


Informed by Cancer Stem Cells of Solid Tumors: Advances in Treatments Targeting Tumor-Promoting Factors and Pathways

 ,
, 
Abstract
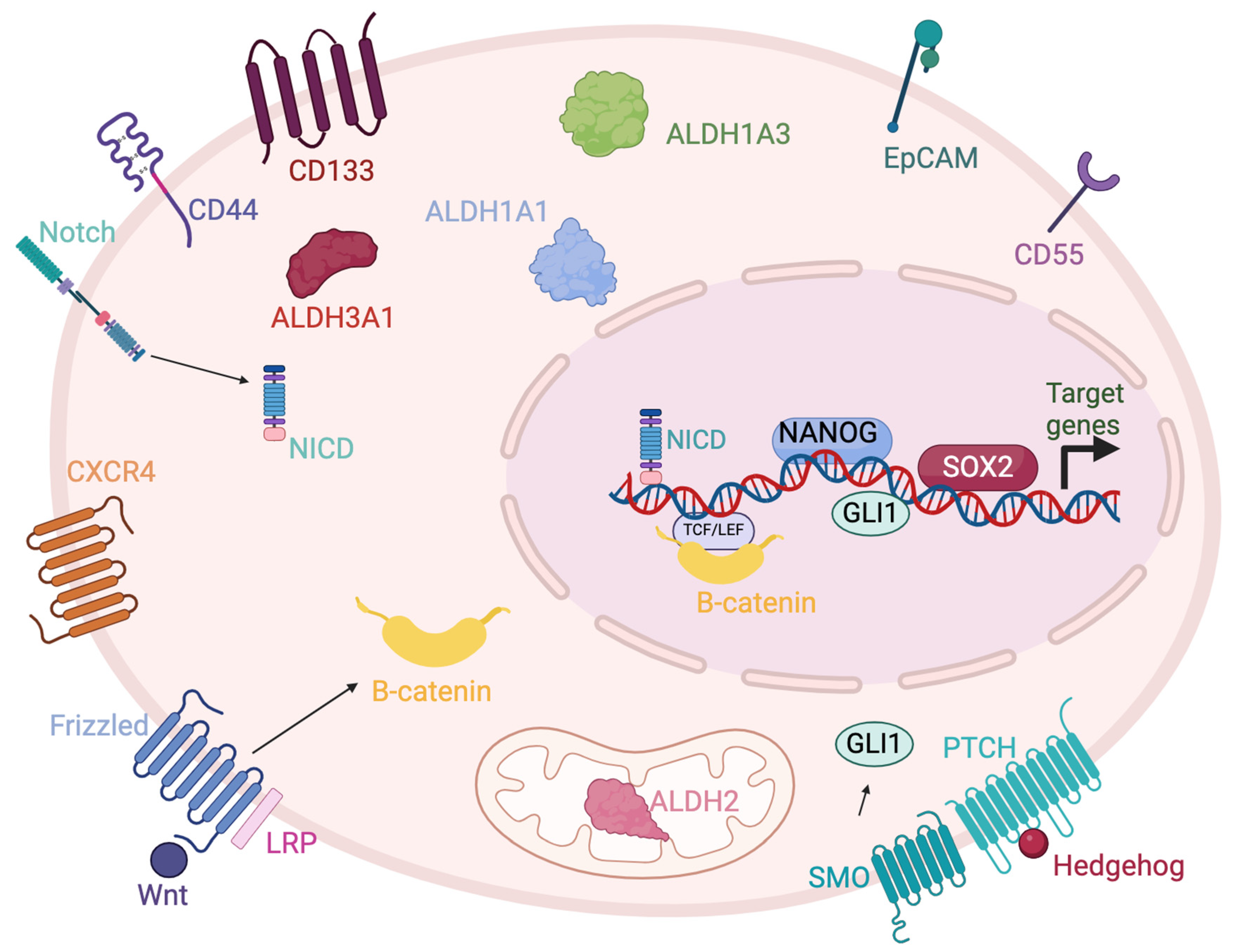
1. Introduction
2. Aldehyde Dehydrogenases
2.1. ALDH1A1
2.2. ALDH1A3
2.3. ALDH2
2.4. ALDH3A1
2.5. Pan-ALDH Inhibitors
3. EpCAM
3.1. Antibodies
3.2. CAR-T Therapies
3.3. Immunotoxins
3.4. Pharmacologic Inhibitors
3.5. Antibody-Drug Conjugates
3.6. Other EpCAM-Targeting Strategies
4. CD44
4.1. Antibodies
4.2. NIR-PIT
4.3. Other CD44-Targeting Strategies
4.4. Pharmacologic Inhibitors
4.5. Peptides
5. CD55
5.1. Antibodies
5.2. Peptides
5.3. Pharmacologic Inhibitors
5.4. Other CD55-Targeting Strategies
6. CXCR4
6.1. Antibodies
6.2. Pharmacologic Inhibitors
6.3. Peptides
7. CD133
7.1. Antibodies
7.2. CAR-T and NK Cells
7.3. BiKEs and Vaccines
7.4. Immunotoxins
7.5. Pharmacologic Inhibitors
7.6. Aptamers and Peptides
7.7. Other CD133-Targeting Strategies
8. Nanog
8.1. PMOs and RNA Interference
8.2. Pharmacologic Inhibition
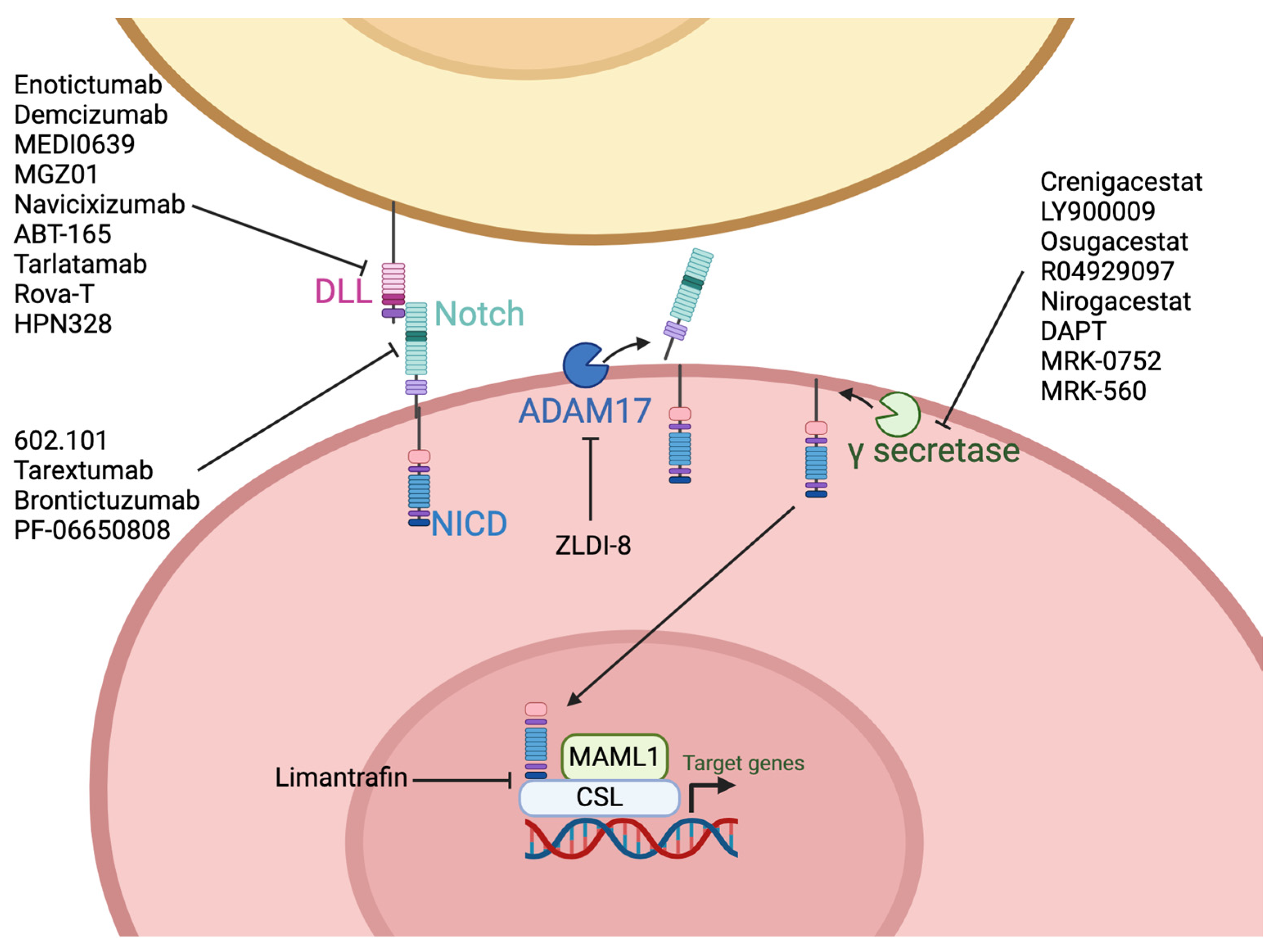
9. Notch
9.1. Pharmacologic Inhibitors
9.2. Antibodies
9.3. T-Cell Engagers
9.4. Antibody Drug Conjugates
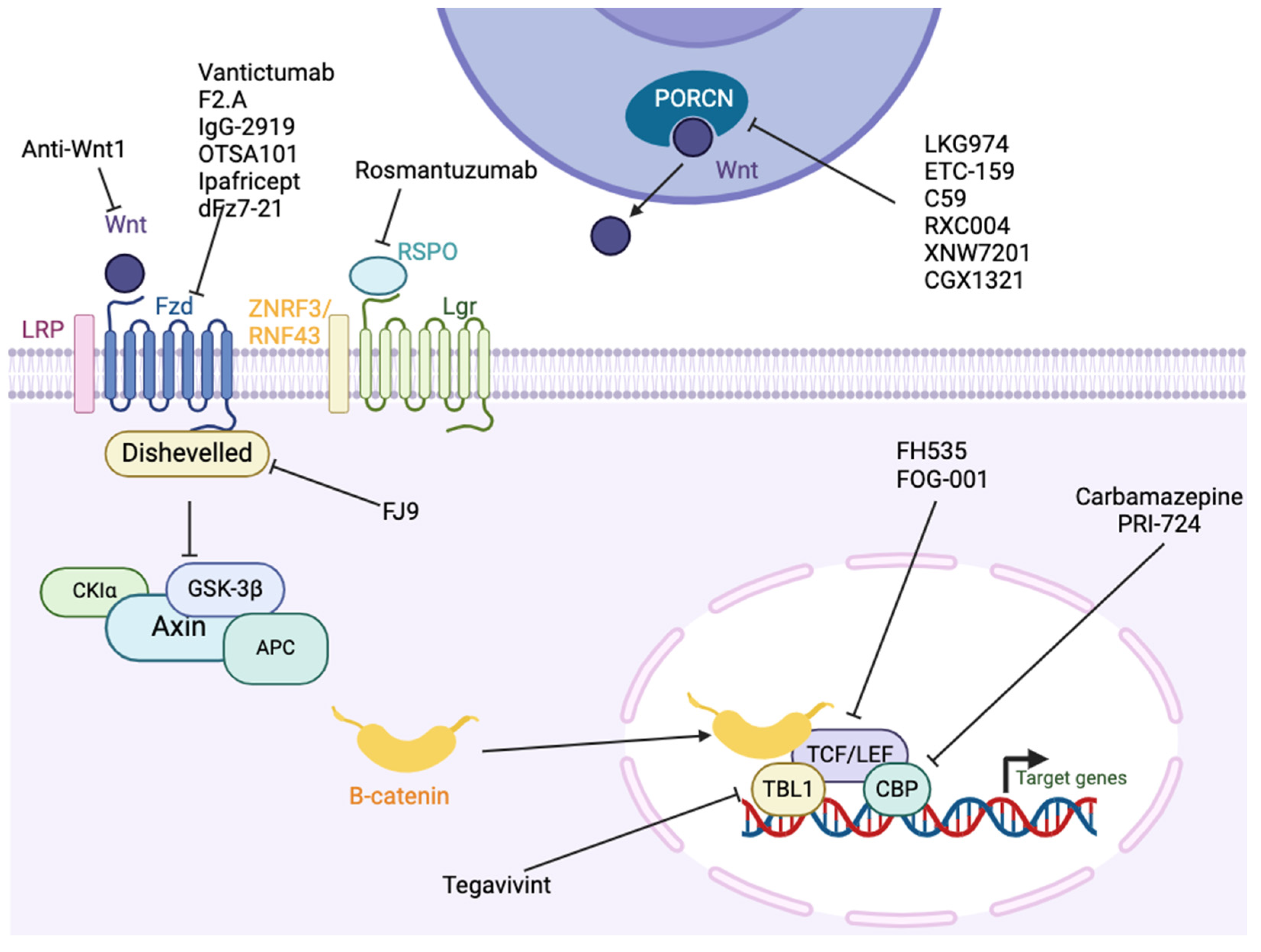
10. Wnt/β-Catenin
10.1. Antibodies
10.2. Pharmacologic Inhibition
11. SOX2
11.1. ZF-ATFs and PIPs
11.2. Peptides
11.3. Immunomodulatory Peptides and Vaccines
11.4. Pharmacologic Inhibition
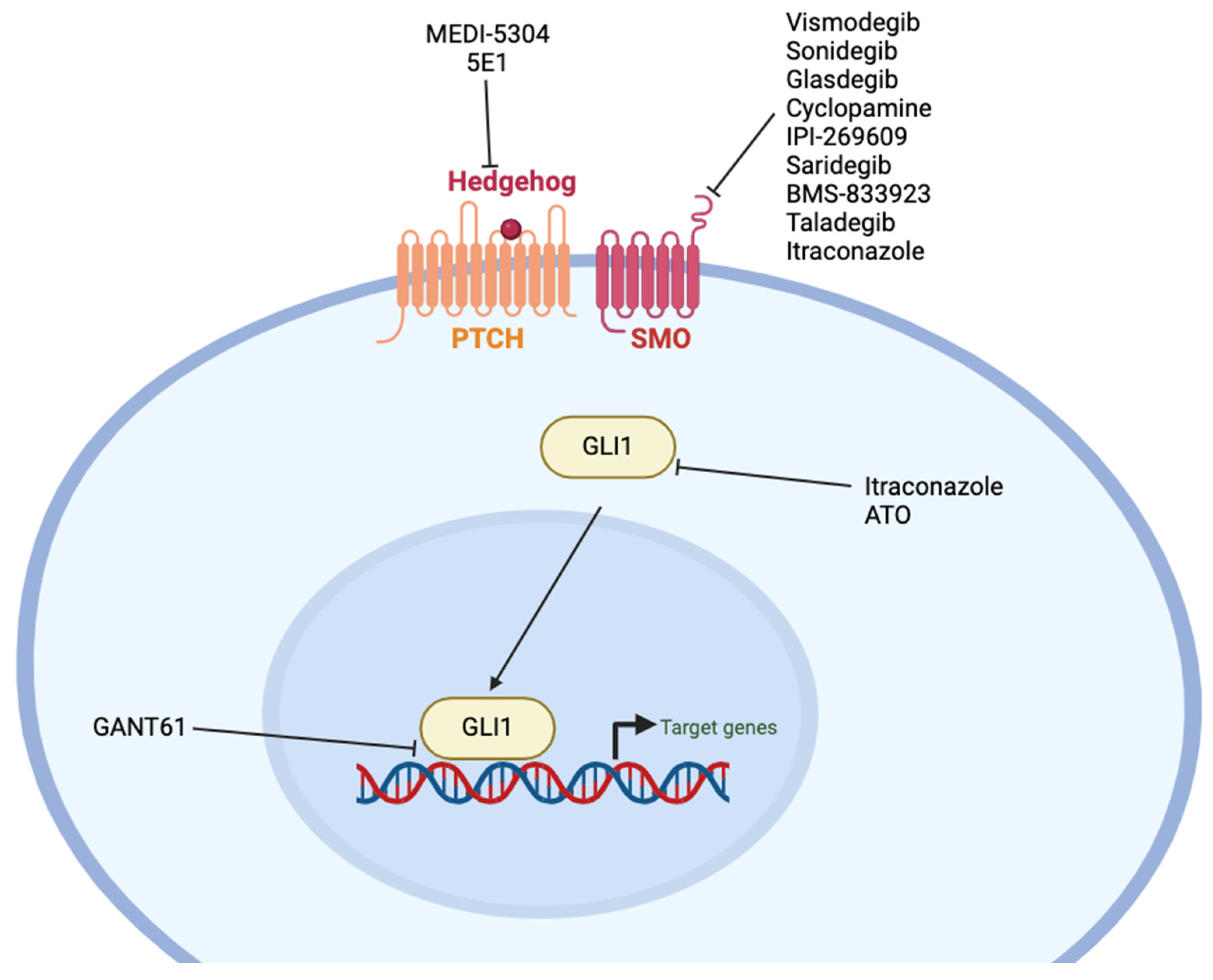
12. Hedgehog
12.1. Pharmacologic Inhibitors
12.2. Antibodies
13. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CSC Target | Drug | Condition | Clinical Status | Reference/Trial Number (if Applicable) |
|---|---|---|---|---|
| ALDH1A1 | NCT-501 | HNSCC, pancreatic, CRC | Preclinical | [42,43,44] |
| CM37 | Ovarian | Preclinical | [45] | |
| Compound 974 | Ovarian | Preclinical | [46] | |
| ALDH1A3 | GA11 | Glioma | Preclinical | [50] |
| MF-7 | Breast | Preclinical | [12] | |
| NR6 | Glioblastoma, CRC | Preclinical | [51] | |
| MCI-INI-3 | Glioma | Preclinical | [52] | |
| YD1701 | CRC | Preclinical | [53] | |
| ALDH2 | CVT-10216 | CRC | Preclinical | [13] |
| Daidzin | CRC | Preclinical | [13] | |
| ALDH3A1 | Dyclonine | SCC, gastric | Preclinical | [60] |
| CB57 | Lung, glioblastoma | Preclinical | [61] | |
| CB29 | Glioblastoma | Preclinical | [62] | |
| EN40 | Lung | Preclinical | [64] | |
| Pan-ALDH | DEAB | Melanoma, pancreatic | Preclinical | [66,67,68] |
| NanoKS100 | Melanoma | Preclinical | [69] | |
| DIMATE | AML | Preclinical | [70,71] | |
| 637A | Ovarian | Preclinical | [72] | |
| Citral | Breast | Preclinical | [73] | |
| Disulfiram | Breast, lung | Preclinical | [73,74,75] | |
| EpCAM | EpAb2-6 | CRC, SCC, pancreatic, lung, | Preclinical | [90,91] |
| Adecatumumab (MT201) | Ovarian, breast, prostate | Phase II | [93,94,95] | |
| AM-928 | Solid tumors | Phase I | NCT05687682 | |
| Solitomab (MT110) | Pancreatic, ovarian, solid tumors, | Phase I | [98,99,100] | |
| Catumaxomab | Malignant ascites | Approval withdrawn in European Union | [101] | |
| EpCAM-CD3 hFc mRNA-LNP | Ovarian | Preclinical | [102] | |
| Anti-EpCAM CAR-T cells | Ovarian, prostate, lung, gastric, pancreatic | Preclinical | [103,104,105,106] | |
| IMC001 | GI tumors | Phase II | [108] NCT05028933, NCT04196465 | |
| VB6-845 | Ovarian, breast, SCLC, CRC, SCC | Phase I | [109,110,111] NCT00481936 | |
| VB4-845 | HNSCC, bladder carcinoma | Phase III | [113,114,115,116] NCT04859751 | |
| SyntOFF | Breast | Preclinical | [119] | |
| chiHEA125-Ama | Pancreatic | Preclinical | [120] | |
| Anti-EpCAM siRNA | Breast, retinoblastoma | Preclinical | [123,124] | |
| CD44 | Bivatuzumab (BIWA-4) | HNSCC | Phase I | [133] NCT02254018 |
| H4C4 | Pancreatic | Preclinical | [134] | |
| IM7 | Breast | Preclinical | [135] | |
| RG7356 (RO5429083) | Leukemia, HNSCC, solid tumors | Phase I | [136,137,138,139,140] NCT01358903, NCT01641250 | |
| Anti-CD44-IR700 NIR-PIT | SCC, CRC, lung | Preclinical | [142,143,144] | |
| rhPRG4 | Breast | Preclinical | [145,146] | |
| Apt#7 | Breast | Preclinical | [149] | |
| CD44-EpCAM aptamer | Ovarian | Preclinical | [150] | |
| ASO 4401 | HCC | Preclinical | [152] | |
| THIQ | HNSCC | Preclinical | [154,155] | |
| JE22-NP | Breast | Preclinical | [156] | |
| Verbascoside | Glioblastoma | Preclinical | [157] | |
| A6 (SPL-108) | Prostate, multiple myeloma, breast, ovarian, leukemia | Phase II | [158,159,160,161,162,163,164,165] NCT00939809, NCT02046928 | |
| CD55 | Anti-CD55 | CRC | Preclinical | [172] |
| MB55 | Lymphoma, leukemia | Preclinical | [173] | |
| CD55 NAb | Neuroblastoma | Preclinical | [168] | |
| 177Lu-anti-CD55 | Lung | Preclinical | [174] | |
| GB262 | Pancreatic | Preclinical | [175] | |
| 105AD7 | CRC, osteosarcoma | Phase II | [176,177,178,179] | |
| PAT-SC1 | Gastric | Phase I | [180] | |
| CD55sp | Cervical | Preclinical | [181] | |
| C-PC/CMC-CD55 | Cervical | Preclinical | [182] | |
| AWT-489 | CRC | Preclinical | [184] | |
| CRISPR/cas9 | Cervical | Preclinical | [171] | |
| siRNA | Breast, ovarian, lung | Preclinical | [185] | |
| CXCR4 | Ulocuplumab (BMS-936564, MDX-1338) | Leukemia, lymphoma, multiple myeloma, breast, Waldenström macroglobulinemia | Phase II | [199,200,201,202,203] NCT01120457, NCT02472977, NCT02305563, NCT03225716, NCT01359657, NCT02666209 |
| PF-06747143 | Leukemia | Preclinical | [204,205,206] | |
| 12G5 | Endometrial, osteosarcoma | Preclinical | [207,208] | |
| ALX-0651 | N/A | Phase I | [209] NCT01374503 | |
| LY2624587 | Lymphoma, leukemia | Phase I | [210] NCT01139788 | |
| Hz515H7 (F50067) | Multiple myeloma, lymphoma, AML | Phase I | [211,213] | |
| MEDI3185 | Multiple myeloma, Burkitt’s lymphoma, ovarian, lung | Preclinical | [212] | |
| Plerixafor (AMD3100, Mozobil®) | Breast, lung, CRC, prostate, pancreatic, | FDA approved for multiple myeloma and lymphoma | [214,215,216,217,218,219,220,221,222,223,224] NCT00694590, NCT05510544, NCT00903968, NCT00906945 | |
| Mavorixafor (X4P-001) | RCC, melanoma, breast, Waldenström’s macroglobulinemia | Phase II | [225,226] NCT02823405, NCT05103917, NCT02667886, NCT02923531, NCT04274738 | |
| USL311 | Glioblastoma | Phase I/II | NCT02765165 | |
| PRX177561 | Glioblastoma | Preclinical | [227] | |
| MSX-122 | Breast, HNSCC, uveal melanoma | Phase I | [228] NCT00591682 | |
| Motixafortide (BL-8040, BKT-140, TNI4001) | AML, breast, pancreatic, solid tumors | Phase II | [218,228,229,230,231,232] NCT01838395, NCT02826486 | |
| LY2510924 | AML, solid tumors, RCC, SCLC | Phase II | [233,234,235] NCT02737072, NCT02652871, NCT01391130, NCT01439568 | |
| CTCE-9908 | Breast, solid tumors | Phase I/II | [236,237] | |
| IS4 | Prostate, melanoma | Preclinical | [238] | |
| CD133 | Anti-CD133 mAb | CRC, breast | Preclinical | [256,257,258] |
| BsAb-CIK | Pancreatic | Preclinical | [259] | |
| 293C3-SDIE | CRC, leukemia | Preclinical | [260,261] | |
| Anti-CD133 CAR NK cells | Ovarian | Preclinical | [262] | |
| Anti-CD133 CAR T cells | SCLC, liver, CRC | Phase II | [263,264,265] NCT02541370, NCT02541370 | |
| 16x133 BiKE | CRC | Preclinical | [266] | |
| 16x15x133 TriKE | CRC, breast, HNSCC, prostate, AML | Preclinical | [267] | |
| 1615EpCAM133 TetraKE | CRC | Preclinical | [268] | |
| ICT-121 | Glioblastoma | Phase I | [269] NCT02049489 | |
| GMI | Lung | Preclinical | [270] | |
| dCD133KDEL | HNSCC, breast, ovarian | Preclinical | [271,272,273] | |
| AC133-saporin | CRC | Preclinical | [274] | |
| Celecoxib | CRC | FDA-approved NSAID for migraines | [275,276] | |
| Trifluridine | CRC | FDA-approved for metastatic CRC | [277] | |
| ACT001 | Lung, glioma | Phase II | [278,279,280] ACTRN12616000228482, NCT05053880 | |
| CD133 aptamers | HCC, breast | Preclinical | [281,282,284] | |
| LS-7 | CRC, breast | Preclinical | [285] | |
| CRISPR/cas9 | CRC | Preclinical | [286] | |
| siRNA | CRC | Preclinical | [287] | |
| AC133 NIR-PIT | Glioma | Preclinical | [288] | |
| Nanog | IGT-PMO | Breast | Preclinical | [302] |
| siRNA | CRC | Preclinical | [305] | |
| SAHA | HNSCC, lymphoma | FDA-approved for T-cell lymphoma | [307,308] | |
| PiB | Prostate | Preclinical | [309] | |
| Resveratrol | Glioblastoma | Preclinical | [313,314] | |
| Aspirin | CRC | FDA-approved NSAID | [315] | |
| Metformin | TNBC | FDA-approved for diabetes mellitus | [316] | |
| DFX, SP10 | Esophageal | Preclinical | [317] | |
| Notch | Crenigacestat (LY3039478) | Breast, CRC, lung, ovarian, glioblastoma, gastric, intrahepatic cholangiocarcinoma, multiple myeloma | Phase I | [327,328,329,330,331] NCT01695005, NCT02836600, NCT03502577 |
| LY900009 | Solid tumors, lymphoma | Phase I | [333] NCT01158404 | |
| Osugacestat (AL101, BMS-906024) | Breast, leukemia, adenoid cystic carcinoma, NSCLC | Phase II | [334,335,336,337,338] NCT03691207, NCT01653470 | |
| RO4929097 (RG473) | Melanoma, CRC, sarcoma, pancreatic adenocarcinoma | Phase II | [340,344] NCT01116687, NCT01120275, NCT01154452, NCT01232829 | |
| Nirogacestat (PF-03084014) | HCC, prostate, dermoid, breast | Phase III | [345,346,347,348,349] NCT01981551, NCT04195399, NCT03785964, NCT01876251 | |
| DAPT | Osteosarcoma, gastric, adenoma | Preclinical | [350,351,352] | |
| MRK-0752 | Breast, ovarian, solid tumors, PDAC, CNS malignancies | Phase II | [326,353,354,355,356,357] NCT01098344 | |
| MRK-560 | Leukemia | Preclinical | [358] | |
| Limantrafin (CB103) | Breast, leukemia | Preclinical | [359,360] | |
| ZLDI-8 | NSCLC, CRC, HCC | Preclinical | [361,362,363] | |
| 602.101 | Breast | Preclinical | [364] | |
| Anti-Notch1 mAb | Lung, CRC | Preclinical | [365] | |
| Tarextumab (OMP-59R5) | Breast, lung, ovarian, pancreatic | Phase II | [366,367,368,369] NCT01277146, NCT01859741, NCT01647828 | |
| Brontictuzumab (OMP-52M51) | Solid tumors, hematologic and lymphoid malignancies, CRC, ACC, | Phase I | [370,371] NCT01778439, NCT01778439 | |
| PF-06650808 | Breast | Phase I | [372] NCT02129205 | |
| Enoticumab (REGN421) | Solid tumors | Phase I | [373] NCT00871559 | |
| Demcizumab (OMP-21M18) | Ovarian, peritoneal, fallopian, NSCLC, pancreatic, solid tumors | Phase I | [374,375] NCT01952249, NCT01189968, NCT01189942, NCT02722954, NCT01952249 | |
| MEDI0639 | Solid tumors | Phase I | [376,377] NCT01952249 | |
| MGZ01 | Breast | Preclinical | [378] | |
| Navicixizumab (OMP-305B83) | Solid tumors, ovarian, peritoneal, fallopian | Phase I | [379,380] NCT02298387, NCT03030287 | |
| ABT-165 | Glioblastoma, CRC | Phase I | [381,382] NCT03368859 | |
| Tarlatamab (AMG 757) | SCLC | Phase II | [383,384,385] NCT03319940, NCT05060016 | |
| HPN328 | SCLC, neuroendocrine | Phase I/II | [386] NCT04471727 | |
| Rova-T | SCLC | Phase III | [387,388] NCT04471727 | |
| Wnt/β-catenin | Vantictumab (OMP-18R5) | CRC, breast, lung, pancreatic, solid tumors, | Phase I | [402,403,404,405] NCT01345201, NCT02005315, |
| F2.A | PDAC | Preclinical | [406] | |
| IgG-2919 | PDAC | Preclinical | [407] | |
| OTSA101 | Synovial sarcoma | Phase I | [408,409,410,411] NCT01469975, NCT04176016 | |
| Ipafricept (OMP-54F28) | Pancreatic, ovarian, solid tumors | Phase I | [413,414,415,416] NCT01608867, NCT02092363, NCT02050178 | |
| Rosmantuzumab (OMP-131R10) | Leukemia, CRC | Phase I | [418,419] NCT02482441 | |
| LGK974 (Wnt974) | HNSCC, CRC, solid tumors | Phase II | [420,421,422,423,424] NCT01351103, NCT0227813, NCT02649530 | |
| ETC-159 | CRC, pancreatic, solid tumors | Phase I | [425,426,427] NCT02521844, NCT02521844 | |
| C59 | Nasopharyngeal carcinoma | Preclinical | [428,429] | |
| RXC004 | Pancreatic, CRC, solid tumors | Phase II | [430,431,432] NCT03447470, NCT04907851, NCT04907539 | |
| XNW7201 | Solid tumors | Phase I | NCT03901950 | |
| CGX1321 | GI tumors | Phase I | [433] NCT02675946 | |
| Tegavivint (BC2059, tegatrabetan) | Desmoid, AML, multiple myeloma | Phase I/II | [434,435,436,437] NCT03459469, NCT04851119 | |
| FH535 | Breast, pancreatic, CRC | Preclinical | [400,438,439,440,441,442] | |
| Doxorubicin | Leukemia | Preclinical | [443] | |
| FOG-001 | Solid tumors | Phase I | NCT05919264 | |
| Carbamazepine | N/A | FDA-approved anti-epileptic | [444] | |
| FJ9 | NSCLC | Preclinical | [445] | |
| dFz7-21 | N/A | Preclinical | [446] | |
| Niclosamide | CRC, ovarian, prostate | FDA-approved anti-helminthic | [447,448,449] NCT02532114, NCT02519582 | |
| SOX2 | ZF-522SKD/ ZF598SKD | Breast | Preclinical | [465] |
| ATF/SOX2 | Lung | Preclinical | [466] | |
| PIP-S2 | N/A | Preclinical | [467] | |
| sP42 | ESCC | Preclinical | [458] | |
| SOX2-iPEP | Breast, ovarian | Preclinical | [468] | |
| 60030 | Oligodendroglioma | Preclinical | [470] | |
| STEMVAC | Breast, NSCLC | Phase II | NCT05242965, NCT05455658, NCT02157051 | |
| Rapamycin | Glioma | FDA-approved for perivascular epithelioid tumors | [473] | |
| MK2206 | ESCC, breast | Phase II | [474,475] NCT01277757 | |
| DC120 | Nasopharyngeal carcinoma | Preclinical | [476] | |
| AZD4547 | Lung | Phase II | [461,477] NCT01791985 | |
| Gefitinib | NSCLC | FDA-approved for NSCLC | [478] | |
| Dasatinib | NSCLC | FDA-approved for CML | [478] | |
| LY294002 | NSCLC | Preclinical | [478] | |
| Erlotinib | NSCLC | FDA-approved for NSCLC | [478] | |
| Gentian violet | Melanoma | FDA-approved antimycotic/antibacterial | [479] | |
| CBP30 | Lung | Preclinical | [480] | |
| Pevonedistat (MLN4924) | Breast, NSCLC, leukemia, multiple myeloma, solid tumors, lymphoma | Phase III | [481,482,483] NCT03268954, NCT03323034, NCT03770260, NCT03965689 | |
| APG-1387 | Nasopharyngeal carcinoma | Preclinical | [462,484] | |
| FT234, FT895 | NSCLC | Preclinical | [485] | |
| CBB1007 | Lung | Preclinical | [486] | |
| Iadademstat (ORY-1001) | Breast, AML, SCLC | Phase II | [487,488] EUDRACT 2013-002447-29, NCT05420636, NCT05546580 | |
| Sonic Hedgehog | Vismodegib (GDC-0449) | Pancreatic, BCC, gastric, ovarian | FDA-approved for BCC | [496,497,498,499,500,501,502] NCT00607724, NCT00833417, NCT03052478, NCT00739661, NCT01088815 |
| Sonidegib (LDE225) | CML, BCC, NSCLC, SCLC, medulloblastoma, breast | FDA-approved for BCC | [503,504,505,506,507,508] NCT01327053, NCT01579929, NCT02027376, NCT01125800, NCT01456676 | |
| Glasdegib (PF-04449913) | AML | FDA-approved for AML | [509,510,511,512] NCT03416179 | |
| Cyclopamine | Breast | Preclinical | [513,514] | |
| IPI-269609 | Pancreatic | Preclinical | [515] | |
| Saridegib (IPI-926) | Medulloblastoma, chondrosarcoma, HNSCC | Phase I | [495,516,517,518,519,520,521] NCT01255800, NCT01383538, NCT01310816 | |
| BMS-833923 (XL139) | Esophageal, ovarian, AML, CML | Phase I | [522,523,524,525] NCT01218477 | |
| Taladegib (LY2940680, ENV-101) | Medulloblastoma, BCC | Phase II | [526,527] NCT01919398, NCT02784795, NCT01226485, NCT05199584 | |
| Hh003 | CRC, pancreatic | Preclinical | [528] | |
| Itraconazole | Medulloblastoma, CRC, breast, biliary tract | FDA-approved anti-fungal | [529,530,531,532,533,534,535,536,537] NCT00769600 | |
| ATO | SCLC, Ewing sarcoma, pancreatic | FDA-approved for acute promyelocytic leukemia | [538,539,540,541,542] | |
| GANT61 | Melanoma, pancreatic, HCC | Preclinical | [492,494,543,544,545] | |
| MEDI-5304 | CRC, pancreatic | Preclinical | [546] | |
| 5E1 | CRC, pancreatic, breast | Preclinical | [546,547,548,549] | |
| α-Ptch1 | Pancreatic | Preclinical | [550] |
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer Stem Cells as Key Drivers of Tumor Progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Chen, K.; Huang, Y.H.; Chen, J.L. Understanding and Targeting Cancer Stem Cells: Therapeutic Implications and Challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, K.; Rasheed, Z.A.; Karisch, R.; Wang, Q.; Kowalski, J.; Susky, E.; Pereira, K.; Karamboulas, C.; Moghal, N.; Rajeshkumar, N.V.; et al. Tumor-Initiating Cells Are Rare in Many Human Tumors. Cell Stem Cell 2010, 7, 279. [Google Scholar] [CrossRef]
- Toledo-Guzmán, M.E.; Bigoni-Ordóñez, G.D.; Hernández, M.I.; Ortiz-Sánchez, E. Cancer Stem Cell Impact on Clinical Oncology. World J. Stem Cells 2018, 10, 183. [Google Scholar] [CrossRef]
- Kurth, I.; Hein, L.; Mäbert, K.; Peitzsch, C.; Koi, L.; Cojoc, M.; Kunz-Schughart, L.; Baumann, M.; Dubrovska, A. Cancer Stem Cell Related Markers of Radioresistance in Head and Neck Squamous Cell Carcinoma. Oncotarget 2015, 6, 34494–34509. [Google Scholar] [CrossRef]
