cool, C.D.; Heise, R.L.; et al. Dichotomous role of integrin-β5 in lung endothelial cells. Pulm. Circ. 2022, 12, e12156. [Google Scholar] [CrossRef] [PubMed]
- Vinaiphat, A.; Pazhanchamy, K.; JebaMercy, G.; Ngan, S.C.; Leow, M.K.; Ho, H.H.; Gao, Y.G.; Lim, K.L.; Richards, A.M.; de Kleijn, D.P.V.; et al. Endothelial Damage Arising From High Salt Hypertension Is Elucidated by Vascular Bed Systematic Profiling. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Li, Z.; Cui, L.; Ban, Y.; Leung, P.C.K.; Li, Y.; Ma, J. Activin A increases human trophoblast invasion by upregulating integrin β1 through ALK4. Faseb. J. 2021, 35, e21220. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Che, J.; Chang, L.; Guo, M.; Bao, X.; Mu, D.; Sun, X.; Zhang, X.; Lu, W.; Xie, J. CD47- and Integrin α4/β1-Comodified-Macrophage-Membrane-Coated Nanoparticles Enable Delivery of Colchicine to Atherosclerotic Plaque. Adv. Healthc. Mater. 2022, 11, e2101788. [Google Scholar] [CrossRef]
- Pang, A.; Cheng, N.; Cui, Y.; Bai, Y.; Hong, Z.; Delaney, M.K.; Zhang, Y.; Chang, C.; Wang, C.; Liu, C.; et al. High-loading Gα(13)-binding EXE peptide nanoparticles prevent thrombosis and protect mice from cardiac ischemia/reperfusion injury. Sci. Transl. Med. 2020, 12, eaaz7287. [Google Scholar] [CrossRef]
- Peters, L.J.F.; Jans, A.; Bartneck, M.; van der Vorst, E.P.C. Immunomodulatory Nanomedicine for the Treatment of Atherosclerosis. J. Clin. Med. 2021, 10, 3185. [Google Scholar] [CrossRef]
- Wilczewska, A.Z.; Niemirowicz, K.; Markiewicz, K.H.; Car, H. Nanoparticles as drug delivery systems. Pharmacol. Rep. 2012, 64, 1020–1037. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, G.; Jin, S.; Xu, L.; Zhao, C.X. Development of High-Drug-Loading Nanoparticles. Chempluschem 2020, 85, 2143–2157. [Google Scholar] [CrossRef] [PubMed]
- Hagemeyer, C.E.; Peter, K. Targeting the platelet integrin GPIIb/IIIa. Curr. Pharm. Des. 2010, 16, 4119–4133. [Google Scholar] [CrossRef] [PubMed]
- Lal, H.; Guleria, R.S.; Foster, D.M.; Lu, G.; Watson, L.E.; Sanghi, S.; Smith, M.; Dostal, D.E. Integrins: Novel therapeutic targets for cardiovascular diseases. Cardiovasc. Hematol. Agents Med. Chem. 2007, 5, 109–132. [Google Scholar] [CrossRef] [PubMed]
- van den Kerkhof, D.L.; van der Meijden, P.E.J.; Hackeng, T.M.; Dijkgraaf, I. Exogenous Integrin αIIbβ3 Inhibitors Revisited: Past, Present and Future Applications. Int. J. Mol. Sci. 2021, 22, 3366. [Google Scholar] [CrossRef]
- Vorchheimer, D.A.; Badimon, J.J.; Fuster, V. Platelet glycoprotein IIb/IIIa receptor antagonists in cardiovascular disease. JAMA 1999, 281, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Harrington, R.A. Controversies surrounding platelet glycoprotein IIb/IIIa inhibitors in percutaneous coronary intervention and acute coronary syndromes. Semin. Thromb. Hemost. 2004, 30, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Breuer, J.; Schneider-Hohendorf, T.; Ostkamp, P.; Herich, S.; Rakhade, S.; Antonijevic, I.; Klotz, L.; Wiendl, H.; Schwab, N. VLA-2 blockade in vivo by vatelizumab induces CD4+FoxP3+ regulatory T cells. Int. Immunol. 2019, 31, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Duplàa, C.; Couffinhal, T.; Dufourcq, P.; Llanas, B.; Moreau, C.; Bonnet, J. The integrin very late antigen-4 is expressed in human smooth muscle cell. Involvement of alpha 4 and vascular cell adhesion molecule-1 during smooth muscle cell differentiation. Circ. Res. 1997, 80, 159–169. [Google Scholar] [CrossRef]
- Lumsden, A.B.; Chen, C.; Hughes, J.D.; Kelly, A.B.; Hanson, S.R.; Harker, L.A. Anti-VLA-4 antibody reduces intimal hyperplasia in the endarterectomized carotid artery in nonhuman primates. J. Vasc. Surg. 1997, 26, 87–93. [Google Scholar] [CrossRef]
- Braun, A.; Dofiles, L.; Rousselle, S.; Guerrero, L.; Gunther, J.; Yednock, T.; Stricker-Krongrad, A.; Messersmith, E. Effects of an alpha-4 integrin inhibitor on restenosis in a new porcine model combining endothelial denudation and stent placement. PLoS ONE 2010, 5, e14314. [Google Scholar] [CrossRef] [PubMed]