- Canino, C.; Luo, Y.Y.; Marcato, P.; Blandino, G.; Pass, H.I.; Cioce, M. A STAT3-NFkB/DDIT3/CEBPβ Axis Modulates ALDH1A3 Expression in Chemoresistant Cell Subpopulations. Oncotarget 2015, 6, 12637. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Giraud, J.; Chambonnier, L.; Dubus, P.; Wittkop, L.; Belleannee, G.; Collet, D.; Soubeyran, I.; Evrard, S.; Rousseau, B.; et al. Characterization of Biomarkers of Tumorigenic and Chemoresistant Cancer Stem Cells in Human Gastric Carcinoma. Clin. Cancer Res. 2017, 23, 1586–1597. [Google Scholar] [CrossRef]
- Alvero, A.B.; Chen, R.; Fu, H.H.; Montagna, M.; Schwartz, P.E.; Rutherford, T.; Silasi, D.A.; Steffensen, K.D.; Waldstrom, M.; Visintin, I.; et al. Molecular Phenotyping of Human Ovarian Cancer Stem Cells Unravels the Mechanisms for Repair and Chemoresistance. Cell Cycle 2009, 8, 158–166. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and Their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, D.; Minata, M.; Ibrahim, A.N.; Yamaguchi, S.; Coviello, V.; Bernstock, J.D.; Harada, S.; Cerione, R.A.; Tannous, B.A.; la Motta, C.; et al. Identification of ALDH1A3 as a Viable Therapeutic Target in Breast Cancer Metastasis-Initiating Cells. Mol. Cancer Ther. 2020, 19, 1134–1147. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.L.; Prince, G.M.S.H.; Batzorig, U.; Huang, C.Y.; Chang, Y.J. ALDH2 Promotes Cancer Stemness and Metastasis in Colorectal Cancer through Activating β-Catenin Signaling. J. Cell Biochem. 2023, 124, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Nie, B.; Pienta, K.J.; Morgan, T.M.; Taichman, R.S. Cancer Stem Cells and Their Role in Metastasis. Pharmacol. Ther. 2013, 138, 285. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; Kriegl, L.; Engel, J.; Kirchner, T.; Jung, A. Prognostic Significance of the Cancer Stem Cell Markers CD133, CD44, and CD166 in Colorectal Cancer. Cancer Investig. 2009, 27, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Qian, X.; Shi, M.; Li, H.; Peng, C.; Ding, X.; Zhang, S.; Zhang, B.; Xu, G.; Lv, Y.; et al. ALDH1A3 Accelerates Pancreatic Cancer Metastasis by Promoting Glucose Metabolism. Front. Oncol. 2020, 10, 915. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer Stem Cells: The Root of Tumor Recurrence and Metastases. Semin. Cancer Biol. 2017, 44, 10–24. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Savic, D.; Dudás, J.; Kvitsaridze, I.; Skvortsov, S.; Riechelmann, H.; Skvortsova, I.I. Cancer Stem Cells and Their Unique Role in Metastatic Spread. Semin. Cancer Biol. 2020, 60, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Dong, J.; Haiech, J.; Kilhoffer, M.C.; Zeniou, M. Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int. 2016, 2016, 1740936. [Google Scholar] [CrossRef]
- Dembinski, J.L.; Krauss, S. Characterization and Functional Analysis of a Slow Cycling Stem Cell-like Subpopulation in Pancreas Adenocarcinoma. Clin. Exp. Metastasis 2009, 26, 611–623. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A Restricted Cell Population Propagates Glioblastoma Growth after Chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Marchitti, S.A.; Brocker, C.; Stagos, D.; Vasiliou, V. Non-P450 Aldehyde Oxidizing Enzymes: The Aldehyde Dehydrogenase Superfamily. Expert Opin. Drug Metab. Toxicol. 2008, 4, 697–720. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W.K. Aldehyde Dehydrogenase: Its Role as a Cancer Stem Cell Marker Comes down to the Specific Isoform. Cell Cycle 2011, 10, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Vasillou, V.; Pappa, A.; Estey, T. Role of Human Aldehyde Dehydrogenases in Endobiotic and Xenobiotic Metabolism. Drug Metab. Rev. 2004, 36, 279–299. [Google Scholar] [CrossRef] [PubMed]
- Lalevée, S.; Anno, Y.N.; Chatagnon, A.; Samarut, E.; Poch, O.; Laudet, V.; Benoit, G.; Lecompte, O.; Rochette-Egly, C. Genome-Wide in Silico Identification of New Conserved and Functional Retinoic Acid Receptor Response Elements (Direct Repeats Separated by 5 Bp). J. Biol. Chem. 2011, 286, 33322–33334. [Google Scholar] [CrossRef] [PubMed]
- Coyle, K.M.; Sultan, M.; Thomas, M.L.; Vaghar-Kashani, A.; Marcato, P. Retinoid Signaling in Cancer and Its Promise for Therapy. J. Carcinog. Mutagen. 2013, 7, 16–18. [Google Scholar] [CrossRef]
- Zhou, L.; Sheng, D.; Wang, D.; Ma, W.; Deng, Q.; Deng, L.; Liu, S. Identification of Cancer-Type Specific Expression Patterns for Active Aldehyde Dehydrogenase (ALDH) Isoforms in ALDEFLUOR Assay. Cell Biol. Toxicol. 2019, 35, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Izumi, K.; Saito, T.; Ohnuki, H.; Terada, M.; Kawano, Y.; Nozawa-Inoue, K.; Saito, C.; Maeda, T. Distinct Expression Patterns and Roles of Aldehyde Dehydrogenases in Normal Oral Mucosa Keratinocytes: Differential Inhibitory Effects of a Pharmacological Inhibitor and RNAi-Mediated Knockdown on Cellular Phenotype and Epithelial Morphology. Histochem. Cell Biol. 2013, 139, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Dallaglio, K.; Chen, Y.; Robinson, W.A.; Robinson, S.E.; McCarter, M.D.; Wang, J.; Gonzalez, R.; Thompson, D.C.; Norris, D.A.; et al. ALDH1A Isozymes Are Markers of Human Melanoma Stem Cells and Potential Therapeutic Targets. Stem Cells 2012, 30, 2100–2113. [Google Scholar] [CrossRef]
- Li, T.; Su, Y.; Mei, Y.; Leng, Q.; Leng, B.; Liu, Z.; Stass, S.A.; Jiang, F. ALDH1A1 Is a Marker for Malignant Prostate Stem Cells and Predictor of Prostate Cancer Patients’ Outcome. Lab. Investig. 2010, 90, 234–244. [Google Scholar] [CrossRef]
- Marcato, P.; Dean, C.A.; Da, P.; Araslanova, R.; Gillis, M.; Joshi, M.; Helyer, L.; Pan, L.; Leidal, A.; Gujar, S.; et al. Aldehyde Dehydrogenase Activity of Breast Cancer Stem Cells Is Primarily Due To Isoform ALDH1A3 and Its Expression Is Predictive of Metastasis. Stem Cells 2011, 29, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde Dehydrogenase 1 Is a Marker for Normal and Malignant Human Colonic Stem Cells (SC) and Tracks SC Overpopulation during Colon Tumorigenesis. Cancer Res. 2009, 69, 3382–3389. [Google Scholar] [CrossRef]
- Terzuoli, E.; Bellan, C.; Aversa, S.; Ciccone, V.; Morbidelli, L.; Giachetti, A.; Donnini, S.; Ziche, M. ALDH3A1 Overexpression in Melanoma and Lung Tumors Drives Cancer Stem Cell Expansion, Impairing Immune Surveillance through Enhanced PD-L1 Output. Cancers 2019, 11, 1963. [Google Scholar] [CrossRef]
- Chen, L.; Wu, M.; Ji, C.; Yuan, M.; Liu, C.; Yin, Q. Silencing Transcription Factor FOXM1 Represses Proliferation, Migration, and Invasion While Inducing Apoptosis of Liver Cancer Stem Cells by Regulating the Expression of ALDH2. IUBMB Life 2020, 72, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.L.; Liu, S.; Cui, W.; Shi, Y.; Liu, Q.; Duan, J.J.; Yu, S.C.; Zhang, X.; Cui, Y.H.; Kung, H.F.; et al. Aldehyde Dehydrogenase 1A1 Circumscribes High Invasive Glioma Cells and Predicts Poor Prognosis. Am. J. Cancer Res. 2015, 5, 1471. [Google Scholar] [PubMed]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal Glioma Stem Cells Are Maintained by Activated Glycolytic Metabolism Involving Aldehyde Dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, R.; Mashima, T.; Kawata, N.; Kumagai, K.; Migita, T.; Sano, T.; Mizunuma, N.; Yamaguchi, K.; Seimiya, H. ALDH1A3-MTOR Axis as a Therapeutic Target for Anticancer Drug-Tolerant Persister Cells in Gastric Cancer. Cancer Sci. 2020, 111, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ren, Y.; Yu, X.; Qian, F.; Bian, B.S.J.; Xiao, H.L.; Wang, W.G.; Xu, S.L.; Yang, J.; Cui, W.; et al. ALDH1A1 Defines Invasive Cancer Stem-like Cells and Predicts Poor Prognosis in Patients with Esophageal Squamous Cell Carcinoma. Mod. Pathol. 2014, 27, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, Q.; Fu, X.; Luo, W. ALDH1A1 Overexpression Is Associated with the Progression and Prognosis in Gastric Cancer. BMC Cancer 2014, 14, 705. [Google Scholar] [CrossRef]
- Meng, E.; Mitra, A.; Tripathi, K.; Finan, M.A.; Scalici, J.; McClellan, S.; Da Silva, L.M.; Reed, E.; Shevde, L.A.; Palle, K.; et al. ALDH1A1 Maintains Ovarian Cancer Stem Cell-like Properties by Altered Regulation of Cell Cycle Checkpoint and DNA Repair Network Signaling. PLoS ONE 2014, 9, e107142. [Google Scholar] [CrossRef]
- Khoury, T.; Ademuyiwa, F.O.; Chandraseekhar, R.; Jabbour, M.; Deleo, A.; Ferrone, S.; Wang, Y.; Wang, X. Aldehyde Dehydrogenase 1A1 Expression in Breast Cancer Is Associated with Stage, Triple Negativity, and Outcome to Neoadjuvant Chemotherapy. Mod. Pathol. 2012, 25, 388–397. [Google Scholar] [CrossRef]
- Kulsum, S.; Sudheendra, H.V.; Pandian, R.; Ravindra, D.R.; Siddappa, G.; Nisheena, R.; Chevour, P.; Ramachandran, B.; Sagar, M.; Jayaprakash, A.; et al. Cancer Stem Cell Mediated Acquired Chemoresistance in Head and Neck Cancer Can Be Abrogated by Aldehyde Dehydrogenase 1 A1 Inhibition. Mol. Carcinog. 2017, 56, 694–711. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.M.; Yasgar, A.; Miller, B.; Lal-Nag, M.; Brimacombe, K.; Hu, X.; Sun, H.; Wang, A.; Xu, X.; Nguyen, K.; et al. Discovery of NCT-501, a Potent and Selective Theophylline-Based Inhibitor of Aldehyde Dehydrogenase 1A1 (ALDH1A1). J. Med. Chem. 2015, 58, 5967. [Google Scholar] [CrossRef] [PubMed]
- Yasgar, A.; Titus, S.A.; Wang, Y.; Danchik, C.; Yang, S.M.; Vasiliou, V.; Jadhav, A.; Maloney, D.J.; Simeonov, A.; Martinez, N.J. A High-Content Assay Enables the Automated Screening and Identification of Small Molecules with Specific ALDH1A1-Inhibitory Activity. PLoS ONE 2017, 12, e0170937. [Google Scholar] [CrossRef] [PubMed]
- Nwani, N.G.; Condello, S.; Wang, Y.; Swetzig, W.M.; Barber, E.; Hurley, T.; Matei, D. A Novel ALDH1A1 Inhibitor Targets Cells with Stem Cell Characteristics in Ovarian Cancer. Cancers 2019, 11, 502. [Google Scholar] [CrossRef] [PubMed]
- Muralikrishnan, V.; Fang, F.; Given, T.C.; Podicheti, R.; Chtcherbinine, M.; Metcalfe, T.X.; Sriramkumar, S.; O’Hagan, H.M.; Hurley, T.D.; Nephew, K.P. A Novel ALDH1A1 Inhibitor Blocks Platinum-Induced Senescence and Stemness in Ovarian Cancer. Cancers 2022, 14, 3437. [Google Scholar] [CrossRef] [PubMed]
- Mclean, M.E.; Maclean, M.R.; Cahill, H.F.; Arun, R.P.; Walker, O.L.; Wasson, M.-C.D.; Fernando, W.; Ponnusamy, P.; Mclean, M.E.; Maclean, M.R.; et al. The Expanding Role of Cancer Stem Cell Marker ALDH1A3 in Cancer and Beyond. Cancers 2023, 15, 492. [Google Scholar] [CrossRef] [PubMed]
- Durinikova, E.; Kozovska, Z.; Poturnajova, M.; Plava, J.; Cierna, Z.; Babelova, A.; Bohovic, R.; Schmidtova, S.; Tomas, M.; Kucerova, L.; et al. ALDH1A3 Upregulation and Spontaneous Metastasis Formation Is Associated with Acquired Chemoresistance in Colorectal Cancer Cells. BMC Cancer 2018, 18, 848. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Dean, C.A.; Liu, R.Z.; Coyle, K.M.; Bydoun, M.; Wallace, M.; Clements, D.; Turner, C.; Mathenge, E.G.; Gujar, S.A.; et al. Aldehyde Dehydrogenase 1A3 Influences Breast Cancer Progression via Differential Retinoic Acid Signaling. Mol. Oncol. 2015, 9, 17–31. [Google Scholar] [CrossRef]
- Cheng, P.; Wang, J.; Waghmare, I.; Sartini, S.; Coviello, V.; Zhang, Z.; Kim, S.H.; Mohyeldin, A.; Pavlyukov, M.S.; Minata, M.; et al. FOXD1-ALDH1A3 Signaling Is a Determinant for the Self-Renewal and Tumorigenicity of Mesenchymal Glioma Stem Cells. Cancer Res. 2016, 76, 7219–7230. [Google Scholar] [CrossRef]
- Gelardi, E.L.M.; Colombo, G.; Picarazzi, F.; Ferraris, D.M.; Mangione, A.; Petrarolo, G.; Aronica, E.; Rizzi, M.; Mori, M.; La Motta, C.; et al. A Selective Competitive Inhibitor of Aldehyde Dehydrogenase 1A3 Hinders Cancer Cell Growth, Invasiveness and Stemness In Vitro. Cancers 2021, 13, 356. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Garavaglia, S.; Ye, Z.; Moretti, A.; Belyaeva, O.V.; Beiser, A.; Ibrahim, M.; Wilk, A.; McClellan, S.; Klyuyeva, A.V.; et al. A Specific Inhibitor of ALDH1A3 Regulates Retinoic Acid Biosynthesis in Glioma Stem Cells. Commun. Biol. 2021, 4, 1420. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.-J.; Wang, D.; Cai, J.; Chen, J.; Zheng, X.-X.; Chen, T.; Wang, J.; Zhang, X.; Yang, Q.-K.; Yu, S.-C. An Aldehyde Dehydrogenase 1A3 Inhibitor Attenuates the Metastasis of Human Colorectal Cancer. Cancer Lett. 2022, 536, 215662. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.O.; Vo, T.H.; Lam, L.H.T.; Le, N.Q.K. ALDH2 as a Potential Stem Cell-Related Biomarker in Lung Adenocarcinoma: Comprehensive Multi-Omics Analysis. Comput. Struct. Biotechnol. J. 2023, 21, 1921. [Google Scholar] [CrossRef] [PubMed]
- Arolfo, M.P.; Overstreet, D.H.; Yao, L.; Fan, P.; Lawrence, A.J.; Tao, G.; Keung, W.M.; Vallee, B.L.; Olive, M.F.; Gass, J.T.; et al. Suppression of Heavy Drinking and Alcohol Seeking by a Selective ALDH-2 Inhibitor. Alcohol. Clin. Exp. Res. 2009, 33, 1935. [Google Scholar] [CrossRef] [PubMed]
- Lowe, E.D.; Gao, G.Y.; Johnson, L.N.; Wing, M.K. Structure of Daidzin, a Naturally Occurring Anti-Alcohol-Addiction Agent, in Complex with Human Mitochondrial Aldehyde Dehydrogenase. J. Med. Chem. 2008, 51, 4482–4487. [Google Scholar] [CrossRef] [PubMed]
- Keung, W.M.; Vallee, B.L. Daidzin and Daidzein Suppress Free-Choice Ethanol Intake by Syrian Golden Hamsters. Proc. Natl. Acad. Sci. USA 1993, 90, 10008–10012. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Chong, R.A.; Yang, Q.; Wei, Y.; Blanco, M.A.; Li, F.; Reiss, M.; Au, J.L.S.; Haffty, B.G.; Kang, Y. MTDH Activation by 8q22 Genomic Gain Promotes Chemoresistance and Metastasis of Poor-Prognosis Breast Cancer. Cancer Cell 2009, 15, 9–20. [Google Scholar] [CrossRef]
- Sládek, N.E.; Kollander, R.; Sreerama, L.; Kiang, D.T. Cellular Levels of Aldehyde Dehydrogenases (ALDH1A1 and ALDH3A1) as Predictors of Therapeutic Responses to Cyclophosphamide-Based Chemotherapy of Breast Cancer: A Retrospective Study. Cancer Chemother. Pharmacol. 2002, 49, 309–321. [Google Scholar] [CrossRef]
- Okazaki, S.; Shintani, S.; Hirata, Y.; Suina, K.; Semba, T.; Yamasaki, J.; Umene, K.; Ishikawa, M.; Saya, H.; Nagano, O. Synthetic Lethality of the ALDH3A1 Inhibitor Dyclonine and XCT Inhibitors in Glutathione Deficiency-Resistant Cancer Cells. Oncotarget 2018, 9, 33832. [Google Scholar] [CrossRef]
- Parajuli, B.; Fishel, M.L.; Hurley, T.D. Selective ALDH3A1 Inhibition by Benzimidazole Analogues Increase Mafosfamide Sensitivity in Cancer Cells. J. Med. Chem. 2014, 57, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, B.; Georgiadis, T.M.; Fishel, M.L.; Hurley, T.D. Development of Selective Inhibitors for Human Aldehyde Dehydrogenase 3A1 (ALDH3A1) for the Enhancement of Cyclophosphamide Cytotoxicity. ChemBioChem 2014, 15, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Khanna, M.; Chen, C.H.; Kimble-Hill, A.; Parajuli, B.; Perez-Miller, S.; Baskaran, S.; Kim, J.; Dria, K.; Vasiliou, V.; Mochly-Rosen, D.; et al. Discovery of a Novel Class of Covalent Inhibitor for Aldehyde Dehydrogenases. J. Biol. Chem. 2011, 286, 43486–43494. [Google Scholar] [CrossRef] [PubMed]
- Counihan, J.L.; Wiggenhorn, A.L.; Anderson, K.E.; Nomura, D.K. Chemoproteomics-Enabled Covalent Ligand Screening Reveals ALDH3A1 as a Lung Cancer Therapy Target. ACS Chem. Biol. 2018, 13, 1970–1977. [Google Scholar] [CrossRef] [PubMed]
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.D.; Vasiliou, V. Aldehyde Dehydrogenase Inhibitors: A Comprehensive Review of the Pharmacology, Mechanism of Action, Substrate Specificity, and Clinical Application. Pharmacol. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.A.; Parajuli, B.; Buchman, C.D.; Dria, K.; Hurley, T.D. N,N-Diethylaminobenzaldehyde (DEAB) as a Substrate and Mechanism-Based Inhibitor for Human ALDH Isoenzymes. Chem. Biol. Interact. 2015, 234, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Huang, Z.M.; Fong, S.; Leong, S.; Jakowatz, J.G.; Charruyer-Reinwald, A.; Wei, M.; Ghadially, R. Targeting ALDH1 to Decrease Tumorigenicity, Growth and Metastasis of Human Melanoma. Melanoma Res. 2015, 25, 138–148. [Google Scholar] [CrossRef] [PubMed]
- N,N Diethylaminobenzaldehyde Targets Aldehyde Dehydrogenase to Eradicate Human Pancreatic Cancer Cells. Available online: https://www.spandidos-publications.com/10.3892/etm.2020.8691 (accessed on 3 December 2023).