- Raab-Westphal, S.; Marshall, J.F.; Goodman, S.L. Integrins as Therapeutic Targets: Successes and Cancers. Cancers 2017, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.J.; Li, M.B.; Wu, X.; Wu, S.; Zhu, W.; Chen, D.; Luo, M.; Eitenmüller, I.; Kampmann, A.; Schaper, J.; et al. Activation of the integrins alpha 5beta 1 and alpha v beta 3 and focal adhesion kinase (FAK) during arteriogenesis. Mol. Cell Biochem. 2009, 322, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Qin, X.; Wang, H.; Miao, R.; Zhang, Y.; Miao, C.; Wang, Z. Effects of integrin α5β1 on the proliferation and migration of human aortic vascular smooth muscle cells. Mol. Med. Rep. 2016, 13, 1147–1155. [Google Scholar] [CrossRef]
- Yurdagul, A., Jr.; Green, J.; Albert, P.; McInnis, M.C.; Mazar, A.P.; Orr, A.W. α5β1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Dev, R.; Doddapattar, P.; Kon, S.; Dhanesha, N.; Chauhan, A.K. Integrin α9 regulates smooth muscle cell phenotype switching and vascular remodeling. JCI Insight 2021, 6, e147134. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Tanaka, Y.; Erdman, J.; Kaneko, Y.; Saito, M.; Higashitani, C.; Smulders, R.; Lademacher, C. ASP5094, a humanized monoclonal antibody against integrin alpha-9, did not show efficacy in patients with rheumatoid arthritis refractory to methotrexate: Results from a phase 2a, randomized, double-blind, placebo-controlled trial. Arthritis Res. Ther. 2020, 22, 252. [Google Scholar] [CrossRef] [PubMed]
- Mawatari, K.; Liu, B.; Kent, K.C. Activation of integrin receptors is required for growth factor-induced smooth muscle cell dysfunction. J. Vasc. Surg. 2000, 31, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Hoshiga, M.; Alpers, C.E.; Smith, L.L.; Giachelli, C.M.; Schwartz, S.M. Alpha-v beta-3 integrin expression in normal and atherosclerotic artery. Circ. Res. 1995, 77, 1129–1135. [Google Scholar] [CrossRef]
- van der Zee, R.; Murohara, T.; Passeri, J.; Kearney, M.; Cheresh, D.A.; Isner, J.M. Reduced intimal thickening following alpha(v)beta3 blockade is associated with smooth muscle cell apoptosis. Cell Adhes. Commun. 1998, 6, 371–379. [Google Scholar] [CrossRef][Green Version]
- Liaw, L.; Skinner, M.P.; Raines, E.W.; Ross, R.; Cheresh, D.A.; Schwartz, S.M.; Giachelli, C.M. The adhesive and migratory effects of osteopontin are mediated via distinct cell surface integrins. Role of alpha v beta 3 in smooth muscle cell migration to osteopontin in vitro. J. Clin. Investig. 1995, 95, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Bendeck, M.P.; Irvin, C.; Reidy, M.; Smith, L.; Mulholland, D.; Horton, M.; Giachelli, C.M. Smooth muscle cell matrix metalloproteinase production is stimulated via alpha(v)beta(3) integrin. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Bae, J.S.; Park, R.W.; Kim, J.E.; Park, J.Y.; Kim, I.S. betaig-h3 triggers signaling pathways mediating adhesion and migration of vascular smooth muscle cells through alphavbeta5 integrin. Exp. Mol. Med. 2006, 38, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zou, J.; Yu, X.; Yin, S.; Tang, C. The antiatherogenic function of kallistatin and its potential mechanism. Acta Biochim. Biophys. Sin. 2020, 52, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.P.; Vaidya, B. Targeted delivery of thrombolytic agents: Role of integrin receptors. Expert. Opin. Drug Deliv. 2009, 6, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhao, X.; O’Brien, K.A.; Stojanovic-Terpo, A.; Delaney, M.K.; Kim, K.; Cho, J.; Lam, S.C.; Du, X. A directional switch of integrin signalling and a new anti-thrombotic strategy. Nature 2013, 503, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Ni, Y.; Yu, H.; Yin, H.; Yang, F.; Li, C.; Sun, D.; Pei, T.; Ma, J.; Deng, L.; et al. Inflammatory endothelium-targeted and cathepsin responsive nanoparticles are effective against atherosclerosis. Theranostics 2022, 12, 4200–4220. [Google Scholar] [CrossRef]
- Slack, R.J.; Macdonald, S.J.F.; Roper, J.A.; Jenkins, R.G.; Hatley, R.J.D. Emerging therapeutic opportunities for integrin inhibitors. Nat. Rev. Drug Discov. 2022, 21, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Grenache, D.G.; Coleman, T.; Semenkovich, C.F.; Santoro, S.A.; Zutter, M.M. Alpha2beta1 integrin and development of atherosclerosis in a mouse model: Assessment of risk. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2104–2109. [Google Scholar] [CrossRef]
- Skinner, M.P.; Raines, E.W.; Ross, R. Dynamic expression of alpha 1 beta 1 and alpha 2 beta 1 integrin receptors by human vascular smooth muscle cells. Alpha 2 beta 1 integrin is required for chemotaxis across type I collagen-coated membranes. Am. J. Pathol. 1994, 145, 1070–1081. [Google Scholar]
- Akanchise, T.; Angelova, A. Ginkgo Biloba and Long COVID: In Vivo and In Vitro Models for the Evaluation of Nanotherapeutic Efficacy. Pharmaceutics 2023, 15, 1562. [Google Scholar] [CrossRef] [PubMed]
- Zerrillo, L.; Gigliobianco, M.R.; D’Atri, D.; Garcia, J.P.; Baldazzi, F.; Ridwan, Y.; Fuentes, G.; Chan, A.; Creemers, L.B.; Censi, R.; et al. PLGA Nanoparticles Grafted with Hyaluronic Acid to Improve Site-Specificity and Drug Dose Delivery in Osteoarthritis Nanotherapy. Nanomaterials 2022, 12, 2248. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Saglietto, A.; Manfredi, R.; Elia, E.; D’Ascenzo, F.; DE Ferrari, G.M.; Biondi-Zoccai, G.; Munzel, T. Cardiovascular disease burden: Italian and global perspectives. Minerva Cardiol. Angiol. 2021, 69, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Bkaily, G.; Simon, Y.; Jazzar, A.; Najibeddine, H.; Normand, A.; Jacques, D. High Na(+) Salt Diet and Remodeling of Vascular Smooth Muscle and Endothelial Cells. Biomedicines 2021, 9, 883. [Google Scholar] [CrossRef] [PubMed]
- Simon, Y.; Jacques, D.; Bkaily, G. High salt-induced morphological and glycocalyx remodeling of human vascular smooth muscle cells is reversible but induces a high sodium salt-like sensitive memory. Can. J. Physiol. Pharmacol. 2023, 101, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Castro, C.; Cordasic, N.; Neureiter, D.; Amann, K.; Marek, I.; Volkert, G.; Stintzing, S.; Jahn, A.; Rascher, W.; Hilgers, K.F.; et al. Under-expression of α8 integrin aggravates experimental atherosclerosis. J. Pathol. 2015, 236, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zheng, J.; Du, Y.; Huang, Y.; Li, J.; Liu, B.; Liu, C.J.; Zhu, Y.; Gao, Y.; Xu, Q.; et al. Cartilage oligomeric matrix protein maintains the contractile phenotype of vascular smooth muscle cells by interacting with alpha(7)beta(1) integrin. Circ. Res. 2010, 106, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.C.; Breuss, J.; Pytela, R.; Kramer, R.H. Functional expression of the alpha 7 integrin receptor in differentiated smooth muscle cells. J. Cell Sci. 1997, 110 Pt 13, 1477–1487. [Google Scholar] [CrossRef]
- Zargham, R.; Touyz, R.M.; Thibault, G. Alpha 8 Integrin overexpression in de-differentiated vascular smooth muscle cells attenuates migratory activity and restores the characteristics of the differentiated phenotype. Atherosclerosis 2007, 195, 303–312. [Google Scholar] [CrossRef]
- Hetherington, I.; Totary-Jain, H. Anti-atherosclerotic therapies: Milestones, challenges, and emerging innovations. Mol. Ther. J. Am. Soc. Gene Ther. 2022, 30, 3106–3117. [Google Scholar] [CrossRef] [PubMed]
- Bonaca, M.P.; Bauersachs, R.M.; Anand, S.S.; Debus, E.S.; Nehler, M.R.; Patel, M.R.; Fanelli, F.; Capell, W.H.; Diao, L.; Jaeger, N.; et al. Rivaroxaban in Peripheral Artery Disease after Revascularization. N. Engl. J. Med. 2020, 382, 1994–2004. [Google Scholar] [CrossRef] [PubMed]
- Howlett, J.G. Nebivolol: Vasodilator properties and evidence for relevance in treatment of cardiovascular disease. Can. J. Cardiol. 2014, 30, S29–S37. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, X.; Yu, L.; Zhao, Y.; Ge, J. Panvascular medicine: An emerging discipline focusing on atherosclerotic diseases. Eur. Heart J. 2022, 43, 4528–4531. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).