- Dinavahi, S.S.; Gowda, R.; Gowda, K.; Bazewicz, C.G.; Chirasani, V.R.; Battu, M.B.; Berg, A.; Dokholyan, N.V.; Amin, S.; Robertson, G.P. Development of a Novel Multi-Isoform ALDH Inhibitor Effective as an Antimelanoma Agent. Mol. Cancer Ther. 2020, 19, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Canuto, R.A.; Muzio, G.; Salvo, R.A.; Maggiora, M.; Trombetta, A.; Chantepie, J.; Fournet, G.; Reichert, U.; Quash, G. The Effect of a Novel Irreversible Inhibitor of Aldehyde Dehydrogenases 1 and 3 on Tumor Cell Growth and Death. Chem. Biol. Interact. 2001, 130–132, 209–218. [Google Scholar] [CrossRef]
- Venton, G.; Pérez-Alea, M.; Baier, C.; Fournet, G.; Quash, G.; Labiad, Y.; Martin, G.; Sanderson, F.; Poullin, P.; Suchon, P.; et al. Aldehyde Dehydrogenases Inhibition Eradicates Leukemia Stem Cells While Sparing Normal Progenitors. Blood Cancer J. 2016, 6, e469. [Google Scholar] [CrossRef]
- Chefetz, I.; Grimley, E.; Yang, K.; Hong, L.; Vinogradova, E.V.; Suciu, R.; Kovalenko, I.; Karnak, D.; Morgan, C.A.; Chtcherbinine, M.; et al. A Pan-ALDH1A Inhibitor Induces Necroptosis in Ovarian Cancer Stem-like Cells. Cell Rep. 2019, 26, 3061–3075.e6. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.L.; de Antueno, R.; Coyle, K.M.; Sultan, M.; Cruickshank, B.M.; Giacomantonio, M.A.; Giacomantonio, C.A.; Duncan, R.; Marcato, P. Citral Reduces Breast Tumor Growth by Inhibiting the Cancer Stem Cell Marker ALDH1A3. Mol. Oncol. 2016, 10, 1485–1496. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, R.; Sagar, R.; Pal, A. Tracing the Journey of Disulfiram: From an Unintended Discovery to a Treatment Option for Alcoholism. J. Ment. Health Hum. Behav. 2015, 20, 41. [Google Scholar] [CrossRef]
- Wang, N.N.; Wang, L.H.; Li, Y.; Fu, S.Y.; Xue, X.; Jia, L.N.; Yuan, X.Z.; Wang, Y.T.; Tang, X.; Yang, J.Y.; et al. Targeting ALDH2 with Disulfiram/Copper Reverses the Resistance of Cancer Cells to Microtubule Inhibitors. Exp. Cell Res. 2018, 362, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, S.V.; Balzar, M.; Winter, M.J.; Bakker, H.A.M.; Briaire-De Bruijn, I.H.; Prins, F.; Fleuren, G.J.; Warnaar, S.O. Epithelial Cell Adhesion Molecule (Ep-CAM) Modulates Cell–Cell Interactions Mediated by Classic Cadherins. J. Cell Biol. 1997, 139, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Munz, M.; Baeuerle, P.A.; Gires, O. The Emerging Role of EpCAM in Cancer and Stem Cell Signaling. Cancer Res. 2009, 69, 5627–5629. [Google Scholar] [CrossRef] [PubMed]
- Ogura, E.; Senzaki, H.; Yoshizawa, K.; Hioki, K.; Tsubura, A. Immunohistochemical Localization of Epithelial Glycoprotein EGP-2 and Carcinoembryonic Antigen in Normal Colonic Mucosa and Colorectal Tumors. Anticancer Res. 1998, 18, 3669–3675. [Google Scholar] [PubMed]
- Herlyn, D.; Herlyn, M.; Steplewski, Z.; Koprowski, H. Monoclonal Antibodies in Cell-Mediated Cytotoxicity against Human Melanoma and Colorectal Carcinoma. Eur. J. Immunol. 1979, 9, 657–659. [Google Scholar] [CrossRef] [PubMed]
- Went, P.T.; Lugli, A.; Meier, S.; Bundi, M.; Mirlacher, M.; Sauter, G.; Dirnhofer, S. Frequent EpCam Protein Expression in Human Carcinomas. Hum. Pathol. 2004, 35, 122–128. [Google Scholar] [CrossRef]
- Lee, C.C.; Yu, C.J.; Panda, S.S.; Chen, K.C.; Liang, K.H.; Huang, W.C.; Wang, Y.S.; Ho, P.C.; Wu, H.C. Epithelial Cell Adhesion Molecule (EpCAM) Regulates HGFR Signaling to Promote Colon Cancer Progression and Metastasis. J. Transl. Med. 2023, 21, 530. [Google Scholar] [CrossRef]
- Hiraga, T.; Ito, S.; Nakamura, H. EpCAM Expression in Breast Cancer Cells Is Associated with Enhanced Bone Metastasis Formation. Int. J. Cancer 2016, 138, 1698–1708. [Google Scholar] [CrossRef] [PubMed]
- Terris, B.; Cavard, C.; Perret, C. EpCAM, a New Marker for Cancer Stem Cells in Hepatocellular Carcinoma. J. Hepatol. 2010, 52, 280–281. [Google Scholar] [CrossRef]
- Yamashita, T.; Ji, J.; Budhu, A.; Forgues, M.; Yang, W.; Wang, H.Y.; Jia, H.; Ye, Q.; Qin, L.X.; Wauthier, E.; et al. EpCAM-Positive Hepatocellular Carcinoma Cells Are Tumor-Initiating Cells with Stem/Progenitor Cell Features. Gastroenterology 2009, 136, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Fan, X.; Fu, B.; Zheng, M.; Zhang, A.; Zhong, K.; Yan, J.; Sun, R.; Tian, Z.; Wei, H. EpCAM Inhibition Sensitizes Chemoresistant Leukemia to Immune Surveillance. Cancer Res. 2017, 77, 482–493. [Google Scholar] [CrossRef]
- Tayama, S.; Motohara, T.; Narantuya, D.; Li, C.; Fujimoto, K.; Sakaguchi, I.; Tashiro, H.; Saya, H.; Nagano, O.; Katabuchi, H. The Impact of EpCAM Expression on Response to Chemotherapy and Clinical Outcomes in Patients with Epithelial Ovarian Cancer. Oncotarget 2017, 8, 44312. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Dombkowski, D.; Meirelles, K.; Pieretti-Vanmarcke, R.; Szotek, P.P.; Chang, H.L.; Preffer, F.I.; Mueller, P.R.; Teixeira, J.; MacLaughlin, D.T.; et al. Müllerian Inhibiting Substance Preferentially Inhibits Stem/Progenitors in Human Ovarian Cancer Cell Lines Compared with Chemotherapeutics. Proc. Natl. Acad. Sci. USA 2010, 107, 18874–18879. [Google Scholar] [CrossRef]
- Ensinger, C.; Kremser, R.; Prommegger, R.; Spizzo, G.; Schmid, K.W. EpCAM Overexpression in Thyroid Carcinomas: A Histopathological Study of 121 Cases. J. Immunother. 2006, 29, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Seligson, D.B.; Pantuck, A.J.; Liu, X.; Huang, Y.; Horvath, S.; Bui, M.H.T.; Han, K.R.; Correa, A.J.L.; Eeva, M.; Tze, S.; et al. Epithelial Cell Adhesion Molecule (KSA) Expression: Pathobiology and Its Role as an Independent Predictor of Survival in Renal Cell Carcinoma. Clin. Cancer Res. 2004, 10, 2659–2669. [Google Scholar] [CrossRef]
- Liao, M.-Y.; Lai, J.-K.; Kuo, M.Y.-P.; Lu, R.-M.; Lin, C.-W.; Cheng, P.-C.; Liang, K.-H.; Wu, H.-C. An Anti-EpCAM Antibody EpAb2-6 for the Treatment of Colon Cancer. Oncotarget 2015, 6, 24947. [Google Scholar] [CrossRef]
- Chen, H.N.; Liang, K.H.; Lai, J.K.; Lan, C.H.; Liao, M.Y.; Hung, S.H.; Chuang, Y.T.; Chen, K.C.; Tsuei, W.W.F.; Wu, H.C. EpCAM Signaling Promotes Tumor Progression and Protein Stability of PD-L1 through the EGFR Pathway. Cancer Res. 2020, 80, 5035–5050. [Google Scholar] [CrossRef]
- Naundorf, S.; Preithner, S.; Mayer, P.; Lippold, S.; Wolf, A.; Hanakam, F.; Fichtner, I.; Kufer, P.; Raum, T.; Riethmüller, G.; et al. In Vitro and in Vivo Activity of MT201, a Fully Human Monoclonal Antibody for Pancarcinoma Treatment. Int. J. Cancer 2002, 100, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Scheulen, M.E.; Dittrich, C.; Obrist, P.; Marschner, N.; Dirix, L.; Schmidt, M.; Rüttinger, D.; Schuler, M.; Reinhardt, C.; et al. An Open-Label, Randomized Phase II Study of Adecatumumab, a Fully Human Anti-EpCAM Antibody, as Monotherapy in Patients with Metastatic Breast Cancer. Ann. Oncol. 2010, 21, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Marschner, N.; Rüttinger, D.; Zugmaier, G.; Nemere, G.; Lehmann, J.; Obrist, P.; Baeuerle, P.A.; Wolf, A.; Schmidt, M.; Abrahamsson, P.A.; et al. Phase II Study of the Human Anti-Epithelial Cell Adhesion Molecule Antibody Adecatumumab in Prostate Cancer Patients with Increasing Serum Levels of Prostate-Specific Antigen after Radical Prostatectomy. Urol. Int. 2010, 85, 386–395. [Google Scholar] [CrossRef]
- Sebastian, M.; Hanusch, C.; Schmidt, M.; Marschner, N.; Oruzio, D.; Wolf, C.; Reinhardt, C.; Eiermann, W.; Rüttinger, D.; Schuler, M. Safety and Antitumor Activity of 3-Weekly Anti-EpCAM Antibody Adecatumumab (MT201) in Combination with Docetaxel for Patients with Metastatic Breast Cancer: Results of a Multicenter Phase Ib Trial. J. Clin. Oncol. 2009, 27 (Suppl. S15), 1009. [Google Scholar] [CrossRef]
- Study Details|Safety of AM-928 Infusion in Advanced Solid Tumors|ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05687682 (accessed on 5 January 2024).
- Tian, Z.; Liu, M.; Zhang, Y.; Wang, X. Bispecific T Cell Engagers: An Emerging Therapy for Management of Hematologic Malignancies. J. Hematol. Oncol. 2021, 14, 75. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.; Dorado, J.; Baeuerle, P.A.; Heeschen, C. EpCAM/CD3-Bispecific T-Cell Engaging Antibody MT110 Eliminates Primary Human Pancreatic Cancer Stem Cells. Clin. Cancer Res. 2012, 18, 465–474. [Google Scholar] [CrossRef]
- English, D.P.; Bellone, S.; Schwab, C.L.; Roque, D.M.; Lopez, S.; Bortolomai, I.; Cocco, E.; Bonazzoli, E.; Chatterjee, S.; Ratner, E.; et al. Solitomab, an Epithelial Cell Adhesion Molecule/CD3 Bispecific Antibody (BiTE), Is Highly Active against Primary Chemotherapy-Resistant Ovarian Cancer Cell Lines in Vitro and Fresh Tumor Cells Ex Vivo. Cancer 2015, 121, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Kebenko, M.; Goebeler, M.E.; Wolf, M.; Hasenburg, A.; Seggewiss-Bernhardt, R.; Ritter, B.; Rautenberg, B.; Atanackovic, D.; Kratzer, A.; Rottman, J.B.; et al. A Multicenter Phase 1 Study of Solitomab (MT110, AMG 110), a Bispecific EpCAM/CD3 T-Cell Engager (BiTE®) Antibody Construct, in Patients with Refractory Solid Tumors. Oncoimmunology 2018, 7, 1450710. [Google Scholar] [CrossRef]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab: Clinical Development and Future Directions. MAbs 2010, 2, 129. [Google Scholar] [CrossRef]
- Golubovskaya, V.; Sienkiewicz, J.; Sun, J.; Huang, Y.; Hu, L.; Zhou, H.; Harto, H.; Xu, S.; Berahovich, R.; Bodmer, W.; et al. MRNA-Lipid Nanoparticle (LNP) Delivery of Humanized EpCAM-CD3 Bispecific Antibody Significantly Blocks Colorectal Cancer Tumor Growth. Cancers 2023, 15, 2860. [Google Scholar] [CrossRef]
- Ang, W.X.; Li, Z.; Chi, Z.; Du, S.-H.; Chen, C.; Tay, J.C.K.; Toh, H.C.; Connolly, J.E.; Xu, X.H.; Wang, S.; et al. Intraperitoneal Immunotherapy with T Cells Stably and Transiently Expressing Anti-EpCAM CAR in Xenograft Models of Peritoneal Carcinomatosis. Oncotarget 2017, 8, 13545–13559. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wu, Y.; Ma, W.; Zhang, S.; Zhang, Y.Q. Adoptive T-Cell Therapy of Prostate Cancer Targeting the Cancer Stem Cell Antigen EpCAM. BMC Immunol. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Karschnia, P.; Cadilha, B.L.; Dede, S.; Lorenz, M.; Seewaldt, N.; Nikolaishvili, E.; Müller, K.; Blobner, J.; Teske, N.; et al. In Vivo Dynamics and Anti-Tumor Effects of EpCAM-Directed CAR T-Cells against Brain Metastases from Lung Cancer. Oncoimmunology 2023, 12, 2163781. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; McCloskey, J.E.; Yang, H.; Puc, J.; Alcaina, Y.; Vedvyas, Y.; Gomez Gallegos, A.A.; Ortiz-Sánchez, E.; de Stanchina, E.; Min, I.M.; et al. Bispecific CAR T Cells against EpCAM and Inducible ICAM-1 Overcome Antigen Heterogeneity and Generate Superior Anti-Tumor Responses. Cancer Immunol. Res. 2021, 9, 1158. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-Negative Myeloid Phenotype Allows Immune Escape of MLL-Rearranged B-ALL from CD19 CAR-T-Cell Therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Fang, W.; Lu, Z.; Tong, C.; Zhang, H.; Ai, G.; Wang, S. EpCAM CAR T (IMC001) for the Treatment of Advanced GI Cancers. J. Clin. Oncol. 2023, 41 (Suppl. S16), 4034. [Google Scholar] [CrossRef]
- Entwistle, J.; Kowalski, M.; Brown, J.; Cizeau, J.; MacDonald, G.C. The Preclinical and Clinical Evaluation of VB6- 845: An Immunotoxin with a de-Immunized Payload for the Systemic Treatment of Solid Tumors. In Antibody-Drug Conjugates and Immunotoxins: From Pre-Clinical Development to Therapeutic Applications; Springer: New York, NY, USA, 2013; pp. 349–367. [Google Scholar] [CrossRef]
- Cizeau, J.; Grenkow, D.M.; Brown, J.G.; Entwistle, J.; MacDonald, G.C. Engineering and Biological Characterization of VB6-845, an Anti-EpCAM Immunotoxin Containing a t-Cell Epitope-Depleted Variant of the Plant Toxin Bouganin. J. Immunother. 2009, 32, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, M.; Brazas, L.; Zaretsky, R.; Rasamoelisolo, M.; MacDonald, G.; Cuthbert, W.; Glover, N. A Phase I Study of VB6–845, an Anti-EpCAM Fusion Protein Targeting Advanced Solid Tumors of Epithelial Origin: Preliminary Results. J. Clin. Oncol. 2008, 26 (Suppl. S15), 14663. [Google Scholar] [CrossRef]
- Brown, J.; Rasamoelisolo, M.; Spearman, M.; Bosc, D.; Cizeau, J.; Entwistle, J.; MacDonald, G.C. Preclinical Assessment of an Anti-EpCAM Immunotoxin: Locoregional Delivery Provides a Safer Alternative to Systemic Administration. Cancer Biother. Radiopharm. 2009, 24, 477–487. [Google Scholar] [CrossRef]
- Dillon, R.L.; Chooniedass, S.; Premsukh, A.; MacDonald, G.; Cizeau, J.; Adams, G.A. Abstract 614: VB4-845 Tumor Cell Killing in a Combination Study with the Anti-PD-1, Nivolumab. Cancer Res. 2017, 77 (Suppl. S13), 614. [Google Scholar] [CrossRef]
- Fitsialos, D.; Quenneville, J.; Rasamoelisolo, M.; Cross, M.; Glover, N.; MacDonald, G. A Phase I Study of VB4–845 in Patients with Advanced, Recurrent Head and Neck Cancer on a Weekly Dosing Scheme. J. Clin. Oncol. 2005, 23 (Suppl. S16), 5569. [Google Scholar] [CrossRef][Green Version]
- MacDonald, G.C.; Rasamoelisolo, M.; Entwistle, J.; Cizeau, J.; Bosc, D.; Cuthbert, W.; Kowalski, M.; Spearman, M.; Glover, N. A Phase I Clinical Study of VB4-845: Weekly Intratumoral Administration of an Anti-EpCAM Recombinant Fusion Protein in Patients with Squamous Cell Carcinoma of the Head and Neck. Drug Des. Devel Ther. 2008, 2, 105. [Google Scholar] [CrossRef]
- Goldberg, I.P.; Lichtbroun, B.; Singer, E.A.; Ghodoussipour, S. Pharmacologic Therapies for Non-Muscle Invasive Bladder Cancer: Current and Future Treatments. Arch. Pharmacol. Ther. 2022, 4, 13. [Google Scholar] [PubMed]
- Lv, M.; Qiu, F.; Li, T.; Sun, Y.; Zhang, C.; Zhu, P.; Qi, X.; Wan, J.; Yang, K.; Zhang, K. Construction, Expression, and Characterization of a Recombinant Immunotoxin Targeting EpCAM. Mediat. Inflamm. 2015, 2015, 460264. [Google Scholar] [CrossRef] [PubMed]
- Tretter, J.Y.; Schorpp, K.; Luxenburger, E.; Trambauer, J.; Steiner, H.; Hadian, K.; Gires, O.; Niessing, D. A High-Content Screen for Small-Molecule Regulators of Epithelial Cell-Adhesion Molecule (EpCAM) Cleavage Yields a Robust Inhibitor. J. Biol. Chem. 2018, 293, 8994–9005. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, R.; Kashyap, R.; Barral, K.; Egea-Jimenez, A.L.; Kovalskyy, D.; Feracci, M.; Garcia, M.; Derviaux, C.; Betzi, S.; Ghossoub, R.; et al. Pharmacological Inhibition of Syntenin PDZ2 Domain Impairs Breast Cancer Cell Activities and Exosome Loading with Syndecan and EpCAM Cargo. J. Extracell. Vesicles 2020, 10, e12039. [Google Scholar] [CrossRef] [PubMed]
- Moldenhauer, G.; Salnikov, A.V.; Lüttgau, S.; Herr, I.; Anderl, J.; Faulstich, H. Therapeutic Potential of Amanitin-Conjugated Anti-Epithelial Cell Adhesion Molecule Monoclonal Antibody Against Pancreatic Carcinoma. JNCI J. Natl. Cancer Inst. 2012, 104, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Lakhin, A.V.; Tarantul, V.Z.; Gening, L.V. Aptamers: Problems, Solutions and Prospects. Acta Naturae 2013, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Zheng, C.; Zhou, S.F.; Qiao, S.; Tran, P.H.L.; Pu, C.; Li, Y.; Kong, L.; Kouzani, A.Z.; Lin, J.; et al. Superior Performance of Aptamer in Tumor Penetration over Antibody: Implication of Aptamer-Based Theranostics in Solid Tumors. Theranostics 2015, 5, 1083. [Google Scholar] [CrossRef]
- Subramanian, N.; Kanwar, J.R.; Athalya, P.K.; Janakiraman, N.; Khetan, V.; Kanwar, R.K.; Eluchuri, S.; Krishnakumar, S. EpCAM Aptamer Mediated Cancer Cell Specific Delivery of EpCAM SiRNA Using Polymeric Nanocomplex. J. Biomed. Sci. 2015, 22, 4. [Google Scholar] [CrossRef]
- Mitra, M.; Kandalam, M.; Rangasamy, J.; Shankar, B.; Maheswari, U.K.; Swaminathan, S.; Krishnakumar, S. Novel Epithelial Cell Adhesion Molecule Antibody Conjugated Polyethyleneimine-Capped Gold Nanoparticles for Enhanced and Targeted Small Interfering RNA Delivery to Retinoblastoma Cells. Mol. Vis. 2013, 19, 1029. [Google Scholar] [PubMed]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From Adhesion Molecules to Signaling Regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef]
- Yu, Q.; Stamenkovic, I. Localization of Matrix Metalloproteinase 9 to the Cell Surface Provides a Mechanism for CD44-Mediated Tumor Invasion. Genes Dev. 1999, 13, 35. [Google Scholar] [CrossRef]
- Zöller, M. CD44: Can a Cancer-Initiating Cell Profit from an Abundantly Expressed Molecule? Nat. Rev. Cancer 2011, 11, 254–267. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells—Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 539291. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Hope, K.J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J.E. Targeting of CD44 Eradicates Human Acute Myeloid Leukemic Stem Cells. Nat. Med. 2006, 12, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Chen, C.; Chang, K.; Karnad, A.; Jagirdar, J.; Kumar, A.P.; Freeman, J.W. CD44 Expression Level and Isoform Contributes to Pancreatic Cancer Cell Plasticity, Invasiveness, and Response to Therapy. Clin. Cancer Res. 2016, 22, 5592–5604. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.W.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.K.; Shimada, Y.; Wang, T.C. Identification of Gastric Cancer Stem Cells Using the Cell Surface Marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef]
- Riechelmann, H.; Sauter, A.; Golze, W.; Hanft, G.; Schroen, C.; Hoermann, K.; Erhardt, T.; Gronau, S. Phase I Trial with the CD44v6-Targeting Immunoconjugate Bivatuzumab Mertansine in Head and Neck Squamous Cell Carcinoma. Oral. Oncol. 2008, 44, 823–829. [Google Scholar] [CrossRef]
- Li, L.; Hao, X.; Qin, J.; Tang, W.; He, F.; Smith, A.; Zhang, M.; Simeone, D.M.; Qiao, X.T.; Chen, Z.N.; et al. Antibody Against CD44s Inhibits Pancreatic Tumor Initiation and Postradiation Recurrence in Mice. Gastroenterology 2014, 146, 1108–1118.e12. [Google Scholar] [CrossRef]
- Uchino, M.; Kojima, H.; Wada, K.; Imada, M.; Onoda, F.; Satofuka, H.; Utsugi, T.; Murakami, Y. Nuclear β-Catenin and CD44 Upregulation Characterize Invasive Cell Populations in Non-Aggressive MCF-7 Breast Cancer Cells. BMC Cancer 2010, 10, 414. [Google Scholar] [CrossRef]
- Hellqvist, E.; Holm, F.; Mason, C.N.; Runza, V.; Weigand, S.; Sadarangani, A.; Jamieson, C.H.M. CD44 Monoclonal Antibody-Enhanced Clearance of Chronic Myeloid Leukemia Stem Cells from The Malignant Niche. Blood 2013, 122, 858. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, C.C.N.; Fecteau, J.F.; Cui, B.; Chen, L.; Zhang, L.; Wu, R.; Rassenti, L.; Lao, F.; Weigand, S.; et al. Targeting Chronic Lymphocytic Leukemia Cells with a Humanized Monoclonal Antibody Specific for CD44. Proc. Natl. Acad. Sci. USA 2013, 110, 6127–6132. [Google Scholar] [CrossRef]
- Perez, A.; Neskey, D.M.; Wen, J.; Goodwin, J.W.; Slingerland, J.; Pereira, L.; Weigand, S.; Franzmann, E.J. Abstract 2521: Targeting CD44 in Head and Neck Squamous Cell Carcinoma (HNSCC) with a New Humanized Antibody RO5429083. Cancer Res. 2012, 72 (Suppl. S8), 2521. [Google Scholar] [CrossRef]
- Menke-van der Houven van Oordt, C.W.; Gomez-Roca, C.; van Herpen, C.; Coveler, A.L.; Mahalingam, D.; Verheul, H.M.W.; van der Graaf, W.T.A.; Christen, R.; Rüttinger, D.; Weigand, S.; et al. First-in-Human Phase I Clinical Trial of RG7356, an Anti-CD44 Humanized Antibody, in Patients with Advanced, CD44-Expressing Solid Tumors. Oncotarget 2016, 7, 80046. [Google Scholar] [CrossRef] [PubMed]
- Vey, N.; Delaunay, J.; Martinelli, G.; Fiedler, W.; Raffoux, E.; Prebet, T.; Gomez-Roca, C.; Papayannidis, C.; Kebenko, M.; Paschka, P.; et al. Phase I Clinical Study of RG7356, an Anti-CD44 Humanized Antibody, in Patients with Acute Myeloid Leukemia. Oncotarget 2016, 7, 32532–32542. [Google Scholar] [CrossRef]
- Mitsunaga, M.; Ogawa, M.; Kosaka, N.; Rosenblum, L.T.; Choyke, P.L.; Kobayashi, H. Cancer-Cell Selective in Vivo Near Infrared Photoimmunotherapy Targeting Specific Membrane Molecules. Nat. Med. 2011, 17, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Nagaya, T.; Nakamura, Y.; Okuyama, S.; Ogata, F.; Maruoka, Y.; Choyke, P.L.; Allen, C.; Kobayashi, H. Syngeneic Mouse Models of Oral Cancer Are Effectively Targetedbyanti-Cd44-BasedNIR-PIT. Mol. Cancer Res. 2017, 15, 1667–1677. [Google Scholar] [CrossRef]
- Nagaya, T.; Friedman, J.; Maruoka, Y.; Ogata, F.; Okuyama, S.; Clavijo, P.E.; Choyke, P.L.; Allen, C.; Kobayashi, H. Host Immunity Following Near-Infrared Photoimmunotherapy Is Enhanced with PD-1 Checkpoint Blockade to Eradicate Established Antigenic Tumors. Cancer Immunol. Res. 2019, 7, 401–413. [Google Scholar] [CrossRef]
- Maruoka, Y.; Furusawa, A.; Okada, R.; Inagaki, F.; Fujimura, D.; Wakiyama, H.; Kato, T.; Nagaya, T.; Choyke, P.L.; Kobayashi, H. Combined CD44- And CD25-Targeted near-Infrared Photoimmunotherapy Selectively Kills Cancer and Regulatory T Cells in Syngeneic Mouse Cancer Models. Cancer Immunol. Res. 2020, 8, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Waller, K.A.; Chin, K.E.; Jay, G.D.; Zhang, L.X.; Teeple, E.; McAllister, S.; Badger, G.J.; Schmidt, T.A.; Fleming, B.C. Intra-Articular Recombinant Human Proteoglycan 4 Mitigates Cartilage Damage after Destabilization of the Medial Meniscus in the Yucatan Minipig. Am. J. Sports Med. 2017, 45, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Chanda, A.; Regmi, S.C.; Karve, K.; Deng, L.; Jay, G.D.; Jirik, F.R.; Schmidt, T.A.; Bonni, S. Recombinant Human PRG4 (RhPRG4) Suppresses Breast Cancer Cell Invasion by Inhibiting TGFβ-Hyaluronan-CD44 Signaling Pathway. PLoS ONE 2019, 14, e0219697. [Google Scholar] [CrossRef] [PubMed]
- Transforming Growth Factor-β1 Induces EMT by the Transactivation of Epidermal Growth Factor Signaling through HA/CD44 in Lung and Breast Cancer Cells. Available online: https://www.spandidos-publications.com/10.3892/ijmm.2015.2222 (accessed on 18 December 2023).
- Rosenberg, J.E.; Bambury, R.M.; Van Allen, E.M.; Drabkin, H.A.; Lara, P.N.; Harzstark, A.L.; Wagle, N.; Figlin, R.A.; Smith, G.W.; Garraway, L.A.; et al. A Phase II Trial of AS1411 (a Novel Nucleolin-Targeted DNA Aptamer) in Metastatic Renal Cell Carcinoma. Investig. New Drugs 2014, 32, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Iida, J.; Clancy, R.; Dorchak, J.; Somiari, R.I.; Somiari, S.; Cutler, M.L.; Mural, R.J.; Shriver, C.D. DNA Aptamers against Exon V10 of CD44 Inhibit Breast Cancer Cell Migration. PLoS ONE 2014, 9, e88712. [Google Scholar] [CrossRef]
- Zheng, J.; Zhao, S.; Yu, X.; Huang, S.; Liu, H.Y. Simultaneous Targeting of CD44 and EpCAM with a Bispecific Aptamer Effectively Inhibits Intraperitoneal Ovarian Cancer Growth. Theranostics 2017, 7, 1373. [Google Scholar] [CrossRef] [PubMed]
- Di Fusco, D.; Dinallo, V.; Marafini, I.; Figliuzzi, M.M.; Romano, B.; Monteleone, G. Antisense Oligonucleotide: Basic Concepts and Therapeutic Application in Inflammatory Bowel Disease. Front. Pharmacol. 2019, 10, 440751. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Choong, P.F.; Poon, L.F.; Zhou, J.; Khng, J.; Jasinghe, V.J.; Palaniyandi, S.; Chen, C.S. Inhibition of CD44 Expression in Hepatocellular Carcinoma Cells Enhances Apoptosis, Chemosensitivity, and Reduces Tumorigenesis and Invasion. Cancer Chemother. Pharmacol. 2008, 62, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Bartolucci, D.; Pession, A.; Hrelia, P.; Tonelli, R. Precision Anti-Cancer Medicines by Oligonucleotide Therapeutics in Clinical Research Targeting Undruggable Proteins and Non-Coding RNAs. Pharmaceutics 2022, 14, 1453. [Google Scholar] [CrossRef]
- Liu, L.K.; Finzel, B.C. Fragment-Based Identification of an Inducible Binding Site on Cell Surface Receptor CD44 for the Design of Protein-Carbohydrate Interaction Inhibitors. J. Med. Chem. 2014, 57, 2714–2725. [Google Scholar] [CrossRef]
- Roy, S.; Kar, M.; Roy, S.; Padhi, S.; Kumar, A.; Thakur, S.; Akhter, Y.; Gatto, G.; Banerjee, B. Inhibition of CD44 Sensitizes Cisplatin-Resistance and Affects Wnt/β-Catenin Signaling in HNSCC Cells. Int. J. Biol. Macromol. 2020, 149, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Espejo-Román, J.M.; Rubio-Ruiz, B.; Cano-Cortés, V.; Cruz-López, O.; Gonzalez-Resines, S.; Domene, C.; Conejo-García, A.; Sánchez-Martín, R.M. Selective Anticancer Therapy Based on a HA-CD44 Interaction Inhibitor Loaded on Polymeric Nanoparticles. Pharmaceutics 2022, 14, 788. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Z.; Chen, C.; Fu, X.; Wang, J.; Fei, X.; Yan, X.; Xu, R. A Low MW Inhibitor of CD44 Dimerization for the Treatment of Glioblastoma. Br. J. Pharmacol. 2020, 177, 3009. [Google Scholar] [CrossRef]
- Finlayson, M. Modulation of CD44 Activity by A6-Peptide. Front. Immunol. 2015, 6, 132943. [Google Scholar] [CrossRef] [PubMed]
- Piotrowicz, R.S.; Damaj, B.B.; Hachicha, M.; Incardona, F.; Howell, S.B.; Finlayson, M. A6 Peptide Activates CD44 Adhesive Activity, Induces FAK and MEK Phosphorylation, and Inhibits the Migration and Metastasis of CD44-Expressing Cells. Mol. Cancer Ther. 2011, 10, 2072–2082. [Google Scholar] [CrossRef] [PubMed]
- Boyd, D.D.; Kim, S.J.; Wang, H.; Jones, T.R.; Gallick, G.E. A Urokinase-Derived Peptide (Å6) Increases Survival of Mice Bearing Orthotopically Grown Prostate Cancer and Reduces Lymph Node Metastasis. Am. J. Pathol. 2003, 162, 619–626. [Google Scholar] [CrossRef]
- Gu, W.; An, J.; Meng, H.; Yu, N.; Zhong, Y.; Meng, F.; Xu, Y.; Cornelissen, J.J.L.M.; Zhong, Z. CD44-Specific A6 Short Peptide Boosts Targetability and Anticancer Efficacy of Polymersomal Epirubicin to Orthotopic Human Multiple Myeloma. Adv. Mater. 2019, 31, 1904742. [Google Scholar] [CrossRef] [PubMed]
- Damaj, B.B.; Incardonna, F.; Piotrowicz, R.; Howell, S.B.; Finlayson, M. Abstract 5120: A6 Peptide Binds to CD44 and Inhibits Migration and Metastasis of CD44+ Cell Lines in in Vitro and in Vivo Studies. Cancer Res. 2010, 70 (Suppl. S8), 5120. [Google Scholar] [CrossRef]
- Gold, M.A.; Brady, W.E.; Lankes, H.A.; Rose, P.G.; Kelley, J.L.; De Geest, K.; Crispens, M.A.; Resnick, K.E.; Howell, S.B. A Phase II Study of a Urokinase-Derived Peptide (A6) in the Treatment of Persistent or Recurrent Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Carcinoma: A Gynecologic Oncology Group Study. Gynecol. Oncol. 2012, 125, 635–639. [Google Scholar] [CrossRef]
- Ghamande, S.A.; Silverman, M.H.; Huh, W.; Behbakht, K.; Ball, G.; Cuasay, L.; Würtz, S.O.; Brunner, N.; Gold, M.A. A Phase 2, Randomized, Double-Blind, Placebo-Controlled Trial of Clinical Activity and Safety of Subcutaneous Å6 in Women with Asymptomatic CA125 Progression after First-Line Chemotherapy of Epithelial Ovarian Cancer. Gynecol. Oncol. 2008, 111, 89–94. [Google Scholar] [CrossRef]
- Yazdian-Robati, R.; Amiri, E.; Kamali, H.; Khosravi, A.; Taghdisi, S.M.; Jaafari, M.R.; Mashreghi, M.; Moosavian, S.A. CD44-Specific Short Peptide A6 Boosts Cellular Uptake and Anticancer Efficacy of PEGylated Liposomal Doxorubicin in Vitro and in Vivo. Cancer Nanotechnol. 2023, 14, 84. [Google Scholar] [CrossRef]
- Bharti, R.; Dey, G.; Lin, F.; Lathia, J.; Reizes, O. CD55 in Cancer: Complementing Functions in a Non-Canonical Manner. Cancer Lett. 2022, 551, 215935. [Google Scholar] [CrossRef]
- Geller, A.; Yan, J. The Role of Membrane Bound Complement Regulatory Proteins in Tumor Development and Cancer Immunotherapy. Front. Immunol. 2019, 10, 440775. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Lin, J.; He, J.; Gao, L.; Lin, S.; Tsang, L.L.; Zhang, H.; He, X.; Wang, G.; Yang, X.; et al. Human Embryonic Stem Cell-Derived Neural Crest Model Unveils CD55 as a Cancer Stem Cell Regulator for Therapeutic Targeting in MYCN-Amplified Neuroblastoma. Neuro Oncol. 2022, 24, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, J.I.; Morii, E.; Liu, Y.; Qiu, Y.; Nakamichi, N.; Jokoji, R.; Miyoshi, Y.; Noguchi, S.; Aozasa, K. Prognostic Significance of CD55 Expression in Breast Cancer. Clin. Cancer Res. 2008, 14, 4780–4786. [Google Scholar] [CrossRef] [PubMed]
- Saygin, C.; Wiechert, A.; Rao, V.S.; Alluri, R.; Connor, E.; Thiagarajan, P.S.; Hale, J.S.; Li, Y.; Chumakova, A.; Jarrar, A.; et al. CD55 Regulates Self-Renewal and Cisplatin Resistance in Endometrioid Tumors. J. Exp. Med. 2017, 214, 2715–2732. [Google Scholar] [CrossRef]
- Leung, T.H.Y.; Tang, H.W.M.; Siu, M.K.Y.; Chan, D.W.; Chan, K.K.L.; Cheung, A.N.Y.; Ngan, H.Y.S. Human Papillomavirus E6 Protein Enriches the CD55(+) Population in Cervical Cancer Cells, Promoting Radioresistance and Cancer Aggressiveness. J. Pathol. 2018, 244, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Dho, S.H.; Cho, E.H.; Lee, J.Y.; Lee, S.; Jung, S.H.; Kim, L.K.; Lim, J.C. A Novel Therapeutic Anti CD55 Monoclonal Antibody Inhibits the Proliferation and Metastasis of Colorectal Cancer Cells. Available online: https://www.spandidos-publications.com/10.3892/or.2019.7337 (accessed on 16 January 2024).
- Macor, P.; Tripodo, C.; Zorzet, S.; Piovan, E.; Bossi, F.; Marzari, R.; Amadori, A.; Tedesco, F. In Vivo Targeting of Human Neutralizing Antibodies against CD55 and CD59 to Lymphoma Cells Increases the Antitumor Activity of Rituximab. Cancer Res. 2007, 67, 10556–10563. [Google Scholar] [CrossRef]
- Dho, S.H.; Kim, S.Y.; Chung, C.; Cho, E.H.; Lee, S.Y.; Kim, J.Y.; Kim, L.K.; Min, S.W.; Lee, J.; Jung, S.H.; et al. Development of a Radionuclide-Labeled Monoclonal Anti-CD55 Antibody with Theranostic Potential in Pleural Metastatic Lung Cancer. Sci. Rep. 2018, 8, 8960. [Google Scholar] [CrossRef]
- Chaudhary, A.K.; Shi, J.; Cai, W.; Wang, B.; Zaman, M.S.; Huang, C.; Lin, J.; Kan, S.Z.; Zhou, J.; Dong, J.; et al. Abstract LB067: Development of a Novel Bispecific Antibody Targeting PD-L1 and CD55 for Cancer Therapy. Cancer Res. 2021, 81 (Suppl. S13), LB067. [Google Scholar] [CrossRef]
- Ullenhag, G.J.; Spendlove, I.; Watson, N.F.S.; Indar, A.A.; Dube, M.; Robins, R.A.; Maxwell-Armstrong, C.; Scholefield, J.H.; Durrant, L.G. A Neoadjuvant/Adjuvant Randomized Trial of Colorectal Cancer Patients Vaccinated with an Anti-Idiotypic Antibody, 105AD7, Mimicking CD55. Clin. Cancer Res. 2006, 12, 7389–7396. [Google Scholar] [CrossRef]
- Pritchard-Jones, K.; Spendlove, I.; Wilton, C.; Whelan, J.; Weeden, S.; Lewis, I.; Hale, J.; Douglas, C.; Pagonis, C.; Campbell, B.; et al. Immune Responses to the 105AD7 Human Anti-Idiotypic Vaccine after Intensive Chemotherapy, for Osteosarcoma. Br. J. Cancer 2005, 92, 1358. [Google Scholar] [CrossRef]
- Denton, G.W.L.; Durrant, L.G.; Hardcastle, J.D.; Austin, E.B.; Sewell, H.F.; Robins, R.A. Clinical Outcome of Colorectal Cancer Patients Treated with Human Monoclonal Anti-Idiotypic Antibody. Int. J. Cancer 1994, 57, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Maxwell-Armstrong, C.A.; Durrant, L.G.; Buckley, T.J.D.; Scholefield, J.H.; Robins, R.A.; Fielding, K.; Monson, J.R.T.; Guillou, P.; Calvert, H.; Carmichael, J.; et al. Randomized Double-Blind Phase II Survival Study Comparing Immunization with the Anti-Idiotypic Monoclonal Antibody 105AD7 against Placebo in Advanced Colorectal Cancer. Br. J. Cancer 2001, 84, 1443–1446. [Google Scholar] [CrossRef]
- Ten-Year Follow-Up of a Prospective Trial for the Targeted Therapy of Gastric Cancer with the Human Monoclonal Antibody PAT-SC1. Available online: https://www.spandidos-publications.com/or/31/3/1059 (accessed on 19 January 2024).
- Liu, G.; Yin, Q.; Ji, H.; Wang, Y.; Liu, H.; Jiang, L.; Zhu, F.; Li, B. A Study on Screening and Antitumor Effect of CD55-Specific Ligand Peptide in Cervical Cancer Cells. Drug Des. Devel Ther. 2018, 12, 3899–3912. [Google Scholar] [CrossRef]
- Liu, G.; Xu, X.; Jiang, L.; Ji, H.; Zhu, F.; Jin, B.; Han, J.; Dong, X.; Yang, F.; Li, B. Targeted Antitumor Mechanism of C-PC/CMC-CD55sp Nanospheres in HeLa Cervical Cancer Cells. Front. Pharmacol. 2020, 11, 906. [Google Scholar] [CrossRef] [PubMed]
- Holla, V.R.; Wang, D.; Brown, J.R.; Mann, J.R.; Katkuri, S.; Dubois, R.N. Prostaglandin E 2 Regulates the Complement Inhibitor CD55/Decay-Accelerating Factor in Colorectal Cancer. J. Biol. Chem. 2005, 280, 476–483. [Google Scholar] [CrossRef]
- Oyama, S.; Fujino, H.; Yamazaki, R.; Okura, I.; Regan, J.W.; Awata, A.; Arai, T.; Murayama, T. A Novel Indole Compound, AWT-489, Inhibits Prostaglandin D2-Induced CD55 Expression by Acting on DP Prostanoid Receptors as an Antagonist in LS174T Human Colon Cancer Cells. Arch. Biochem. Biophys. 2014, 541, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, S.; Cinci, M.; Hasmann, M.; Fehring, V.; Kirschfink, M. Lipoplex Mediated Silencing of Membrane Regulators (CD46, CD55 and CD59) Enhances Complement-Dependent Anti-Tumor Activity of Trastuzumab and Pertuzumab. Mol. Oncol. 2013, 7, 580–594. [Google Scholar] [CrossRef]
- Pawig, L.; Klasen, C.; Weber, C.; Bernhagen, J.; Noels, H. Diversity and Inter-Connections in the CXCR4 Chemokine Receptor/Ligand Family: Molecular Perspectives. Front. Immunol. 2015, 6, 156039. [Google Scholar] [CrossRef]
- Schiraldi, M.; Raucci, A.; Muñoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 Promotes Recruitment of Inflammatory Cells to Damaged Tissues by Forming a Complex with CXCL12 and Signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Behnam Azad, B.; Nimmagadda, S. The Intricate Role of CXCR4 in Cancer. Adv. Cancer Res. 2014, 124, 31. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, N.; Zhang, T.T.; Nakanishi, T. Involvement of CXCR4 in Normal and Abnormal Development. Cells 2019, 8, 185. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Le, K.; Xu, M.; Ming, J.; Yang, W.; Zhang, Q.; Lu, L.; Xi, Z.; Ruan, S.; Huang, T. CXCR4 Antagonist AMD3100 Reverses the Resistance to Tamoxifen in Breast Cancer via Inhibiting AKT Phosphorylation. Mol. Ther. Oncolytics 2020, 18, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A Symbiotic Bridge Linking Cancer Cells and Their Stromal Neighbors in Oncogenic Communication Networks. Oncogene 2016, 35, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Jiang, R.; Zhang, C.; Feng, Z.; Wang, X. The Regulatory Role of Cancer Stem Cell Marker Gene CXCR4 in the Growth and Metastasis of Gastric Cancer. npj Precis. Oncol. 2023, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Chiwaki, F.; Takahashi, R.U.; Aoyagi, K.; Yanagihara, K.; Nishimura, T.; Tamaoki, M.; Komatsu, M.; Komatsuzaki, R.; Matsusaki, K.; et al. Identification and Characterization of CXCR4-Positive Gastric Cancer Stem Cells. PLoS ONE 2015, 10, e0130808. [Google Scholar] [CrossRef]
- Dubrovska, A.; Elliott, J.; Salamone, R.J.; Telegeev, G.D.; Stakhovsky, A.E.; Schepotin, I.B.; Yan, F.; Wang, Y.; Bouchez, L.C.; Kularatne, S.A.; et al. CXCR4 Expression in Prostate Cancer Progenitor Cells. PLoS ONE 2012, 7, e31226. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.; Dalterio, C.; Camerlingo, R.; Tirino, V.; Consales, C.; Riccio, A.; Ieranò, C.; Cecere, S.C.; Losito, N.S.; Greggi, S.; et al. Identification of a Distinct Population of CD133+CXCR4+ Cancer Stem Cells in Ovarian Cancer. Sci. Rep. 2015, 5, 10357. [Google Scholar] [CrossRef]
- Dubrovska, A.; Hartung, A.; Bouchez, L.C.; Walker, J.R.; Reddy, V.A.; Cho, C.Y.; Schultz, P.G. CXCR4 Activation Maintains a Stem Cell Population in Tamoxifen-Resistant Breast Cancer Cells through AhR Signaling. Br. J. Cancer 2012, 107, 43–52. [Google Scholar] [CrossRef]
- Crees, Z.D.; Rettig, M.P.; Jayasinghe, R.G.; Stockerl-Goldstein, K.; Larson, S.M.; Arpad, I.; Milone, G.A.; Martino, M.; Stiff, P.; Sborov, D.; et al. Motixafortide and G-CSF to Mobilize Hematopoietic Stem Cells for Autologous Transplantation in Multiple Myeloma: A Randomized Phase 3 Trial. Nat. Med. 2023, 29, 869–879. [Google Scholar] [CrossRef]
- Kuhne, M.R.; Mulvey, T.; Belanger, B.; Chen, S.; Pan, C.; Chong, C.; Cao, F.; Niekro, W.; Kempe, T.; Henning, K.A.; et al. BMS-936564/MDX-1338: A Fully Human Anti-CXCR4 Antibody Induces Apoptosis in Vitro and Shows Antitumor Activity in Vivo in Hematologic Malignancies. Clin. Cancer Res. 2013, 19, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, M.K.; Kumar, D.; Jones, H.; Amaya-Chanaga, C.I.; Choi, M.Y.; Melo-Cardenas, J.; Ale-Ali, A.; Kuhne, M.R.; Sabbatini, P.; Cohen, L.J.; et al. Ulocuplumab (BMS-936564/MDX1338): A Fully Human Anti-CXCR4 Antibody Induces Cell Death in Chronic Lymphocytic Leukemia Mediated through a Reactive Oxygen Species-Dependent Pathway. Oncotarget 2016, 7, 2809–2822. [Google Scholar] [CrossRef]
- Azad, B.B.; Chatterjee, S.; Lesniak, W.G.; Lisok, A.; Pullambhatla, M.; Bhujwalla, Z.M.; Pomper, M.G.; Nimmagadda, S. A Fully Human CXCR4 Antibody Demonstrates Diagnostic Utility and Therapeutic Efficacy in Solid Tumor Xenografts. Oncotarget 2016, 7, 12344. [Google Scholar] [CrossRef][Green Version]
- Roccaro, A.M.; Mishima, Y.; Sacco, A.; Moschetta, M.; Tai, Y.T.; Shi, J.; Zhang, Y.; Reagan, M.R.; Huynh, D.; Kawano, Y.; et al. CXCR4 Regulates Extra-Medullary Myeloma through Epithelial-Mesenchymal-Transition-like Transcriptional Activation. Cell Rep. 2015, 12, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P.; Meid, K.; Hunter, Z.R.; Flynn, C.A.; Sarosiek, S.R.; Leventoff, C.R.; White, T.P.; Cao, Y.; Roccaro, A.M.; Sacco, A.; et al. Phase 1 Study of Ibrutinib and the CXCR4 Antagonist Ulocuplumab in CXCR4-Mutated Waldenström Macroglobulinemia. Blood 2021, 138, 1535–1539. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Liu, C.J.; Redd, R.A.; Perez, R.P.; Baz, R.; Zavidij, O.; Sklavenitis-Pistofidis, R.; Richardson, P.G.; Anderson, K.C.; Laubach, J.; et al. A Phase Ib/II Trial of the First-in-Class Anti-CXCR4 Antibody Ulocuplumab in Combination with Lenalidomide or Bortezomib Plus Dexamethasone in Relapsed Multiple Myeloma. Clin. Cancer Res. 2020, 26, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Gu, Y.; Pascual, B.; Yan, Z.; Hallin, M.; Zhang, C.; Fan, C.; Wang, W.; Lam, J.; Spilker, M.E.; et al. A Novel CXCR4 Antagonist IgG1 Antibody (PF-06747143) for the Treatment of Hematologic Malignancies. Blood Adv. 2017, 1, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Saavedra, E.; Tang, R.; Gu, Y.; Lappin, P.; Trajkovic, D.; Liu, S.H.; Smeal, T.; Fantin, V.; De Botton, S.; et al. Targeting Primary Acute Myeloid Leukemia with a New CXCR4 Antagonist IgG1 Antibody (PF-06747143). Sci. Rep. 2017, 7, 7305. [Google Scholar] [CrossRef]
- Kashyap, M.K.; Amaya-Chanaga, C.I.; Kumar, D.; Simmons, B.; Huser, N.; Gu, Y.; Hallin, M.; Lindquist, K.; Yafawi, R.; Choi, M.Y.; et al. Targeting the CXCR4 Pathway Using a Novel Anti-CXCR4 IgG1 Antibody (PF-06747143) in Chronic Lymphocytic Leukemia. J. Hematol. Oncol. 2017, 10, 112. [Google Scholar] [CrossRef]
- Gelmini, S.; Mangoni, M.; Castiglione, F.; Beltrami, C.; Pieralli, A.; Andersson, K.L.; Fambrini, M.; Taddei, G.L.; Serio, M.; Orlando, C. The CXCR4/CXCL12 Axis in Endometrial Cancer. Clin. Exp. Metastasis 2009, 26, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, P.; Arlt, M.J.E.; Campanile, C.; Husmann, K.; Gvozdenovic, A.; Apuzzo, T.; Thelen, M.; Born, W.; Fuchs, B. CXCR4 Antibody Treatment Suppresses Metastatic Spread to the Lung of Intratibial Human Osteosarcoma Xenografts in Mice. Clin. Exp. Metastasis 2014, 31, 339–349. [Google Scholar] [CrossRef] [PubMed]
- De Pauw, T.; De Mey, L.; Debacker, J.M.; Raes, G.; Van Ginderachter, J.A.; De Groof, T.W.M.; Devoogdt, N. Current Status and Future Expectations of Nanobodies in Oncology Trials. Expert Opin. Investig. Drugs 2023, 32, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Bin Peng, S.; Zhang, X.; Paul, D.; Kays, L.M.; Ye, M.; Vaillancourt, P.; Dowless, M.; Stancato, L.F.; Stewart, J.; Uhlik, M.T.; et al. Inhibition of CXCR4 by LY2624587, a Fully Humanized Anti-CXCR4 Antibody Induces Apoptosis of Hematologic Malignancies. PLoS ONE 2016, 11, e0150585. [Google Scholar] [CrossRef]
- Broussas, M.; Boute, N.; Akla, B.; Berger, S.; Beau-Larvor, C.; Champion, T.; Robert, A.; Beck, A.; Haeuw, J.F.; Goetsch, L.; et al. A New Anti-CXCR4 Antibody That Blocks the CXCR4/SDF-1 Axis and Mobilizes Effector Cells. Mol. Cancer Ther. 2016, 15, 1890–1899. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Wang, Y.; Steiner, P.; Mazzola, A.-M.; Wetzel, L.; Passino, M.; McDermott, B.; Huang, K.; Bedian, V.; Greenberg, N. Abstract 5462: MEDI3185, a Potent Anti-CXCR4 Antibody, Inhibits Tumor Cell Migration, Signaling and Tumor Growth in Preclinical Models. Cancer Res. 2013, 73 (Suppl. S8), 5462. [Google Scholar] [CrossRef]
- Fouquet, G.; Guidez, S.; Richez, V.; Stoppa, A.-M.; Le Tourneau, C.; Macro, M.; Gruchet, C.; Bobin, A.; Moya, N.; Systchenko, T.; et al. Phase I Dose-Escalation Study of F50067, a Humanized Anti-CXCR4 Monoclonal Antibody Alone and in Combination with Lenalidomide and Low-Dose Dexamethasone, in Relapsed or Refractory Multiple Myeloma. Oncotarget 2018, 9, 23890–23899. [Google Scholar] [CrossRef] [PubMed]
- Chaudary, N.; Pintilie, M.; Jelveh, S.; Lindsay, P.; Hill, R.P.; Milosevic, M. Plerixafor Improves Primary Tumor Response and Reduces Metastases in Cervical Cancer Treated with Radio-Chemotherapy. Clin. Cancer Res. 2017, 23, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Andritsos, L.A.; Byrd, J.C.; Cheverton, P.; Wu, J.; Sivina, M.; Kipps, T.J.; Burger, J.A. A Multicenter Phase 1 Study of Plerixafor and Rituximab in Patients with Chronic Lymphocytic Leukemia. Leuk. Lymphoma 2019, 60, 3461–3469. [Google Scholar] [CrossRef]
- Heckmann, D.; Maier, P.; Laufs, S.; Wenz, F.; Zeller, W.J.; Fruehauf, S.; Allgayer, H. CXCR4 Expression and Treatment with SDF-1α or Plerixafor Modulate Proliferation and Chemosensitivity of Colon Cancer Cells. Transl. Oncol. 2013, 6, 124. [Google Scholar] [CrossRef]
- Taromi, S.; Kayser, G.; Catusse, J.; von Elverfeldt, D.; Reichardt, W.; Braun, F.; Weber, W.A.; Zeiser, R.; Burger, M. CXCR4 Antagonists Suppress Small Cell Lung Cancer Progression. Oncotarget 2016, 7, 85185. [Google Scholar] [CrossRef] [PubMed]
- Lefort, S.; Thuleau, A.; Kieffer, Y.; Sirven, P.; Bieche, I.; Marangoni, E.; Vincent-Salomon, A.; Mechta-Grigoriou, F. CXCR4 Inhibitors Could Benefit to HER2 but Not to Triple-Negative Breast Cancer Patients. Oncogene 2017, 36, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Saur, D.; Seidler, B.; Schneider, G.; Algül, H.; Beck, R.; Senekowitsch-Schmidtke, R.; Schwaiger, M.; Schmid, R.M. CXCR4 Expression Increases Liver and Lung Metastasis in a Mouse Model of Pancreatic Cancer. Gastroenterology 2005, 129, 1237–1250. [Google Scholar] [CrossRef]
- Rubin, J.B.; Kung, A.L.; Klein, R.S.; Chan, J.A.; Sun, Y.P.; Schmidt, K.; Kieran, M.W.; Luster, A.D.; Segal, R.A. A Small-Molecule Antagonist of CXCR4 Inhibits Intracranial Growth of Primary Brain Tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 13513–13518. [Google Scholar] [CrossRef] [PubMed]
- Domanska, U.M.; Timmer-Bosscha, H.; Nagengast, W.B.; Oude Munnink, T.H.; Kruizinga, R.C.; Ananias, H.J.K.; Kliphuis, N.M.; Huls, G.; De Vries, E.G.E.; De Jong, I.J.; et al. CXCR4 Inhibition with AMD3100 Sensitizes Prostate Cancer to Docetaxel Chemotherapy. Neoplasia 2012, 14, 709. [Google Scholar] [CrossRef]
- Chen, I.X.; Chauhan, V.P.; Posada, J.; Ng, M.R.; Wu, M.W.; Adstamongkonkul, P.; Huang, P.; Lindeman, N.; Langer, R.; Jain, R.K. Blocking CXCR4 Alleviates Desmoplasia, Increases T-Lymphocyte Infiltration, and Improves Immunotherapy in Metastatic Breast Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 4558–4566. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Atkins, M.B.; Rose, T.L.; Alter, R.S.; Ju, Y.; Niland, K.; Wang, Y.; Arbeit, R.; Parasuraman, S.; Gan, L.; et al. A Phase 1b Trial of the CXCR4 Inhibitor Mavorixafor and Nivolumab in Advanced Renal Cell Carcinoma Patients with No Prior Response to Nivolumab Monotherapy. Investig. New Drugs 2021, 39, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Wang, Y.; Pierce, R.H.; Campbell, J.S.; Yushak, M.; Milhem, M.; Ross, M.; Niland, K.; Arbeit, R.D.; Parasuraman, S.; et al. Mavorixafor, an Orally Bioavailable CXCR4 Antagonist, Increases Immune Cell Infiltration and Inflammatory Status of Tumor Microenvironment in Patients with Melanoma. Cancer Res. Commun. 2022, 2, 904–913. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Marampon, F.; Colapietro, A.; Delle Monache, S.; Sferra, R.; Vitale, F.; Richardson, P.J.; Patient, L.; Burbidge, S.; et al. The Brain-Penetrating CXCR4 Antagonist, PRX177561, Increases the Antitumor Effects of Bevacizumab and Sunitinib in Preclinical Models of Human Glioblastoma. J. Hematol. Oncol. 2017, 10, 5. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Colapietro, A.; Vitale, F.; Vetuschi, A.; Pompili, S.; Rossi, G.; Marampon, F.; Richardson, P.J.; Patient, L.; et al. The Novel CXCR4 Antagonist, PRX177561, Reduces Tumor Cell Proliferation and Accelerates Cancer Stem Cell Differentiation in Glioblastoma Preclinical Models. Tumor Biol. 2017, 39, 1010428317695528. [Google Scholar] [CrossRef]
- Liang, Z.; Zhan, W.; Zhu, A.; Yoon, Y.; Lin, S.; Sasaki, M.; Klapproth, J.M.A.; Yang, H.; Grossniklaus, H.E.; Xu, J.; et al. Development of a Unique Small Molecule Modulator of CXCR4. PLoS ONE 2012, 7, e34038. [Google Scholar] [CrossRef] [PubMed]
- Rebolledo-Bustillo, M.; Garcia-Gomez, D.; Dávila, E.M.; Castro, M.E.; Caballero, N.A.; Melendez, F.J.; Baizabal-Aguirre, V.M.; Sanchez-Gaytan, B.L.; Perez-Aguilar, J.M. Structural Basis of the Binding Mode of the Antineoplastic Compound Motixafortide (BL-8040) in the CXCR4 Chemokine Receptor. Int. J. Mol. Sci. 2023, 24, 4393. [Google Scholar] [CrossRef] [PubMed]
- Borthakur, G.; Ofran, Y.; Tallman, M.S.; Foran, J.; Uy, G.L.; DiPersio, J.F.; Showel, M.M.; Shimoni, A.; Nagler, A.; Rowe, J.M.; et al. BL-8040 CXCR4 Antagonist Is Safe and Demonstrates Antileukemic Activity in Combination with Cytarabine for the Treatment of Relapsed/Refractory Acute Myelogenous Leukemia: An Open-Label Safety and Efficacy Phase 2a Study. Cancer 2021, 127, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.; Klein, S.; Bulvik, B.; Wald, H.; Weiss, I.D.; Olam, D.; Weiss, L.; Beider, K.; Eizenberg, O.; Wald, O.; et al. The CXCR4 Inhibitor BL-8040 Induces the Apoptosis of AML Blasts by Downregulating ERK, BCL-2, MCL-1 and Cyclin-D1 via Altered MiR-15a/16-1 Expression. Leukemia 2017, 31, 2336–2346. [Google Scholar] [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 Antagonist, in Combination with Pembrolizumab and Chemotherapy for Pancreatic Cancer: The COMBAT Trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Bockorny, B.; Macarulla, T.; Semenisty, V.; Borazanci, E.; Feliu, J.; Ponz-Sarvise, M.; Abad, D.G.; Oberstein, P.; Alistar, A.; Muñoz, A.; et al. Motixafortide and Pembrolizumab Combined to Nanoliposomal Irinotecan, Fluorouracil, and Folinic Acid in Metastatic Pancreatic Cancer: The COMBAT/KEYNOTE-202 Trial. Clin. Cancer Res. 2021, 27, 5020–5027. [Google Scholar] [CrossRef] [PubMed]
- Galsky, M.D.; Vogelzang, N.J.; Conkling, P.; Polzer, J.; Roberson, S.; Stille, J.R.; Saleh, M.; Thornton, D. A Phase I Trial of LY2510924, a CXCR4 Peptide Antagonist, in Patients with Advanced Cancer. Clin. Cancer Res. 2014, 20, 3581–3588. [Google Scholar] [CrossRef] [PubMed]
- Boddu, P.; Borthakur, G.; Koneru, M.; Huang, X.; Naqvi, K.; Wierda, W.; Bose, P.; Jabbour, E.; Estrov, Z.; Burger, J.; et al. Initial Report of a Phase I Study of LY2510924, Idarubicin, and Cytarabine in Relapsed/Refractory Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 369. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, M.H.; Messersmith, W.; Kindler, H.; Zhang, W.; Pitou, C.; Szpurka, A.M.; Wang, D.; Peng, S.-B.; Vangerow, B.; Khan, A.A.; et al. Safety and Pharmacokinetics of CXCR4 Peptide Antagonist, LY2510924, in Combination with Durvalumab in Advanced Refractory Solid Tumors. J. Pancreat. Cancer 2020, 6, 21. [Google Scholar] [CrossRef]
- Huang, E.H.; Singh, B.; Cristofanilli, M.; Gelovani, J.; Wei, C.; Vincent, L.; Cook, K.R.; Lucci, A. A CXCR4 Antagonist CTCE-9908 Inhibits Primary Tumor Growth and Metastasis of Breast Cancer. J. Surg. Res. 2009, 155, 231–236. [Google Scholar] [CrossRef]
- Phase I/II Study of CTCE-9908, a Novel Anticancer Agent that Inhibits CXCR4, in Patients with Advanced Solid Cancers|Molecular Cancer Therapeutics|American Association for Cancer Research. Available online: https://aacrjournals.org/mct/article/6/11_Supplement/A153/240072/Phase-I-II-study-of-CTCE-9908-a-novel-anticancer (accessed on 26 February 2024).
- Hamshaw, I.; Cominetti, M.M.D.; Lai, W.Y.; Searcey, M.; Mueller, A. The Development of Potent, Competitive CXCR4 Antagonists for the Prevention of Cancer Metastasis. Biochem. Pharmacol. 2023, 218, 115921. [Google Scholar] [CrossRef] [PubMed]
- Barzegar Behrooz, A.; Syahir, A.; Ahmad, S. CD133: Beyond a Cancer Stem Cell Biomarker. J. Drug Target. 2019, 27, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Glumac, P.M.; LeBeau, A.M. The Role of CD133 in Cancer: A Concise Review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Bauer, N.; Fonseca, A.V.; Florek, M.; Freund, D.; Jászai, J.; Bornhäuser, M.; Fargeas, C.A.; Corbeil, D. New Insights into the Cell Biology of Hematopoietic Progenitors by Studying Prominin-1 (CD133). Cells Tissues Organs 2008, 188, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Zacchigna, S.; Oh, H.; Wilsch-Bräuninger, M.; Missol-Kolka, E.; Jászai, J.; Jansen, S.; Tanimoto, N.; Tonagel, F.; Seeliger, M.; Huttner, W.B.; et al. Loss of the Cholesterol-Binding Protein Prominin-1/CD133 Causes Disk Dysmorphogenesis and Photoreceptor Degeneration. J. Neurosci. 2009, 29, 2297–2308. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous Stem Cells Can Arise from Pediatric Brain Tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef] [PubMed]
- Identification of a Cancer Stem Cell in Human Brain Tumors|Cancer Research|American Association for Cancer Research. Available online: https://aacrjournals.org/cancerres/article/63/18/5821/510370/Identification-of-a-Cancer-Stem-Cell-in-Human (accessed on 22 February 2024).
- Yuan, X.; Curtin, J.; Xiong, Y.; Liu, G.; Waschsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Yu, J.S. Isolation of Cancer Stem Cells from Adult Glioblastoma Multiforme. Oncogene 2004, 23, 9392–9400. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of Human Brain Tumor Initiating Cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Vander Griend, D.J.; Karthaus, W.L.; Dalrymple, S.; Meeker, A.; DeMarzo, A.M.; Isaacs, J.T. The Role of CD133 in Normal Human Prostate Stem Cells and Malignant Cancer-Initiating Cells. Cancer Res. 2008, 68, 9703–9711. [Google Scholar] [CrossRef]
- Ma, S.; Tang, K.H.; Chan, Y.P.; Lee, T.K.; Kwan, P.S.; Castilho, A.; Ng, I.; Man, K.; Wong, N.; To, K.F.; et al. MiR-130b Promotes CD133(+) Liver Tumor-Initiating Cell Growth and Self-Renewal via Tumor Protein 53-Induced Nuclear Protein 1. Cell Stem Cell 2010, 7, 694–707. [Google Scholar] [CrossRef]
- Fang, D.D.; Kim, Y.J.; Lee, C.N.; Aggarwal, S.; McKinnon, K.; Mesmer, D.; Norton, J.; Birse, C.E.; He, T.; Ruben, S.M.; et al. Expansion of CD133+ Colon Cancer Cultures Retaining Stem Cell Properties to Enable Cancer Stem Cell Target Discovery. Br. J. Cancer 2010, 102, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and Expansion of the Tumorigenic Lung Cancer Stem Cell Population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shi, S.; Yen, Y.; Brown, J.; Ta, J.Q.; Le, A.D. A Subpopulation of CD133+ Cancer Stem-like Cells Characterized in Human Oral Squamous Cell Carcinoma Confer Resistance to Chemotherapy. Cancer Lett. 2010, 289, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Joseph, C.; Arshad, M.; Kurozomi, S.; Althobiti, M.; Miligy, I.M.; Al-izzi, S.; Toss, M.S.; Goh, F.Q.; Johnston, S.J.; Martin, S.G.; et al. Overexpression of the Cancer Stem Cell Marker CD133 Confers a Poor Prognosis in Invasive Breast Cancer. Breast Cancer Res. Treat. 2019, 174, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Song, X.; Chen, Z.; Li, X.; Li, M.; Liu, H.; Li, J. CD133 Expression and the Prognosis of Colorectal Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2013, 8, e56380. [Google Scholar] [CrossRef] [PubMed]
- da Costa, W.H.; Rocha, R.M.; da Cunha, I.W.; da Fonseca, F.P.; Guimaraes, G.C.; de Cassio Zequi, S. CD133 Immunohistochemical Expression Predicts Progression and Cancer-Related Death in Renal Cell Carcinoma. World J. Urol. 2012, 30, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Fleming, J.B.; Wang, H.; Abbruzzese, J.L.; Choi, W.; Kopetz, S.; McConkey, D.J.; Evans, D.B.; Gallick, G.E. ALDH Activity Selectively Defines an Enhanced Tumor-Initiating Cell Population Relative to CD133 Expression in Human Pancreatic Adenocarcinoma. PLoS ONE 2011, 6, e20636. [Google Scholar] [CrossRef] [PubMed]
- Mohd-Zahid, M.H.; Zulkifli, S.N.; Che Abdullah, C.A.; Lim, J.; Fakurazi, S.; Wong, K.K.; Zakaria, A.D.; Ismail, N.; Uskoković, V.; Mohamud, R.; et al. Gold Nanoparticles Conjugated with Anti-CD133 Monoclonal Antibody and 5-Fluorouracil Chemotherapeutic Agent as Nanocarriers for Cancer Cell Targeting. RSC Adv. 2021, 11, 16131. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.K.; Roger, E.; Toti, U.; Niu, L.; Ohlfest, J.R.; Panyam, J. CD133-Targeted Paclitaxel Delivery Inhibits Local Tumor Recurrence in a Mouse Model of Breast Cancer. J. Control. Release 2013, 171, 280–287. [Google Scholar] [CrossRef]
- Ning, S.T.; Lee, S.Y.; Wei, M.F.; Peng, C.L.; Lin, S.Y.F.; Tsai, M.H.; Lee, P.C.; Shih, Y.H.; Lin, C.Y.; Luo, T.Y.; et al. Targeting Colorectal Cancer Stem-Like Cells with Anti-CD133 Antibody-Conjugated SN-38 Nanoparticles. ACS Appl. Mater. Interfaces 2016, 8, 17793–17804. [Google Scholar] [CrossRef]
- Huang, J.; Li, C.; Wang, Y.; Lv, H.; Guo, Y.; Dai, H.; Wicha, M.S.; Chang, A.E.; Li, Q. Cytokine-Induced Killer (CIK) Cells Bound with Anti-CD3/Anti-CD133 Bispecific Antibodies Target CD133high Cancer Stem Cells in Vitro and in Vivo. Clin. Immunol. 2013, 149, 156–168. [Google Scholar] [CrossRef]
- Riegg, F.; Lutz, M.S.; Schmied, B.J.; Heitmann, J.S.; Queudeville, M.; Lang, P.; Jung, G.; Salih, H.R.; Märklin, M.A. FC-Optimized CD133 Antibody for Induction of NK Cell Reactivity against B Cell Acute Lymphoblastic Leukemia. Cancers 2021, 13, 1632. [Google Scholar] [CrossRef]
- Schmied, B.J.; Riegg, F.; Zekri, L.; Grosse-Hovest, L.; Bühring, H.J.; Jung, G.; Salih, H.R. An Fc-Optimized CD133 Antibody for Induction of Natural Killer Cell Reactivity against Colorectal Cancer. Cancers 2019, 11, 789. [Google Scholar] [CrossRef]
- Klapdor, R.; Wang, S.; Hacker, U.; Büning, H.; Morgan, M.; Dörk, T.; Hillemanns, P.; Schambach, A. Improved Killing of Ovarian Cancer Stem Cells by Combining a Novel Chimeric Antigen Receptor–Based Immunotherapy and Chemotherapy. Hum. Gene Ther. 2017, 28, 886–896. [Google Scholar] [CrossRef]
- Taromi, S.; Firat, E.; Simonis, A.; Braun, L.M.; Apostolova, P.; Elze, M.; Passlick, B.; Schumacher, A.; Lagies, S.; Frey, A.; et al. Enhanced AC133-Specific CAR T Cell Therapy Induces Durable Remissions in Mice with Metastatic Small Cell Lung Cancer. Cancer Lett. 2022, 538, 215697. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-Directed CAR T Cells for Advanced Metastasis Malignancies: A Phase I Trial. Oncoimmunology 2018, 7, e1440169. [Google Scholar] [CrossRef]
- Dai, H.; Tong, C.; Shi, D.; Chen, M.; Guo, Y.; Chen, D.; Han, X.; Wang, H.; Wang, Y.; Shen, P. Efficacy and Biomarker Analysis of CD133-Directed CAR T Cells in Advanced Hepatocellular Carcinoma: A Single-Arm, Open-Label, Phase II Trial. Oncoimmunology 2020, 9, 1846926. [Google Scholar] [CrossRef]
- Schmohl, J.U.; Gleason, M.K.; Dougherty, P.R.; Miller, J.S.; Vallera, D.A. Heterodimeric Bispecific Single Chain Variable Fragments (ScFv) Killer Engagers (BiKEs) Enhance NK-Cell Activity Against CD133+ Colorectal Cancer Cells. Target. Oncol. 2016, 11, 353–361. [Google Scholar] [CrossRef]
- Schmohl, J.U.; Felices, M.; Oh, F.; Lenvik, A.J.; Lebeau, A.M.; Panyam, J.; Miller, J.S.; Vallera, D.A. Engineering of Anti-CD133 Trispecific Molecule Capable of Inducing NK Expansion and Driving Antibody-Dependent Cell-Mediated Cytotoxicity. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2017, 49, 1140–1152. [Google Scholar] [CrossRef]
- Schmohl, J.U.; Felices, M.; Todhunter, D.; Taras, E.; Miller, J.S.; Vallera, D.A. Tetraspecific ScFv Construct Provides NK Cell Mediated ADCC and Self-Sustaining Stimuli via Insertion of IL-15 as a Cross-Linker. Oncotarget 2016, 7, 73830. [Google Scholar] [CrossRef]
- Rudnick, J.D.; Fink, K.L.; Landolfi, J.C.; Markert, J.; Piccioni, D.E.; Glantz, M.J.; Swanson, S.J.; Gringeri, A.; Yu, J. Immunological Targeting of CD133 in Recurrent Glioblastoma: A Multi-Center Phase I Translational and Clinical Study of Autologous CD133 Dendritic Cell Immunotherapy. J. Clin. Oncol. 2017, 35 (Suppl. S15), 2059. [Google Scholar] [CrossRef]
- Hsin, I.L.; Chiu, L.Y.; Ou, C.C.; Wu, W.J.; Sheu, G.T.; Ko, J.L. CD133 Inhibition via Autophagic Degradation in Pemetrexed-Resistant Lung Cancer Cells by GMI, a Fungal Immunomodulatory Protein from Ganoderma Microsporum. Br. J. Cancer 2020, 123, 449–458. [Google Scholar] [CrossRef]
- Waldron, N.N.; Kaufman, D.S.; Oh, S.; Inde, Z.; Hexum, M.K.; Ohlfest, J.R.; Vallera, D.A. Targeting Tumor-Initiating Cancer Cells with DCD133KDEL Shows Impressive Tumor Reductions in a Xenotransplant Model of Human Head and Neck Cancer. Mol. Cancer Ther. 2011, 10, 1829–1838. [Google Scholar] [CrossRef]
- Ohlfest, J.R.; Zellmer, D.M.; Panyam, J.; Swaminathan, S.K.; Oh, S.; Waldron, N.N.; Toma, S.; Vallera, D.A. Immunotoxin Targeting CD133+ Breast Carcinoma Cells. Drug Deliv. Transl. Res. 2013, 3, 195–204. [Google Scholar] [CrossRef]
- Skubitz, A.P.N.; Taras, E.P.; Boylan, K.L.M.; Waldron, N.N.; Oh, S.; Panoskaltsis-Mortari, A.; Vallera, D.A. Targeting CD133 in an in Vivo Ovarian Cancer Model Reduces Ovarian Cancer Progression. Gynecol. Oncol. 2013, 130, 579–587. [Google Scholar] [CrossRef]
- Bostad, M.; Olsen, C.E.; Peng, Q.; Berg, K.; Høgset, A.; Selbo, P.K. Light-Controlled Endosomal Escape of the Novel CD133-Targeting Immunotoxin AC133–Saporin by Photochemical Internalization—A Minimally Invasive Cancer Stem Cell-Targeting Strategy. J. Control. Release 2015, 206, 37–48. [Google Scholar] [CrossRef]
- Deng, Y.; Su, Q.; Mo, J.; Fu, X.; Zhang, Y.; Lin, E.H. Celecoxib Downregulates CD133 Expression through Inhibition of the Wnt Signaling Pathway in Colon Cancer Cells. Cancer Investig. 2013, 31, 97–102. [Google Scholar] [CrossRef]
- Jung, K.H.; Lee, J.H.; Kim, M.; Lee, E.J.; Cho, Y.S.; Lee, K.H. Celecoxib-Induced Modulation of Colon Cancer CD133 Expression Occurs through AKT Inhibition and Is Monitored by 89Zr Immuno-PET. Mol. Imaging 2022, 2022, 4906934. [Google Scholar] [CrossRef]
- Tsunekuni, K.; Konno, M.; Haraguchi, N.; Koseki, J.; Asai, A.; Matsuoka, K.; Kobunai, T.; Takechi, T.; Doki, Y.; Mori, M.; et al. CD44/CD133-Positive Colorectal Cancer Stem Cells Are Sensitive to Trifluridine Exposure. Sci. Rep. 2019, 9, 14861. [Google Scholar] [CrossRef]
- Deng, H.; Liu, H.; Yang, G.; Wang, D.; Luo, Y.; Li, C.; Qi, Z.; Liu, Z.; Wang, P.; Jia, Y.; et al. ACT001 Inhibited CD133 Transcription by Targeting and Inducing Olig2 Ubiquitination Degradation. Oncogenesis 2023, 12, 19. [Google Scholar] [CrossRef]
- Xi, X.; Liu, N.; Wang, Q.; Chu, Y.; Yin, Z.; Ding, Y.; Lu, Y. ACT001, a Novel PAI-1 Inhibitor, Exerts Synergistic Effects in Combination with Cisplatin by Inhibiting PI3K/AKT Pathway in Glioma. Cell Death Dis. 2019, 10, 757. [Google Scholar] [CrossRef]
- Lickliter, J.D.; Jennens, R.; Lemech, C.R.; Kichenadasse, G.; Cai, D.; Su, S.Y.-C. Phase 1 Dose-Escalation Study of ACT001 in Patients with Recurrent Glioblastoma and Other Advanced Solid Tumors. J. Clin. Oncol. 2021, 39 (Suppl. S15), 2037. [Google Scholar] [CrossRef]
- Yin, W.; Pham, C.V.; Wang, T.; Al Shamaileh, H.; Chowdhury, R.; Patel, S.; Li, Y.; Kong, L.; Hou, Y.; Zhu, Y.; et al. Inhibition of Autophagy Promotes the Elimination of Liver Cancer Stem Cells by CD133 Aptamer-Targeted Delivery of Doxorubicin. Biomolecules 2022, 12, 1623. [Google Scholar] [CrossRef]
- Zhou, G.; Da Won Bae, S.; Nguyen, R.; Huo, X.; Han, S.; Zhang, Z.; Hebbard, L.; Duan, W.; Eslam, M.; Liddle, C.; et al. An Aptamer-Based Drug Delivery Agent (CD133-Apt-Dox) Selectively and Effectively Kills Liver Cancer Stem-like Cells. Cancer Lett. 2021, 501, 124–132. [Google Scholar] [CrossRef]
- Fang, H.; Xie, J.; Zhang, M.; Zhao, Z.; Wan, Y.; Yao, Y. MiRNA-21 Promotes Proliferation and Invasion of Triple-Negative Breast Cancer Cells through Targeting PTEN. Am. J. Transl. Res. 2017, 9, 953. [Google Scholar]
- Yin, H.; Xiong, G.; Guo, S.; Xu, C.; Xu, R.; Guo, P.; Shu, D. Delivery of Anti-MiRNA for Triple-Negative Breast Cancer Therapy Using RNA Nanoparticles Targeting Stem Cell Marker CD133. Mol. Ther. 2019, 27, 1252–1261. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, C.; Liu, G.; Liu, H.; Zhou, C.; Lu, Y.; Zhou, C.; Yuan, L.; Li, X. A Novel Mouse CD133 Binding-Peptide Screened by Phage Display Inhibits Cancer Cell Motility in Vitro. Clin. Exp. Metastasis 2012, 29, 185–196. [Google Scholar] [CrossRef]
- Li, W.; Cho, M.Y.; Lee, S.; Jang, M.; Park, J.; Park, R. CRISPR-Cas9 Mediated CD133 Knockout Inhibits Colon Cancer Invasion through Reduced Epithelial-Mesenchymal Transition. PLoS ONE 2019, 14, e0220860. [Google Scholar] [CrossRef]
- Asadzadeh, Z.; Mansoori, B.; Mohammadi, A.; Kazemi, T.; Mokhtarzadeh, A.; Shanehbandi, D.; Hemmat, N.; Derakhshani, A.; Brunetti, O.; Safaei, S.; et al. The Combination Effect of Prominin1 (CD133) Suppression and Oxaliplatin Treatment in Colorectal Cancer Therapy. Biomed. Pharmacother. 2021, 137, 111364. [Google Scholar] [CrossRef]
- Jing, H.; Weidensteiner, C.; Reichardt, W.; Gaedicke, S.; Zhu, X.; Grosu, A.L.; Kobayashi, H.; Niedermann, G. Imaging and Selective Elimination of Glioblastoma Stem Cells with Theranostic Near-Infrared-Labeled CD133-Specific Antibodies. Theranostics 2016, 6, 862. [Google Scholar] [CrossRef]
- Cavaleri, F.; Schöler, H.R. Nanog: A New Recruit to the Embryonic Stem Cell Orchestra. Cell 2003, 113, 551–552. [Google Scholar] [CrossRef]
- Jeter, C.R.; Liu, B.; Liu, X.; Chen, X.; Liu, C.; Calhoun-Davis, T.; Repass, J.; Zaehres, H.; Shen, J.J.; Tang, D.G. NANOG Promotes Cancer Stem Cell Characteristics and Prostate Cancer Resistance to Androgen Deprivation. Oncogene 2011, 30, 3833–3845. [Google Scholar] [CrossRef]
- Rodrigo, J.P.; Villaronga, M.Á.; Menéndez, S.T.; Hermida-Prado, F.; Quer, M.; Vilaseca, I.; Allonca, E.; Pedregal Mallo, D.; Astudillo, A.; García-Pedrero, J.M. A Novel Role for Nanog As An Early Cancer Risk Marker In Patients with Laryngeal Precancerous Lesions. Sci. Rep. 2017, 7, 11110. [Google Scholar] [CrossRef]
- Lu, X.; Mazur, S.J.; Lin, T.; Appella, E.; Xu, Y. The Pluripotency Factor Nanog Promotes Breast Cancer Tumorigenesis and Metastasis. Oncogene 2013, 33, 2655–2664. [Google Scholar] [CrossRef]
- Vasefifar, P.; Motafakkerazad, R.; Maleki, L.A.; Najafi, S.; Ghrobaninezhad, F.; Najafzadeh, B.; Alemohammad, H.; Amini, M.; Baghbanzadeh, A.; Baradaran, B. Nanog, as a Key Cancer Stem Cell Marker in Tumor Progression. Gene 2022, 827, 146448. [Google Scholar] [CrossRef]
- Begicevic, R.R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Liu, J.; Chen, S.; Fang, C.; Zhang, X.; Luo, Z. Prognostic Significance of NANOG Expression in Solid Tumors: A Meta-Analysis. Onco Targets Ther. 2018, 11, 5515–5526. [Google Scholar] [CrossRef]
- Jeter, C.R.; Badeaux, M.; Choy, G.; Chandra, D.; Patrawala, L.; Liu, C.; Calhoun-Davis, T.; Zaehres, H.; Daley, G.Q.; Tang, D.G. Functional Evidence That the Self-Renewal Gene NANOG Regulates Human Tumor Development. Stem Cells 2009, 27, 993–1005. [Google Scholar] [CrossRef]
- Ibrahim, E.E.; Babaei-Jadidi, R.; Saadeddin, A.; Spencer-Dene, B.; Hossaini, S.; Abuzinadah, M.; Li, N.; Fadhil, W.; Ilyas, M.; Bonnet, D.; et al. Embryonic NANOG Activity Defines Colorectal Cancer Stem Cells and Modulates through AP1- and TCF-Dependent Mechanisms. Stem Cells 2012, 30, 2076–2087. [Google Scholar] [CrossRef]
- Shan, J.; Shen, J.; Liu, L.; Xia, F.; Xu, C.; Duan, G.; Xu, Y.; Ma, Q.; Yang, Z.; Zhang, Q.; et al. Nanog Regulates Self-Renewal of Cancer Stem Cells through the Insulin-like Growth Factor Pathway in Human Hepatocellular Carcinoma. Hepatology 2012, 56, 1004–1014. [Google Scholar] [CrossRef]
- Qin, J.; Liu, X.; Laffin, B.; Chen, X.; Choy, G.; Jeter, C.R.; Calhoun-Davis, T.; Li, H.; Palapattu, G.S.; Pang, S.; et al. The PSA-/Lo Prostate Cancer Cell Population Harbors Self-Renewing Long-Term Tumor-Propagating Cells That Resist Castration. Cell Stem Cell 2012, 10, 556–569. [Google Scholar] [CrossRef]
- Liu, Y.; Clem, B.; Zuba-Surma, E.K.; El-Naggar, S.; Telang, S.; Jenson, A.B.; Wang, Y.; Shao, H.; Ratajczak, M.Z.; Chesney, J.; et al. Mouse Fibroblasts Lacking RB1 Function Form Spheres and Undergo Reprogramming to a Cancer Stem Cell Phenotype. Cell Stem Cell 2009, 4, 336–347. [Google Scholar] [CrossRef]
- Xiang, T.; Long, H.; He, L.; Han, X.; Lin, K.; Liang, Z.; Zhuo, W.; Xie, R.; Zhu, B. Interleukin-17 Produced by Tumor Microenvironment Promotes Self-Renewal of CD133+ Cancer Stem-like Cells in Ovarian Cancer. Oncogene 2013, 34, 165–176. [Google Scholar] [CrossRef]
- Kundu, J.; Banerjee, P.; Bose, C.; Das, U.; Ghosh, U.; Sinha, S. Internal Oligoguanidinium Transporter: Mercury-Free Scalable Synthesis, Improvement of Cellular Localization, Endosomal Escape, Mitochondrial Localization, and Conjugation with Antisense Morpholino for NANOG Inhibition to Induce Chemosensitization of Taxol in MCF-7 Cells. Bioconjug. Chem. 2020, 31, 2367–2382. [Google Scholar] [CrossRef]
- Summerton, J. Morpholino Antisense Oligomers: The Case for an RNase H-Independent Structural Type. Biochim. Biophys. Acta 1999, 1489, 141–158. [Google Scholar] [CrossRef]
- Godfrey, C.; Desviat, L.R.; Smedsrød, B.; Piétri-Rouxel, F.; Denti, M.A.; Disterer, P.; Lorain, S.; Nogales-Gadea, G.; Sardone, V.; Anwar, R.; et al. Delivery Is Key: Lessons Learnt from Developing Splice-switching Antisense Therapies. EMBO Mol. Med. 2017, 9, 545. [Google Scholar] [CrossRef]
- Khosravi, N.; Shahgoli, V.K.; Amini, M.; Safaei, S.; Mokhtarzadeh, A.; Mansoori, B.; Derakhshani, A.; Baghbanzadeh, A.; Baradaran, B. Suppression of Nanog Inhibited Cell Migration and Increased the Sensitivity of Colorectal Cancer Cells to 5-Fluorouracil. Eur. J. Pharmacol. 2021, 894, 173871. [Google Scholar] [CrossRef]
- Baltus, G.A.; Kowalski, M.P.; Tutter, A.V.; Kadam, S. A Positive Regulatory Role for the MSin3A-HDAC Complex in Pluripotency through Nanog and Sox2. J. Biol. Chem. 2009, 284, 6998–7006. [Google Scholar] [CrossRef]
- Marks, P.A. Discovery and Development of SAHA as an Anticancer Agent. Oncogene 2007, 26, 1351–1356. [Google Scholar] [CrossRef]
- Kumar, B.; Yadav, A.; Lang, J.C.; Teknos, T.N.; Kumar, P. Suberoylanilide Hydroxamic Acid (SAHA) Reverses Chemoresistance in Head and Neck Cancer Cells by Targeting Cancer Stem Cells via the Downregulation of Nanog. Genes Cancer 2015, 6, 169. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, M.; Zhu, Y.; Dai, X.; Dang, F.; Ren, J.; Ren, S.; Shulga, Y.V.; Beca, F.; Gan, W.; et al. SPOP Promotes Nanog Destruction to Suppress Stem Cell Traits and Prostate Cancer Progression. Dev. Cell 2019, 48, 329–344.e5. [Google Scholar] [CrossRef]
- Moretto-Zita, M.; Jin, H.; Shen, Z.; Zhao, T.; Briggs, S.P.; Xu, Y. Phosphorylation Stabilizes Nanog by Promoting Its Interaction with Pin1. Proc. Natl. Acad. Sci. USA 2010, 107, 13312–13317. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Takamiya, M.; Takahashi, M.; Miyashita, H.; Ikeda, H.; Terada, T.; Matsuo, Y.; Shirouzu, M.; Yokoyama, S.; Fujimori, F.; et al. Pin1 and Par14 Peptidyl Prolyl Isomerase Inhibitors Block Cell Proliferation. Chem. Biol. 2003, 10, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Rustighi, A.; Zannini, A.; Tiberi, L.; Sommaggio, R.; Piazza, S.; Sorrentino, G.; Nuzzo, S.; Tuscano, A.; Eterno, V.; Benvenuti, F.; et al. Prolyl-Isomerase Pin1 Controls Normal and Cancer Stem Cells of the Breast. EMBO Mol. Med. 2014, 6, 99–119. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Okada, M.; Shibuya, K.; Watanabe, E.; Seino, S.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; Kitanaka, C. Resveratrol Promotes Proteasome-Dependent Degradation of Nanog via P53 Activation and Induces Differentiation of Glioma Stem Cells. Stem Cell Res. 2013, 11, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Sethi, G.; Um, J.Y.; Shanmugam, M.K.; Arfuso, F.; Kumar, A.P.; Bishayee, A.; Ahn, K.S. The Role of Resveratrol in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2589. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, B.; Wang, J.; Li, J.; Gong, Y.; Li, S.; Wang, C.; Cui, B.; Xue, X.; Yang, M.; et al. Reduction of NANOG Mediates the Inhibitory Effect of Aspirin on Tumor Growth and Stemness in Colorectal Cancer. Cell Physiol. Biochem. 2017, 44, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Liu, W.; Tala, T.; Wang, H.; Li, F.; Zhang, H.; Wu, Y.; Kong, Y.; Zhou, Z.; Wang, C.; et al. Metformin Suppresses Triple-Negative Breast Cancer Stem Cells by Targeting KLF5 for Degradation. Cell Discov. 2017, 3, 17010. [Google Scholar] [CrossRef] [PubMed]
- Narusaka, T.; Ohara, T.; Noma, K.; Nishiwaki, N.; Katsura, Y.; Kato, T.; Sato, H.; Tomono, Y.; Kikuchi, S.; Tazawa, H.; et al. Nanog Is a Promising Chemoresistant Stemness Marker and Therapeutic Target by Iron Chelators for Esophageal Cancer. Int. J. Cancer 2021, 149, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch Signaling Pathway: Architecture, Disease, and Therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. 2017, 12, 245. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in Oncology: The Path Forward. Nat. Rev. Drug Discov. 2020, 20, 125–144. [Google Scholar] [CrossRef]
- Hu, Y.Y.; Zheng, M.H.; Cheng, G.; Li, L.; Liang, L.; Gao, F.; Wei, Y.N.; Fu, L.A.; Han, H. Notch Signaling Contributes to the Maintenance of Both Normal Neural Stem Cells and Patient-Derived Glioma Stem Cells. BMC Cancer 2011, 11, 82. [Google Scholar] [CrossRef]
- Xiao, W.; Gao, Z.; Duan, Y.; Yuan, W.; Ke, Y. Notch Signaling Plays a Crucial Role in Cancer Stem-like Cells Maintaining Stemness and Mediating Chemotaxis in Renal Cell Carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 41. [Google Scholar] [CrossRef]
- Abel, E.V.; Kim, E.J.; Wu, J.; Hynes, M.; Bednar, F.; Proctor, E.; Wang, L.; Dziubinski, M.L.; Simeone, D.M. The Notch Pathway Is Important in Maintaining the Cancer Stem Cell Population in Pancreatic Cancer. PLoS ONE 2014, 9, e91983. [Google Scholar] [CrossRef]
- D’Angelo, R.C.; Ouzounova, M.; Davis, A.; Choi, D.; Tchuenkam, S.M.; Kim, G.; Luther, T.; Quraishi, A.A.; Senbabaoglu, Y.; Conley, S.J.; et al. Notch Reporter Activity in Breast Cancer Cell Lines Identifies a Subset of Cells with Stem Cell Activity. Mol. Cancer Ther. 2015, 14, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Maillard, I. Therapeutic Targeting of Notch Signaling: From Cancer to Inflammatory Disorders. Front. Cell Dev. Biol. 2021, 9, 649205. [Google Scholar] [CrossRef]
- Bender, M.H.; Gao, H.; Capen, A.R.; Clay, J.M.; Hipskind, P.A.; Reel, J.K.; Zamek-Gliszczynski, M.J.; Manro, J.R.; Benhadji, K.; Patel, B.K.R. Abstract 1131: Novel Inhibitor of Notch Signaling for the Treatment of Cancer. Cancer Res. 2013, 73 (Suppl. S8), 1131. [Google Scholar] [CrossRef]
- Mancarella, S.; Serino, G.; Dituri, F.; Cigliano, A.; Ribback, S.; Wang, J.; Chen, X.; Calvisi, D.F.; Giannelli, G. Crenigacestat, a Selective NOTCH1 Inhibitor, Reduces Intrahepatic Cholangiocarcinoma Progression by Blocking VEGFA/DLL4/MMP13 Axis. Cell Death Differ. 2020, 27, 2330–2343. [Google Scholar] [CrossRef]
- Massard, C.; Azaro, A.; Soria, J.C.; Lassen, U.; Le Tourneau, C.; Sarker, D.; Smith, C.; Ohnmacht, U.; Oakley, G.; Patel, B.K.R.; et al. First-in-Human Study of LY3039478, an Oral Notch Signaling Inhibitor in Advanced or Metastatic Cancer. Ann. Oncol. 2018, 29, 1911–1917. [Google Scholar] [CrossRef]
- Doi, T.; Tajimi, M.; Mori, J.; Asou, H.; Inoue, K.; Benhadji, K.A.; Naito, Y. A Phase 1 Study of Crenigacestat (LY3039478), the Notch Inhibitor, in Japanese Patients with Advanced Solid Tumors. Investig. New Drugs 2021, 39, 469. [Google Scholar] [CrossRef] [PubMed]
- Even, C.; Lassen, U.; Merchan, J.; Le Tourneau, C.; Soria, J.C.; Ferte, C.; Ricci, F.; Diener, J.T.; Yuen, E.; Smith, C.; et al. Safety and Clinical Activity of the Notch Inhibitor, Crenigacestat (LY3039478), in an Open-Label Phase I Trial Expansion Cohort of Advanced or Metastatic Adenoid Cystic Carcinoma. Investig. New Drugs 2020, 38, 402. [Google Scholar] [CrossRef] [PubMed]
- Cowan, A.J.; Pont, M.J.; Sather, B.D.; Turtle, C.J.; Till, B.G.; Libby, E.N.; Coffey, D.G.; Tuazon, S.A.; Wood, B.; Gooley, T.; et al. γ-Secretase Inhibitor in Combination with BCMA Chimeric Antigen Receptor T-Cell Immunotherapy for Individuals with Relapsed or Refractory Multiple Myeloma: A Phase 1, First-in-Human Trial. Lancet Oncol. 2023, 24, 811–822. [Google Scholar] [CrossRef]
- Pant, S.; Jones, S.F.; Kurkjian, C.D.; Infante, J.R.; Moore, K.N.; Burris, H.A.; McMeekin, D.S.; Benhadji, K.A.; Patel, B.K.R.; Frenzel, M.J.; et al. A First-in-Human Phase I Study of the Oral Notch Inhibitor, LY900009, in Patients with Advanced Cancer. Eur. J. Cancer 2016, 56, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gavai, A.V.; Quesnelle, C.; Norris, D.; Han, W.C.; Gill, P.; Shan, W.; Balog, A.; Chen, K.; Tebben, A.; Rampulla, R.; et al. Discovery of Clinical Candidate BMS-906024: A Potent-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors. ACS Med. Chem. Lett. 2015, 6, 523. [Google Scholar] [CrossRef] [PubMed]
- Ferrarotto, R.; Mishra, V.; Herz, E.; Yaacov, A.; Solomon, O.; Rauch, R.; Mondshine, A.; Motin, M.; Leibovich-Rivkin, T.; Davis, M.; et al. AL101, a Gamma-Secretase Inhibitor, Has Potent Antitumor Activity against Adenoid Cystic Carcinoma with Activated NOTCH Signaling. Cell Death Dis. 2022, 13, 678. [Google Scholar] [CrossRef] [PubMed]
- Zweidler-McKay, P.A.; DeAngelo, D.J.; Douer, D.; Dombret, H.; Ottmann, O.G.; Vey, N.; Thomas, D.A.; Zhu, L.; Huang, F.; Bajaj, G.; et al. The Safety and Activity of BMS-906024, a Gamma Secretase Inhibitor (GSI) with Anti-Notch Activity, in Patients with Relapsed T-Cell Acute Lymphoblastic Leukemia (T-ALL): Initial Results of a Phase 1 Trial. Blood 2014, 124, 968. [Google Scholar] [CrossRef]
- Ferrarotto, R.; Ho, A.L.; Wirth, L.J.; Dekel, E.; Walker, R.W.; Vergara-Silva, A.L. ACCURACY: Phase (P) 2 Trial of AL101, a Pan-Notch Inhibitor, in Patients (Pts) with Recurrent/Metastatic (R/M) Adenoid Cystic Carcinoma (ACC) with Notch Activating Mutations (Notch Act Mut). J. Clin. Oncol. 2019, 37 (Suppl. S15), TPS6098. [Google Scholar] [CrossRef]
- Morgan, K.M.; Fischer, B.S.; Lee, F.Y.; Shah, J.J.; Bertino, J.R.; Rosenfeld, J.; Singh, A.; Khiabanian, H.; Pine, S.R. Gamma Secretase Inhibition by BMS-906024 Enhances Efficacy of Paclitaxel in Lung Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 2759. [Google Scholar] [CrossRef]
- Luistro, L.; He, W.; Smith, M.; Packman, K.; Vilenchik, M.; Carvajal, D.; Roberts, J.; Cai, J.; Berkofsky-Fessler, W.; Hilton, H.; et al. Preclinical Profile of a Potent γ-Secretase Inhibitor Targeting Notch Signaling with in Vivo Efficacy and Pharmacodynamic Properties. Cancer Res. 2009, 69, 7672–7680. [Google Scholar] [CrossRef]
- Huynh, C.; Poliseno, L.; Segura, M.F.; Medicherla, R.; Haimovic, A.; Menendez, S.; Shang, S.; Pavlick, A.; Shao, Y.; Darvishian, F.; et al. The Novel Gamma Secretase Inhibitor RO4929097 Reduces the Tumor Initiating Potential of Melanoma. PLoS ONE 2011, 6, e25264. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Yeatman, T.; Weber, J.; Coppola, D.; Schell, M.J.; Han, G.; Almhanna, K.; Kim, R.; Valone, T.; Jump, H.; et al. A Phase II Study of RO4929097 in Metastatic Colorectal Cancer. Eur. J. Cancer 2012, 48, 997. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Moon, J.; Redman, B.G.; Chidiac, T.; Flaherty, L.E.; Zha, Y.; Othus, M.; Ribas, A.; Sondak, V.K.; Gajewski, T.F.; et al. Phase 2 Study of RO4929097, a Gamma-Secretase Inhibitor, in Metastatic Melanoma: SWOG 0933. Cancer 2015, 121, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.M.; Rosenbaum, E.; Wu, N.; Dickson, M.A.; Sheikh, T.N.; D’Angelo, S.P.; Chi, P.; Keohan, M.L.; Erinjeri, J.P.; Antonescu, C.R.; et al. A Phase Ib/II Randomized Study of RO4929097, a Gamma-Secretase or Notch Inhibitor with or without Vismodegib, a Hedgehog Inhibitor, in Advanced Sarcoma. Clin. Cancer Res. 2022, 28, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- De Jesus-Acosta, A.; Laheru, D.; Maitra, A.; Arcaroli, J.; Rudek, M.A.; Dasari, A.; Blatchford, P.J.; Quackenbush, K.; Messersmith, W. A Phase II Study of the Gamma Secretase Inhibitor RO4929097 in Patients with Previously Treated Metastatic Pancreatic Adenocarcinoma. Investig. New Drugs 2014, 32, 739. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.X.; Xu, A.; Zhang, C.C.; Olson, P.; Chen, L.; Lee, T.K.; Cheung, T.T.; Lo, C.M.; Wang, X.Q. Notch Inhibitor PF-03084014 Inhibits Hepatocellular Carcinoma Growth and Metastasis via Suppression of Cancer Stemness Due to Reduced Activation of Notch1–Stat3. Mol. Cancer Ther. 2017, 16, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Dai, J.; Keller, J.M.; Mizokami, A.; Xia, S.; Keller, E.T. Notch Pathway Inhibition Using PF-03084014, a γ-Secretase Inhibitor (GSI), Enhances the Anti-Tumor Effect of Docetaxel in Prostate Cancer. Clin. Cancer Res. 2015, 21, 4619. [Google Scholar] [CrossRef]
- Kummar, S.; Coyne, G.O.S.; Do, K.T.; Turkbey, B.; Meltzer, P.S.; Polley, E.; Choyke, P.L.; Meehan, R.; Vilimas, R.; Horneffer, Y.; et al. Clinical Activity of the γ-Secretase Inhibitor PF-03084014 in Adults with Desmoid Tumors (Aggressive Fibromatosis). J. Clin. Oncol. 2017, 35, 1561. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.; Ratan, R.; Alcindor, T.; Schöffski, P.; van der Graaf, W.T.; Wilky, B.A.; Riedel, R.F.; Lim, A.; Smith, L.M.; Moody, S.; et al. Nirogacestat, a γ-Secretase Inhibitor for Desmoid Tumors. N. Engl. J. Med. 2023, 388, 898–912. [Google Scholar] [CrossRef]
- Locatelli, M.A.; Aftimos, P.; Claire Dees, E.; LoRusso, P.M.; Pegram, M.D.; Awada, A.; Huang, B.; Cesari, R.; Jiang, Y.; Shaik, M.N.; et al. Phase I Study of the Gamma Secretase Inhibitor PF-03084014 in Combination with Docetaxel in Patients with Advanced Triple-Negative Breast Cancer. Oncotarget 2017, 8, 2320. [Google Scholar] [CrossRef]
- Dai, G.; Deng, S.; Guo, W.; Yu, L.; Yang, J.; Zhou, S.; Gao, T. Notch Pathway Inhibition Using DAPT, a γ-Secretase Inhibitor (GSI), Enhances the Antitumor Effect of Cisplatin in Resistant Osteosarcoma. Mol. Carcinog. 2019, 58, 3–18. [Google Scholar] [CrossRef]
- Barat, S.; Chen, X.; Bui, K.C.; Bozko, P.; Götze, J.; Christgen, M.; Krech, T.; Malek, N.P.; Plentz, R.R. Gamma-Secretase Inhibitor IX (GSI) Impairs Concomitant Activation of Notch and Wnt-Beta-Catenin Pathways in CD44+ Gastric Cancer Stem Cells. Stem Cells Transl. Med. 2017, 6, 819. [Google Scholar] [CrossRef]
- Feng, J.; Wang, J.; Liu, Q.; Li, J.; Zhang, Q.; Zhuang, Z.; Yao, X.; Liu, C.; Li, Y.; Cao, L.; et al. DAPT, a γ-Secretase Inhibitor, Suppresses Tumorigenesis, and Progression of Growth Hormone-Producing Adenomas by Targeting Notch Signaling. Front. Oncol. 2019, 9, 809. [Google Scholar] [CrossRef]
- Schott, A.F.; Landis, M.D.; Dontu, G.; Griffith, K.A.; Layman, R.M.; Krop, I.; Paskett, L.A.; Wong, H.; Dobrolecki, L.E.; Lewis, M.T.; et al. Preclinical and Clinical Studies of Gamma Secretase Inhibitors with Docetaxel on Human Breast Tumors. Clin. Cancer Res. 2013, 19, 1512. [Google Scholar] [CrossRef]
- Chen, X.; Gong, L.; Ou, R.; Zheng, Z.; Chen, J.; Xie, F.; Huang, X.; Qiu, J.; Zhang, W.; Jiang, Q.; et al. Sequential Combination Therapy of Ovarian Cancer with Cisplatin and γ-Secretase Inhibitor MK-0752. Gynecol. Oncol. 2016, 140, 537–544. [Google Scholar] [CrossRef]
- Krop, I.; Demuth, T.; Guthrie, T.; Wen, P.Y.; Mason, W.P.; Chinnaiyan, P.; Butowski, N.; Groves, M.D.; Kesari, S.; Freedman, S.J.; et al. Phase I Pharmacologic and Pharmacodynamic Study of the Gamma Secretase (Notch) Inhibitor MK-0752 in Adult Patients with Advanced Solid Tumors. J. Clin. Oncol. 2012, 30, 2307–2313. [Google Scholar] [CrossRef]
- Cook, N.; Basu, B.; Smith, D.M.; Gopinathan, A.; Evans, J.; Steward, W.P.; Palmer, D.; Propper, D.; Venugopal, B.; Hategan, M.; et al. A Phase I Trial of the γ-Secretase Inhibitor MK-0752 in Combination with Gemcitabine in Patients with Pancreatic Ductal Adenocarcinoma. Br. J. Cancer 2018, 118, 793–801. [Google Scholar] [CrossRef]
- Hoffman, L.M.; Fouladi, M.; Olson, J.; Daryani, V.M.; Stewart, C.F.; Wetmore, C.; Kocak, M.; Onar-Thomas, A.; Wagner, L.; Gururangan, S.; et al. Phase I Trial of Weekly MK-0752 in Children with Refractory Central Nervous System Malignancies: A Pediatric Brain Tumor Consortium Study. Childs Nerv. Syst. 2015, 31, 1283. [Google Scholar] [CrossRef]
- Habets, R.A.; De Bock, C.E.; Serneels, L.; Lodewijckx, I.; Verbeke, D.; Nittner, D.; Narlawar, R.; Demeyer, S.; Dooley, J.; Liston, A.; et al. Safe Targeting of T Cell Acute Lymphoblastic Leukemia by Pathology-Specific NOTCH Inhibition. Sci. Transl. Med. 2019, 11, 6246. [Google Scholar] [CrossRef]
- Lehal, R.; Zaric, J.; Vigolo, M.; Urech, C.; Frismantas, V.; Zangger, N.; Cao, L.; Berger, A.; Chicote, I.; Loubéry, S.; et al. Pharmacological Disruption of the Notch Transcription Factor Complex. Proc. Natl. Acad. Sci. USA 2020, 117, 16292–16301. [Google Scholar] [CrossRef]
- Vigolo, M.; Urech, C.; Lamy, S.; Monticone, G.; Zabaleta, J.; Hossain, F.; Wyczechowska, D.; Del Valle, L.; O’Regan, R.M.; Miele, L.; et al. The Efficacy of CB-103, a First-in-Class Transcriptional Notch Inhibitor, in Preclinical Models of Breast Cancer. Cancers 2023, 15, 3957. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.Y.; Zu, Y.X.; Jiang, X.W.; Sun, X.T.; Liu, T.Y.; Li, R.L.; Wu, Q.; Zhang, Y.S.; Zhao, Q.C. Novel ADAM-17 Inhibitor ZLDI-8 Inhibits the Proliferation and Metastasis of Chemo-Resistant Non-Small-Cell Lung Cancer by Reversing Notch and Epithelial Mesenchymal Transition in Vitro and in Vivo. Pharmacol. Res. 2019, 148, 104406. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, D.; Jiang, Q.; Cao, S.; Sun, H.; Chai, Y.; Li, X.; Ren, T.; Yang, R.; Feng, F.; et al. Novel ADAM-17 Inhibitor ZLDI-8 Enhances the in Vitro and in Vivo Chemotherapeutic Effects of Sorafenib on Hepatocellular Carcinoma Cells. Cell Death Dis. 2018, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Li, D.D.; Zhao, C.H.; Ding, H.W.; Wu, Q.; Ren, T.S.; Wang, J.; Chen, C.Q.; Zhao, Q.C. A Novel Inhibitor of ADAM17 Sensitizes Colorectal Cancer Cells to 5-Fluorouracil by Reversing Notch and Epithelial-Mesenchymal Transition in Vitro and in Vivo. Cell Prolif. 2018, 51, e12480. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Paranjape, A.N.; Rangarajan, A.; Dighe, R.R. A Monoclonal Antibody against Human Notch1 Ligand-Binding Domain Depletes Subpopulation of Putative Breast Cancer Stem-like Cells. Mol. Cancer Ther. 2012, 11, 77–86. [Google Scholar] [CrossRef]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; De Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic Antibody Targeting of Individual Notch Receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef]
- Yen, W.C.; Fischer, M.M.; Axelrod, F.; Bond, C.; Cain, J.; Cancilla, B.; Henner, W.R.; Meisner, R.; Sato, A.; Shah, J.; et al. Targeting Notch Signaling with a Notch2/Notch3 Antagonist (Tarextumab) Inhibits Tumor Growth and Decreases Tumor-Initiating Cell Frequency. Clin. Cancer Res. 2015, 21, 2084–2095. [Google Scholar] [CrossRef]
- Pietanza, M.C.; Spira, A.I.; Jotte, R.M.; Gadgeel, S.M.; Mita, A.C.; Hart, L.L.; Gluck, W.L.; Chiang, A.C.; Liu, S.V.; Kapoun, A.M.; et al. Final Results of Phase Ib of Tarextumab (TRXT, OMP-59R5, Anti-Notch2/3) in Combination with Etoposide and Platinum (EP) in Patients (Pts) with Untreated Extensive-Stage Small-Cell Lung Cancer (ED-SCLC). J. Clin. Oncol. 2015, 33 (Suppl. S15), 7508. [Google Scholar] [CrossRef]
- Smith, D.C.; Chugh, R.; Patnaik, A.; Papadopoulos, K.P.; Wang, M.; Kapoun, A.M.; Xu, L.; Dupont, J.; Stagg, R.J.; Tolcher, A. A Phase 1 Dose Escalation and Expansion Study of Tarextumab (OMP-59R5) in Patients with Solid Tumors. Investig. New Drugs 2019, 37, 722. [Google Scholar] [CrossRef]
- Hu, Z.I.; Bendell, J.C.; Bullock, A.; LoConte, N.K.; Hatoum, H.; Ritch, P.; Hool, H.; Leach, J.W.; Sanchez, J.; Sohal, D.P.S.; et al. A Randomized Phase II Trial of Nab-Paclitaxel and Gemcitabine with Tarextumab or Placebo in Patients with Untreated Metastatic Pancreatic Cancer. Cancer Med. 2019, 8, 5148–5157. [Google Scholar] [CrossRef]
- Ferrarotto, R.; Eckhardt, G.; Patnaik, A.; LoRusso, P.; Faoro, L.; Heymach, J.V.; Kapoun, A.M.; Xu, L.; Munster, P. A Phase I Dose-Escalation and Dose-Expansion Study of Brontictuzumab in Subjects with Selected Solid Tumors. Ann. Oncol. 2018, 29, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Casulo, C.; Ruan, J.; Dang, N.H.; Gore, L.; Diefenbach, C.; Beaven, A.W.; Castro, J.E.; Porcu, P.; Faoro, L.; Dupont, J.; et al. Safety and Preliminary Efficacy Results of a Phase I First-in-Human Study of the Novel Notch-1 Targeting Antibody Brontictuzumab (OMP-52M51) Administered Intravenously to Patients with Hematologic Malignancies. Blood 2016, 128, 5108. [Google Scholar] [CrossRef]
- Rosen, L.S.; Wesolowski, R.; Baffa, R.; Liao, K.H.; Hua, S.Y.; Gibson, B.L.; Pirie-Shepherd, S.; Tolcher, A.W. A Phase I, Dose-Escalation Study of PF-06650808, an Anti-Notch3 Antibody–Drug Conjugate, in Patients with Breast Cancer and Other Advanced Solid Tumors. Investig. New Drugs 2020, 38, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, E.G.; LoRusso, P.; Strother, R.M.; Diamond, J.R.; Younger, A.; Messersmith, W.A.; Adriaens, L.; Liu, L.; Kao, R.J.; DiCioccio, A.T.; et al. A Phase I First-in-Human Study of Enoticumab (REGN421), a Fully Human Delta-like Ligand 4 (Dll4) Monoclonal Antibody in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 2695–2703. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Handley, K.F.; Burger, R.; Molin, G.Z.D.; Stagg, R.; Sood, A.K.; Moore, K.N. Demcizumab Combined with Paclitaxel for Platinum-Resistant Ovarian, Primary Peritoneal, and Fallopian Tube Cancer: The SIERRA Open-Label Phase Ib Trial. Gynecol. Oncol. 2020, 157, 386–391. [Google Scholar] [CrossRef] [PubMed]
- McKeage, M.J.; Kotasek, D.; Markman, B.; Hidalgo, M.; Millward, M.J.; Jameson, M.B.; Harris, D.L.; Stagg, R.J.; Kapoun, A.M.; Xu, L.; et al. Phase IB Trial of the Anti-Cancer Stem Cell DLL4-Binding Agent Demcizumab with Pemetrexed and Carboplatin as First-Line Treatment of Metastatic Non-Squamous NSCLC. Target. Oncol. 2018, 13, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.W.; Ross, S.; Veldman-Jones, M.; Foltz, I.N.; Clavette, B.C.; Manchulenko, K.; Eberlein, C.; Kendrew, J.; Petteruti, P.; Cho, S.; et al. MEDI0639: A Novel Therapeutic Antibody Targeting Dll4 Modulates Endothelial Cell Function and Angiogenesis in Vivo. Mol. Cancer Ther. 2012, 11, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Dowlati, A.; Naing, A.; Gribbin, M.J.; Jenkins, D.W.; Chang, L.L.; Lai, D.W.; Smith, D.C. Phase I Study of MEDI0639 in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2015, 33 (Suppl. S15), 3024. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Z.; Jia, X.; Wang, L.; Chen, Z.; Wang, S.; Wang, M.; Zhang, J.; Wu, M. MMGZ01, an Anti-DLL4 Monoclonal Antibody, Promotes Nonfunctional Vessels and Inhibits Breast Tumor Growth. Cancer Lett. 2016, 372, 118–127. [Google Scholar] [CrossRef]
- Jimeno, A.; Moore, K.N.; Gordon, M.; Chugh, R.; Diamond, J.R.; Aljumaily, R.; Mendelson, D.; Kapoun, A.M.; Xu, L.; Stagg, R.; et al. A First-in-Human Phase 1a Study of the Bispecific Anti-DLL4/Anti-VEGF Antibody Navicixizumab (OMP-305B83) in Patients with Previously Treated Solid Tumors. Investig. New Drugs 2019, 37, 461–472. [Google Scholar] [CrossRef]
- Fu, S.; Corr, B.R.; Culm-Merdek, K.; Mockbee, C.; Youssoufian, H.; Stagg, R.; Naumann, R.W.; Wenham, R.M.; Rosengarten, R.D.; Benjamin, L.; et al. Phase Ib Study of Navicixizumab Plus Paclitaxel in Patients with Platinum-Resistant Ovarian, Primary Peritoneal, or Fallopian Tube Cancer. J. Clin. Oncol. 2022, 40, 2568. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hickson, J.A.; Ambrosi, D.J.; Haasch, D.L.; Foster-Duke, K.D.; Eaton, L.J.; DiGiammarino, E.L.; Panchal, S.C.; Jiang, F.; Mudd, S.R.; et al. Abt-165, a Dual Variable Domain Immunoglobulin (Dvd-Ig) Targeting Dll4 and Vegf, Demonstrates Superior Efficacy and Favorable Safety Profiles in Preclinical Models. Mol. Cancer Ther. 2018, 17, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.; Strickler, J.; Gordon, M.; Barve, M.; Wang, L.; Yue, H.; Motwani, M.; Kasichayanula, S.; Naumovski, L.; Hamilton, E. Phase 1b Open-Label Study Evaluating the Safety, Pharmacokinetics, and Preliminary Efficacy of ABT-165 plus FOLFIRI in Patients with Second-Line (2L) Colorectal Cancer (CRC). Ann. Oncol. 2018, 29, v66. [Google Scholar] [CrossRef]
- Giffin, M.J.; Cooke, K.; Lobenhofer, E.K.; Estrada, J.; Zhan, J.; Deegen, P.; Thomas, M.; Murawsky, C.M.; Werner, J.; Liu, S.; et al. AMG 757, a Half-Life Extended, DLL3-Targeted Bispecific T-Cell Engager, Shows High Potency and Sensitivity in Preclinical Models of Small-Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 1526–1537. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Champiat, S.; Lai, W.V.; Izumi, H.; Govindan, R.; Boyer, M.; Hummel, H.D.; Borghaei, H.; Johnson, M.L.; Steeghs, N.; et al. Tarlatamab, a First-in-Class DLL3-Targeted Bispecific T-Cell Engager, in Recurrent Small-Cell Lung Cancer: An Open-Label, Phase I Study. J. Clin. Oncol. 2023, 41, 2893. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.-J.; Cho, B.C.; Felip, E.; Korantzis, I.; Ohashi, K.; Majem, M.; Juan-Vidal, O.; Handzhiev, S.; Izumi, H.; Lee, J.-S.; et al. Tarlatamab for Patients with Previously Treated Small-Cell Lung Cancer. N. Engl. J. Med. 2023, 389, 2063–2075. [Google Scholar] [CrossRef]
- Johnson, M.L.; Dy, G.K.; Mamdani, H.; Dowlati, A.; Schoenfeld, A.J.; Pacheco, J.M.; Sanborn, R.E.; Menon, S.P.; Santiago, L.; Yaron, Y.; et al. Interim Results of an Ongoing Phase 1/2a Study of HPN328, a Tri-Specific, Half-Life Extended, DLL3-Targeting, T-Cell Engager, in Patients with Small Cell Lung Cancer and Other Neuroendocrine Cancers. J. Clin. Oncol. 2022, 40 (Suppl. S16), 8566. [Google Scholar] [CrossRef]
- Hann, C.L.; Burns, T.F.; Dowlati, A.; Morgensztern, D.; Ward, P.J.; Koch, M.M.; Chen, C.; Ludwig, C.; Patel, M.; Nimeiri, H.; et al. A Phase 1 Study Evaluating Rovalpituzumab Tesirine in Frontline Treatment of Patients with Extensive-Stage SCLC. J. Thorac. Oncol. 2021, 16, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Saunders, L.R.; Bankovich, A.J.; Anderson, W.C.; Aujay, M.A.; Bheddah, S.; Black, K.A.; Desai, R.; Escarpe, P.A.; Hampl, J.; Laysang, A.; et al. A DLL3-Targeted Antibody-Drug Conjugate Eradicates High-Grade Pulmonary Neuroendocrine Tumor-Initiating Cells in Vivo. Sci. Transl. Med. 2015, 7, 302ra136. [Google Scholar] [CrossRef]
- Johnson, M.L.; Zvirbule, Z.; Laktionov, K.; Helland, A.; Cho, B.C.; Gutierrez, V.; Colinet, B.; Lena, H.; Wolf, M.; Gottfried, M.; et al. Rovalpituzumab Tesirine as a Maintenance Therapy After First-Line Platinum-Based Chemotherapy in Patients with Extensive-Stage–SCLC: Results From the Phase 3 MERU Study. J. Thorac. Oncol. 2021, 16, 1570–1581. [Google Scholar] [CrossRef]
- Morgensztern, D.; Besse, B.; Greillier, L.; Santana-Davila, R.; Ready, N.; Hann, C.L.; Glisson, B.S.; Farago, A.F.; Dowlati, A.; Rudin, C.M.; et al. Efficacy and Safety of Rovalpituzumab Tesirine in Third-Line and beyond Patients with DLL3-Expressing, Relapsed/Refractory Small-Cell Lung Cancer: Results from the Phase II TrINITY Study. Clin. Cancer Res. 2019, 25, 6958–6966. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-Catenin Signaling: Function, Biological Mechanisms, and Therapeutic Opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/β-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Yu, C.; Li, F.; Zuo, Y.; Wang, Y.; Yao, L.; Wu, C.; Wang, C.; Ye, L. Wnt/β-Catenin Signaling in Cancers and Targeted Therapies. Signal Transduct. Target. Ther. 2021, 6, 307. [Google Scholar] [CrossRef] [PubMed]
- Willert, K.; Brown, J.D.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R.; Nusse, R. Wnt Proteins Are Lipid-Modified and Can Act as Stem Cell Growth Factors. Nature 2003, 423, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Baarsma, H.A.; Königshoff, M. ‘WNT-Er Is Coming’: WNT Signaling in Chronic Lung Diseases. Thorax 2017, 72, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, W.-M.; Zhang, X.-L.; He, H.-Q.; Sun, X.-L.; Zeng, H.; Xu, X.-F.; Huang, L.; Zhu, Z.; Zhang, L.; et al. AGE/RAGE Promotes Thecalcification of Human Aortic Smooth Muscle Cells via the Wnt/β-Catenin Axis. Am. J. Transl. Res. 2016, 8, 4644. [Google Scholar] [PubMed]
- González-Fernández, C.; Gonzalez, P.; Andres-Benito, P.; Ferrer, I.; Rodríguez, F.J. Wnt Signaling Alterations in the Human Spinal Cord of Amyotrophic Lateral Sclerosis Cases: Spotlight on Fz2 and Wnt5a. Mol. Neurobiol. 2019, 56, 6777–6791. [Google Scholar] [CrossRef]
- Vermeulen, L.; De Sousa E Melo, F.; Van Der Heijden, M.; Cameron, K.; De Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt Activity Defines Colon Cancer Stem Cells and Is Regulated by the Microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Guo, W.; Lasky, J.L.; Chang, C.J.; Mosessian, S.; Lewis, X.; Xiao, Y.; Yeh, J.E.; Chen, J.Y.; Iruela-Arispe, M.L.; Varella-Garcia, M.; et al. Multi-Genetic Events Collaboratively Contribute to Pten-Null Leukaemia Stem-Cell Formation. Nature 2008, 453, 529–533. [Google Scholar] [CrossRef]
- Jang, G.B.; Kim, J.Y.; Cho, S.D.; Park, K.S.; Jung, J.Y.; Lee, H.Y.; Hong, I.S.; Nam, J.S. Blockade of Wnt/β-Catenin Signaling Suppresses Breast Cancer Metastasis by Inhibiting CSC-like Phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Koo, B.S.; Kim, J.M.; Huang, S.; Rho, Y.S.; Bae, W.J.; Kang, H.J.; Kim, Y.S.; Moon, J.H.; Lim, Y.C. Wnt/β-Catenin Signaling Maintains Self-Renewal and Tumorigenicity of Head and Neck Squamous Cell Carcinoma Stem-like Cells by Activating Oct4. J. Pathol. 2014, 234, 99–107. [Google Scholar] [CrossRef]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt Pathway Inhibition via the Targeting of Frizzled Receptors Results in Decreased Growth and Tumorigenicity of Human Tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.C.; Rosen, L.S.; Chugh, R.; Goldman, J.W.; Xu, L.; Kapoun, A.; Brachmann, R.K.; Dupont, J.; Stagg, R.J.; Tolcher, A.W.; et al. First-in-Human Evaluation of the Human Monoclonal Antibody Vantictumab (OMP-18R5; Anti-Frizzled) Targeting the WNT Pathway in a Phase I Study for Patients with Advanced Solid Tumors. J. Clin. Oncol. 2013, 31 (Suppl. S15), 2540. [Google Scholar] [CrossRef]
- Davis, S.L.; Cardin, D.B.; Shahda, S.; Lenz, H.-J.; Dotan, E.; O’Neil, B.; Kapoun, A.M.; Stagg, R.J.; Berlin, J.; Messersmith, W.A.; et al. A Phase Ib Dose Escalation Study of Vantictumab (VAN) in Combination with Nab-Paclitaxel (Nab-P) and Gemcitabine (G) in Patients with Previously Untreated Stage IV Pancreatic Cancer. J. Clin. Oncol. 2019, 37 (Suppl. S4), 249. [Google Scholar] [CrossRef]
- Diamond, J.R.; Becerra, C.; Richards, D.; Mita, A.; Osborne, C.; O’Shaughnessy, J.; Zhang, C.; Henner, R.; Kapoun, A.M.; Xu, L.; et al. Phase Ib Clinical Trial of the Anti-Frizzled Antibody Vantictumab (OMP-18R5) plus Paclitaxel in Patients with Locally Advanced or Metastatic HER2-Negative Breast Cancer. Breast Cancer Res. Treat. 2020, 184, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, Z.; Adams, J.J.; Blazer, L.L.; Gakhal, A.K.; Jarvik, N.; Steinhart, Z.; Robitaille, M.; Mascall, K.; Pan, J.; Angers, S.; et al. A Synthetic Anti-Frizzled Antibody Engineered for Broadened Specificity Exhibits Enhanced Anti-Tumor Properties. MAbs 2018, 10, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Steinhart, Z.; Pavlovic, Z.; Chandrashekhar, M.; Hart, T.; Wang, X.; Zhang, X.; Robitaille, M.; Brown, K.R.; Jaksani, S.; Overmeer, R.; et al. Genome-Wide CRISPR Screens Reveal a Wnt–FZD5 Signaling Circuit as a Druggable Vulnerability of RNF43-Mutant Pancreatic Tumors. Nat. Med. 2016, 23, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Li, H.K.; Sugyo, A.; Tsuji, A.B.; Morokoshi, Y.; Minegishi, K.; Nagatsu, K.; Kanda, H.; Harada, Y.; Nagayama, S.; Katagiri, T.; et al. α-Particle Therapy for Synovial Sarcoma in the Mouse Using an Astatine-211-Labeled Antibody against Frizzled Homolog 10. Cancer Sci. 2018, 109, 2302–2309. [Google Scholar] [CrossRef]
- Sudo, H.; Tsuji, A.B.; Sugyo, A.; Harada, Y.; Nagayama, S.; Katagiri, T.; Nakamura, Y.; Higashi, T. FZD10-targeted A-radioimmunotherapy with 225Ac-labeled OTSA101 Achieves Complete Remission in a Synovial Sarcoma Model. Cancer Sci. 2022, 113, 721. [Google Scholar] [CrossRef]
- Giraudet, A.-L.; Badel, J.-N.; Cassier, P.; Desuzinges, C.; Kriza, D.; Perol, D.; Blay, J.-Y. SYNFRIZZ-A Phase Ia/Ib of a Radiolabelled Monoclonal AB for the Treatment of Relapsing Synovial Sarcoma. J. Nucl. Med. 2014, 55 (Suppl. S1), 223. [Google Scholar]
- Giraudet, A.L.; Cassier, P.A.; Iwao-Fukukawa, C.; Garin, G.; Badel, J.N.; Kryza, D.; Chabaud, S.; Gilles-Afchain, L.; Clapisson, G.; Desuzinges, C.; et al. A First-in-Human Study Investigating Biodistribution, Safety and Recommended Dose of a New Radiolabeled MAb Targeting FZD10 in Metastatic Synovial Sarcoma Patients. BMC Cancer 2018, 18, 646. [Google Scholar] [CrossRef]
- Le, P.N.; McDermott, J.D.; Jimeno, A. Targeting the Wnt Pathway in Human Cancers: Therapeutic Targeting with a Focus on OMP-54F28. Pharmacol. Ther. 2015, 146, 1–11. [Google Scholar] [CrossRef]
- Fischer, M.M.; Cancilla, B.; Yeung, V.P.; Cattaruzza, F.; Chartier, C.; Murriel, C.L.; Cain, J.; Tam, R.; Cheng, C.Y.; Evans, J.W.; et al. WNT Antagonists Exhibit Unique Combinatorial Antitumor Activity with Taxanes by Potentiating Mitotic Cell Death. Sci. Adv. 2017, 3, e1700090. [Google Scholar] [CrossRef]
- Jimeno, A.; Gordon, M.; Chugh, R.; Messersmith, W.; Mendelson, D.; Dupont, J.; Stagg, R.; Kapoun, A.M.; Xu, L.; Uttamsingh, S.; et al. A First-in-Human Phase I Study of the Anticancer Stem Cell Agent Ipafricept (OMP-54F28), a Decoy Receptor for Wnt Ligands, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 7490–7497. [Google Scholar] [CrossRef]
- Moore, K.N.; Gunderson, C.C.; Sabbatini, P.; McMeekin, D.S.; Mantia-Smaldone, G.; Burger, R.A.; Morgan, M.A.; Kapoun, A.M.; Brachmann, R.K.; Stagg, R.; et al. A Phase 1b Dose Escalation Study of Ipafricept (OMP54F28) in Combination with Paclitaxel and Carboplatin in Patients with Recurrent Platinum-Sensitive Ovarian Cancer. Gynecol. Oncol. 2019, 154, 294–301. [Google Scholar] [CrossRef]
- Dotan, E.; Cardin, D.B.; Lenz, H.J.; Messersmith, W.; O’Neil, B.; Cohen, S.J.; Denlinger, C.S.; Shahda, S.; Astsaturov, I.; Kapoun, A.M.; et al. Phase Ib Study of Wnt Inhibitor Ipafricept with Gemcitabine and Nab-Paclitaxel in Patients with Previously Untreated Stage IV Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 5348–5357. [Google Scholar] [CrossRef]
- He, B.; You, L.; Uematsu, K.; Xu, Z.; Lee, A.Y.; Matsangou, M.; McCormick, F.; Jablons, D.M. A Monoclonal Antibody against Wnt-1 Induces Apoptosis in Human Cancer Cells. Neoplasia 2004, 6, 7. [Google Scholar] [CrossRef]
- Salik, B.; Yi, H.; Hassan, N.; Santiappillai, N.; Vick, B.; Connerty, P.; Duly, A.; Trahair, T.; Woo, A.J.; Beck, D.; et al. Targeting RSPO3-LGR4 Signaling for Leukemia Stem Cell Eradication in Acute Myeloid Leukemia. Cancer Cell 2020, 38, 263–278.e6. [Google Scholar] [CrossRef]
- Bendell, J.; Eckhardt, G.S.; Hochster, H.S.; Morris, V.K.; Strickler, J.; Kapoun, A.M.; Wang, M.; Xu, L.; McGuire, K.; Dupont, J.; et al. Initial Results from a Phase 1a/b Study of OMP-131R10, a First-in-Class Anti-RSPO3 Antibody, in Advanced Solid Tumors and Previously Treated Metastatic Colorectal Cancer (CRC). Eur. J. Cancer 2016, 69, S29–S30. [Google Scholar] [CrossRef]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-Driven Cancer through the Inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Schatoff, E.M.; Murphy, C.; Zafra, M.P.; Wilkinson, J.E.; Elemento, O.; Dow, L.E. R-Spondin Chromosome Rearrangements Drive Wnt-Dependent Tumor Initiation and Maintenance in the Intestine. Nat. Commun. 2017, 8, 15945. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, M.; Tabatabae Far, M.A.; Mirzaei, H.; Ghasemi, F. Evaluation of Antitumor Effects of Aspirin and LGK974 Drugs on Cellular Signaling Pathways, Cell Cycle and Apoptosis in Colorectal Cancer Cell Lines Compared to Oxaliplatin Drug. Fundam. Clin. Pharmacol. 2020, 34, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Argilés, G.; Connolly, R.M.; Vaishampayan, U.; de Jonge, M.; Garralda, E.; Giannakis, M.; Smith, D.C.; Dobson, J.R.; McLaughlin, M.E.; et al. Phase 1 Study of Single-Agent WNT974, a First-in-Class Porcupine Inhibitor, in Patients with Advanced Solid Tumors. Br. J. Cancer 2021, 125, 28–37. [Google Scholar] [CrossRef]
- Tabernero, J.; Van Cutsem, E.; Garralda, E.; Tai, D.; De Braud, F.; Geva, R.; van Bussel, M.T.J.; Fiorella Dotti, K.; Elez, E.; de Miguel, M.J.; et al. A Phase Ib/II Study of WNT974 + Encorafenib + Cetuximab in Patients with BRAF V600E-Mutant KRAS Wild-Type Metastatic Colorectal Cancer. Oncologist 2023, 28, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; Ke, Z.; Harmston, N.; Ho, S.Y.; Frois, A.O.; Alam, J.; Jeyaraj, D.A.; Pendharkar, V.; Ghosh, K.; Virshup, I.H.; et al. Wnt Addiction of Genetically Defined Cancers Reversed by PORCN Inhibition. Oncogene 2016, 35, 2197–2207. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Tan, D.S.; Subbiah, V.; Weekes, C.D.; Teneggi, V.; Diermayr, V.; Ethirajulu, K.; Yeo, P.; Chen, D.; Blanchard, S.; et al. First-in-Human Phase 1 Study of ETC-159 an Oral PORCN Inhbitor in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2017, 35 (Suppl. S15), 2584. [Google Scholar] [CrossRef]
- Tan, D.S.P.; Ng, M.C.H.; Subbiah, V.; Messersmith, W.A.; Strickler, J.H.; Diermayr, V.; Cometa, J.; Blanchard, S.; Nellore, R.; Pendharkar, V.; et al. A Phase 1B Dose Escalation Study of ETC-159 in Combination with Pembrolizumab in Advanced or Metastatic Solid Tumors. J. Clin. Oncol. 2023, 41 (Suppl. S16), 2601. [Google Scholar] [CrossRef]
- Proffitt, K.D.; Madan, B.; Ke, Z.; Pendharkar, V.; Ding, L.; Lee, M.A.; Hannoush, R.N.; Virshup, D.M. Pharmacological Inhibition of the Wnt Acyltransferase PORCN Prevents Growth of WNT-Driven Mammary Cancer. Cancer Res. 2013, 73, 502–507. [Google Scholar] [CrossRef]
- Cheng, Y.; Phoon, Y.P.; Jin, X.; Chong, S.Y.S.; Ip, J.C.Y.; Wong, B.W.Y.; Lung, M.L. Wnt-C59 Arrests Stemness and Suppresses Growth of Nasopharyngeal Carcinoma in Mice by Inhibiting the Wnt Pathway in the Tumor Microenvironment. Oncotarget 2015, 6, 14428. [Google Scholar] [CrossRef][Green Version]
- Phillips, C.; Bhamra, I.; Eagle, C.; Flanagan, E.; Armer, R.; Jones, C.D.; Bingham, M.; Calcraft, P.; Edmenson Cook, A.; Thompson, B.; et al. The Wnt Pathway Inhibitor RXC004 Blocks Tumor Growth and Reverses Immune Evasion in Wnt Ligand–Dependent Cancer Models. Cancer Res. Commun. 2022, 2, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.; Woodcock, S.; Bhamra, I.; Jones, C.; Armer, R.; Robertson, J.; Phillips, C. Abstract 2566: Pre-Clinical Efficacy of the Wnt Pathway Inhibitor RXC004 in Combination with Anti-Cancer Therapies. Cancer Res. 2022, 82 (Suppl. S12), 2566. [Google Scholar] [CrossRef]
- Cook, N.; Blagden, S.; Lopez, J.; Sarker, D.; Greystoke, A.; Harris, N.; Kazmi, F.; Naderi, A.; Nintos, G.; Franco, A.O.; et al. 517MO Phase I Study of the Porcupine (PORCN) Inhibitor RXC004 in Patients with Advanced Solid Tumors. Ann. Oncol. 2021, 32, S586–S587. [Google Scholar] [CrossRef]
- Giannakis, M.; Le, D.T.; Pishvaian, M.J.; Weinberg, B.A.; Papadopoulos, K.P.; Shen, L.; Gong, J.; Li, J.; Strickler, J.H.; Zhou, A.; et al. Phase 1 Study of WNT Pathway Porcupine Inhibitor CGX1321 and Phase 1b Study of CGX1321 + Pembrolizumab (Pembro) in Patients (Pts) with Advanced Gastrointestinal (GI) Tumors. J. Clin. Oncol. 2023, 41 (Suppl. S16), 3514. [Google Scholar] [CrossRef]
- Fiskus, W.; Sharma, S.; Saha, S.; Shah, B.; Devaraj, S.G.T.; Sun, B.; Horrigan, S.; Leveque, C.; Zu, Y.; Iyer, S.; et al. Pre-Clinical Efficacy of Combined Therapy with Novel β-Catenin Antagonist BC2059 and Histone Deacetylase Inhibitor against AML Cells. Leukemia 2015, 29, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Braggio, D.A.; de Faria, F.C.C.; Koller, D.; Jin, F.; Zewdu, A.; Lopez, G.; Batte, K.; Casadei, L.; Welliver, M.; Horrigan, S.K.; et al. Preclinical Efficacy of the Wnt/β-Catenin Pathway Inhibitor BC2059 for the Treatment of Desmoid Tumors. PLoS ONE 2022, 17, e0276047. [Google Scholar] [CrossRef] [PubMed]
- Savvidou, I.; Khong, T.; Cuddihy, A.; McLean, C.; Horrigan, S.; Spencer, A. β-Catenin Inhibitor BC2059 Is Efficacious as Monotherapy or in Combination with Proteasome Inhibitor Bortezomib in Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 1765–1778. [Google Scholar] [CrossRef] [PubMed]
- Cranmer, L.D.; Razak, A.R.A.; Ratan, R.; Choy, E.; George, S.; Liebner, D.A.; Stenehjem, D.D.; Gounder, M.M. Results of a Phase I Dose Escalation and Expansion Study of Tegavivint (BC2059), a First-in-Class TBL1 Inhibitor for Patients with Progressive, Unresectable Desmoid Tumor. J. Clin. Oncol. 2022, 40 (Suppl. S16), 11523. [Google Scholar] [CrossRef]
- Handeli, S.; Simon, J.A. A Small-Molecule Inhibitor of Tcf/β-Catenin Signaling down-Regulates PPARγ and PPARδ Activities. Mol. Cancer Ther. 2008, 7, 521–529. [Google Scholar] [CrossRef]
- Iida, J.; Dorchak, J.; Lehman, J.R.; Clancy, R.; Luo, C.; Chen, Y.; Somiari, S.; Ellsworth, R.E.; Hu, H.; Mural, R.J.; et al. FH535 Inhibited Migration and Growth of Breast Cancer Cells. PLoS ONE 2012, 7, e44418. [Google Scholar] [CrossRef]
- Wu, M.Y.; Liang, R.R.; Chen, K.; Shen, M.; Tian, Y.L.; Li, D.M.; Duan, W.M.; Gui, Q.; Gong, F.R.; Lian, L.; et al. FH535 Inhibited Metastasis and Growth of Pancreatic Cancer Cells. Onco Targets Ther. 2015, 8, 1651–1670. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhi, Q.; Shen, M.; Gong, F.R.; Zhou, B.P.; Lian, L.; Shen, B.; Chen, K.; Duan, W.; Wu, M.Y.; et al. FH535, a β-Catenin Pathway Inhibitor, Represses Pancreatic Cancer Xenograft Growth and Angiogenesis. Oncotarget 2016, 7, 47145. [Google Scholar] [CrossRef][Green Version]
- Chen, Y.; Rao, X.; Huang, K.; Jiang, X.; Wang, H.; Teng, L. FH535 Inhibits Proliferation and Motility of Colon Cancer Cells by Targeting Wnt/β-Catenin Signaling Pathway. J. Cancer 2017, 8, 3142. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.M.; Tao, F.; Roy, A.; Lin, T.; He, X.C.; Chen, S.; Lu, X.; Nemechek, J.; Ruan, L.; Yu, X.; et al. Overcoming Wnt–β-Catenin Dependent Anticancer Therapy Resistance in Leukaemia Stem Cells. Nat. Cell Biol. 2020, 22, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ren, J.; Hillier, J.; Lu, W.; Jones, E.Y. Antiepileptic Drug Carbamazepine Binds to a Novel Pocket on the Wnt Receptor Frizzled-8. J. Med. Chem. 2020, 63, 3252–3260. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; You, L.; Xu, Z.; Uematsu, K.; Shan, J.; He, B.; Mikami, I.; Edmondson, L.R.; Neale, G.; Zheng, J.; et al. An Antagonist of Dishevelled Protein-Protein Interaction Suppresses β-Catenin–Dependent Tumor Cell Growth. Cancer Res. 2007, 67, 573–579. [Google Scholar] [CrossRef]
- Nile, A.H.; De Sousa E Melo, F.; Mukund, S.; Piskol, R.; Hansen, S.; Zhou, L.; Zhang, Y.; Fu, Y.; Gogol, E.B.; Kömüves, L.G.; et al. A Selective Peptide Inhibitor of Frizzled 7 Receptors Disrupts Intestinal Stem Cells. Nat. Chem. Biol. 2018, 14, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Osada, T.; Chen, M.; Yang, X.Y.; Spasojevic, I.; Vandeusen, J.B.; Hsu, D.; Clary, B.M.; Clay, T.M.; Chen, W.; Morse, M.A.; et al. Antihelminth Compound Niclosamide Downregulates Wnt Signaling and Elicits Antitumor Responses in Tumors with Activating APC Mutations. Cancer Res. 2011, 71, 4172–4182. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.K.; Bai, M.Y.; Hu, T.M.; Wang, Y.C.; Chao, T.K.; Weng, S.J.; Huang, R.L.; Su, P.H.; Lai, H.C. Preclinical Evaluation of a Nanoformulated Antihelminthic, Niclosamide, in Ovarian Cancer. Oncotarget 2016, 7, 8993. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Haugk, K.; McKiernan, J.S.; Gulati, R.; Cheng, H.H.; Maes, J.L.; Dumpit, R.F.; Nelson, P.S.; Montgomery, B.; McCune, J.S.; et al. A Phase I Study of Niclosamide in Combination with Enzalutamide in Men with Castration-Resistant Prostate Cancer. PLoS ONE 2018, 13, e0198389. [Google Scholar] [CrossRef]
- Chen, B.; Dodge, M.E.; Tang, W.; Lu, J.; Ma, Z.; Fan, C.W.; Wei, S.; Hao, W.; Kilgore, J.; Williams, N.S.; et al. Small Molecule–Mediated Disruption of Wnt-Dependent Signaling in Tissue Regeneration and Cancer. Nat. Chem. Biol. 2009, 5, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H.; Chiorean, E.G.; Kwak, E.L.; Lenz, H.-J.; Nadler, P.I.; Wood, D.L.; Fujimori, M.; Inada, T.; Kouji, H.; McWilliams, R.R. Final Results of a Phase Ib Dose-Escalation Study of PRI-724, a CBP/Beta-Catenin Modulator, plus Gemcitabine (GEM) in Patients with Advanced Pancreatic Adenocarcinoma (APC) as Second-Line Therapy after FOLFIRINOX or FOLFOX. J. Clin. Oncol. 2016, 34 (Suppl. S15), e15721. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Ning, Y.; Yang, D.; Cole, S.; Kahn, M.; Zoghbi, M.; Berg, J.; Fujimori, M.; Inada, T.; Kouji, H.; et al. A Phase I First-in-Human Study of PRI-724 in Patients (Pts) with Advanced Solid Tumors. J. Clin. Oncol. 2013, 31 (Suppl. S15), 2501. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An Embryonic Stem Cell–like Gene Expression Signature in Poorly Differentiated Aggressive Human Tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, T.; Lengerke, C. SOX2 Protein Biochemistry in Stemness, Reprogramming, and Cancer: The PI3K/AKT/SOX2 Axis and Beyond. Oncogene 2019, 39, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Avilion, A.A.; Nicolis, S.K.; Pevny, L.H.; Perez, L.; Vivian, N.; Lovell-Badge, R. Multipotent Cell Lineages in Early Mouse Development Depend on SOX2 Function. Genes Dev. 2003, 17, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Sarkar, A.; Yram, M.A.; Polo, J.M.; Bronson, R.; Sengupta, S.; Seandel, M.; Geijsen, N.; Hochedlinger, K. Sox2(+) Adult Stem and Progenitor Cells Are Important for Tissue Regeneration and Survival of Mice. Cell Stem Cell 2011, 9, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Xie, F.; Zhao, T.; Zhang, R.; Gao, A.; Chen, Y.; Li, H.; Zhang, S.; Xiao, Z.; Li, J.; et al. Targeting SOX2 Protein with Peptide Aptamers for Therapeutic Gains against Esophageal Squamous Cell Carcinoma. Mol. Ther. 2020, 28, 901–913. [Google Scholar] [CrossRef]
- Al Mamun, M.; Mannoor, K.; Cao, J.; Qadri, F.; Song, X. SOX2 in Cancer Stemness: Tumor Malignancy and Therapeutic Potentials. J. Mol. Cell Biol. 2020, 12, 85. [Google Scholar] [CrossRef]
- Zhang, S.; Xiong, X.; Sun, Y. Functional Characterization of SOX2 as an Anticancer Target. Signal Transduct. Target. Ther. 2020, 5, 135. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ji, W.; Yu, Y.; Li, Z.; Niu, X.; Xia, W.; Lu, S. FGFR1-ERK1/2-SOX2 Axis Promotes Cell Proliferation, Epithelial-Mesenchymal Transition, and Metastasis in FGFR1-Amplified Lung Cancer. Oncogene 2018, 37, 5340–5354. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Yu, Y.; Li, Z.L.; Chen, M.Y.; Deng, R.; Huang, X.; Wang, G.F.; Zhang, M.X.; Yang, Q.; Ravichandran, S.; et al. XIAP Limits Autophagic Degradation of Sox2 and Is A Therapeutic Target in Nasopharyngeal Carcinoma Stem Cells. Theranostics 2018, 8, 1494. [Google Scholar] [CrossRef]
- Boumahdi, S.; Driessens, G.; Lapouge, G.; Rorive, S.; Nassar, D.; Le Mercier, M.; Delatte, B.; Caauwe, A.; Lenglez, S.; Nkusi, E.; et al. SOX2 Controls Tumor Initiation and Cancer Stem-Cell Functions in Squamous-Cell Carcinoma. Nature 2014, 511, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Huang, S.; Chen, S.; Chen, J.; Wang, Z.; Wang, Y.; Zheng, H. SOX2 Promotes Chemoresistance, Cancer Stem Cells Properties, and Epithelial–Mesenchymal Transition by β-Catenin and Beclin1/Autophagy Signaling in Colorectal Cancer. Cell Death Dis. 2021, 12, 449. [Google Scholar] [CrossRef] [PubMed]
- Stolzenburg, S.; Rots, M.G.; Beltran, A.S.; Rivenbark, A.G.; Yuan, X.; Qian, H.; Strahl, B.D.; Blancafort, P. Targeted Silencing of the Oncogenic Transcription Factor SOX2 in Breast Cancer. Nucleic Acids Res. 2012, 40, 6725. [Google Scholar] [CrossRef] [PubMed]
- Yokota, E.; Yamatsuji, T.; Takaoka, M.; Haisa, M.; Takigawa, N.; Miyake, N.; Ikeda, T.; Mori, T.; Ohno, S.; Sera, T.; et al. Targeted Silencing of SOX2 by an Artificial Transcription Factor Showed Antitumor Effect in Lung and Esophageal Squamous Cell Carcinoma. Oncotarget 2017, 8, 103063–103076. [Google Scholar] [CrossRef]
- Taniguchi, J.; Pandian, G.N.; Hidaka, T.; Hashiya, K.; Bando, T.; Kim, K.K.; Sugiyama, H. A Synthetic DNA-Binding Inhibitor of SOX2 Guides Human Induced Pluripotent Stem Cells to Differentiate into Mesoderm. Nucleic Acids Res. 2017, 45, 9219. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Wang, E.; Sorolla, A.; Kan, Y.J.; Malik, A.; Batra, J.; Young, K.A.; Tie, W.J.; Blancafort, P.; Mancera, R.L. Design and Characterization of a Cell-Penetrating Peptide Derived from the Sox2 Transcription Factor. Int. J. Mol. Sci. 2021, 22, 9354. [Google Scholar] [CrossRef]
- Schmitz, M.; Temme, A.; Senner, V.; Ebner, R.; Schwind, S.; Stevanovic, S.; Wehner, R.; Schackert, G.; Schackert, H.K.; Fussel, M.; et al. Identification of SOX2 as a Novel Glioma-Associated Antigen and Potential Target for T Cell-Based Immunotherapy. Br. J. Cancer 2007, 96, 1293–1301. [Google Scholar] [CrossRef]
- Favaro, R.; Appolloni, I.; Pellegatta, S.; Sanga, A.B.; Pagella, P.; Gambini, E.; Pisati, F.; Ottolenghi, S.; Foti, M.; Finocchiaro, G.; et al. Sox2 Is Required to Maintain Cancer Stem Cells in a Mouse Model of High-Grade Oligodendroglioma. Cancer Res. 2014, 74, 1833–1844. [Google Scholar] [CrossRef] [PubMed]
- Garros-Regulez, L.; Garcia, I.; Carrasco-Garcia, E.; Lantero, A.; Aldaz, P.; Moreno-Cugnon, L.; Arrizabalaga, O.; Undabeitia, J.; Torres-Bayona, S.; Villanua, J.; et al. Targeting SOX2 as a Therapeutic Strategy in Glioblastoma. Front. Oncol. 2016, 6, 225912. [Google Scholar] [CrossRef]
- Mohamed, M.A.; Elkhateeb, W.A.; Daba, G.M. Rapamycin Golden Jubilee and Still the Miraculous Drug: A Potent Immunosuppressant, Antitumor, Rejuvenative Agent, and Potential Contributor in COVID-19 Treatment. Bioresour. Bioprocess 2022, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Garros-Regulez, L.; Aldaz, P.; Arrizabalaga, O.; Moncho-Amor, V.; Carrasco-Garcia, E.; Manterola, L.; Moreno-Cugnon, L.; Barrena, C.; Villanua, J.; Ruiz, I.; et al. MTOR Inhibition Decreases SOX2-SOX9 Mediated Glioma Stem Cell Activity and Temozolomide Resistance. Expert Opin. Ther. Targets 2016, 20, 393–405. [Google Scholar] [CrossRef]
- Wang, Z.; Kang, L.; Zhang, H.; Huang, Y.; Fang, L.; Li, M.; Brown, P.J.; Arrowsmith, C.H.; Li, J.; Wong, J. AKT Drives SOX2 Overexpression and Cancer Cell Stemness in Esophageal Cancer by Protecting SOX2 from UBR5-Mediated Degradation. Oncogene 2019, 38, 5250–5264. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II Trial of AKT Inhibitor MK-2206 in Patients with Advanced Breast Cancer Who Have Tumors with PIK3CA or AKT Mutations, and/or PTEN Loss/PTEN Mutation. Breast Cancer Res. 2019, 21, 78. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Ji, J.; Deng, R.; Tang, J.; Yang, F.; Feng, G.K.; Chen, W.D.; Wu, X.Q.; Qian, X.J.; Ding, K.; et al. DC120, a Novel AKT Inhibitor, Preferentially Suppresses Nasopharyngeal Carcinoma Cancer Stem-like Cells by Downregulating Sox2. Oncotarget 2015, 6, 6944. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coombes, R.C.; Badman, P.D.; Lozano-Kuehne, J.P.; Liu, X.; Macpherson, I.R.; Zubairi, I.; Baird, R.D.; Rosenfeld, N.; Garcia-Corbacho, J.; Cresti, N.; et al. Results of the Phase IIa RADICAL Trial of the FGFR Inhibitor AZD4547 in Endocrine Resistant Breast Cancer. Nat. Commun. 2022, 13, 3246. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Trevino, J.; Bora-Singhal, N.; Coppola, D.; Haura, E.; Altiok, S.; Chellappan, S.P. EGFR/Src/Akt Signaling Modulates Sox2 Expression and Self-Renewal of Stem-like Side-Population Cells in Non-Small Cell Lung Cancer. Mol. Cancer 2012, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Pietrobono, S.; Morandi, A.; Gagliardi, S.; Gerlini, G.; Borgognoni, L.; Chiarugi, P.; Arbiser, J.L.; Stecca, B. Down-Regulation of SOX2 Underlies the Inhibitory Effects of the Triphenylmethane Gentian Violet on Melanoma Cell Self-Renewal and Survival. J. Investig. Dermatol. 2016, 136, 2059–2069. [Google Scholar] [CrossRef]
- Kim, B.R.; Coyaud, E.; Laurent, E.M.N.; St-Germain, J.; Van De Laar, E.; Tsao, M.S.; Raught, B.; Moghal, N. Identification of the SOX2 Interactome by BioID Reveals EP300 as a Mediator of SOX2-Dependent Squamous Differentiation and Lung Squamous Cell Carcinoma Growth. Mol. Cell. Proteom. 2017, 16, 1864–1888. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Xie, C.M.; Li, H.; Tan, M.; Chen, G.; Schiff, R.; Xiong, X.; Sun, Y. The FBXW2–MSX2–SOX2 Axis Regulates Stem Cell Property and Drug Resistance of Cancer Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 20528–20538. [Google Scholar] [CrossRef] [PubMed]
- Swords, R.T.; Erba, H.P.; DeAngelo, D.J.; Smith, P.G.; Pickard, M.D.; Dezube, B.J.; Giles, F.J.; Medeiros, B.C. The Novel, Investigational NEDD8-Activating Enzyme Inhibitor MLN4924 In Adult Patients with Acute Myeloid Leukemia (AML) or High-Grade Myelodysplastic Syndromes (MDS): A Phase 1 Study. Blood 2010, 116, 658. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Griffin, P.; Kelly, K.R.; Carew, J.S. MLN4924: A Novel First-in-Class Inhibitor of NEDD8-Activating Enzyme for Cancer Therapy. Expert Opin. Investig. Drugs 2012, 21, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Luo, Q.; Yan, X.; Yuan, L.; Yi, H.; Zhang, L.; Li, B.; Zhang, Y.; Sun, J.; Qiu, M.Z.; et al. A Novel SMAC Mimetic APG-1387 Exhibits Dual Antitumor Effect on HBV-Positive Hepatocellular Carcinoma with High Expression of CIAP2 by Inducing Apoptosis and Enhancing Innate Anti-Tumor Immunity. Biochem. Pharmacol. 2018, 154, 127–135. [Google Scholar] [CrossRef]
- Bora-Singhal, N.; Mohankumar, D.; Saha, B.; Colin, C.M.; Lee, J.Y.; Martin, M.W.; Zheng, X.; Coppola, D.; Chellappan, S. Novel HDAC11 Inhibitors Suppress Lung Adenocarcinoma Stem Cell Self-Renewal and Overcome Drug Resistance by Suppressing Sox2. Sci. Rep. 2020, 10, 4722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, F.; Wang, J.; Yin, F.; Xu, Z.; Qi, D.; Wu, X.; Cao, Y.; Liang, W.; Liu, Y.; et al. Pluripotent Stem Cell Protein Sox2 Confers Sensitivity to LSD1 Inhibition in Cancer Cells. Cell Rep. 2013, 5, 445–457. [Google Scholar] [CrossRef]
- Cuyàs, E.; Gumuzio, J.; Verdura, S.; Brunet, J.; Bosch-Barrera, J.; Martin-Castillo, B.; Alarcón, T.; Encinar, J.A.; Martin, Á.G.; Menendez, J.A. The LSD1 Inhibitor Iadademstat (ORY-1001) Targets SOX2-Driven Breast Cancer Stem Cells: A Potential Epigenetic Therapy in Luminal-B and HER2-Positive Breast Cancer Subtypes. Aging 2020, 12, 4794. [Google Scholar] [CrossRef]
- Somervaille, T.; Salamero, O.; Montesinos, P.; Willekens, C.; Perez Simon, J.A.; Pigneux, A.; Recher, C.; Popat, R.; Molinero, C.; Mascaro, C.; et al. Safety, Phamacokinetics (PK), Pharmacodynamics (PD) and Preliminary Activity in Acute Leukemia of Ory-1001, a First-in-Class Inhibitor of Lysine-Specific Histone Demethylase 1A (LSD1/KDM1A): Initial Results from a First-in-Human Phase 1 Study. Blood 2016, 128, 4060. [Google Scholar] [CrossRef]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef]
- Sari, I.N.; Phi, L.T.H.; Jun, N.; Wijaya, Y.T.; Lee, S.; Kwon, H.Y. Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells 2018, 7, 208. [Google Scholar] [CrossRef]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog Signaling and Bmi-1 Regulate Self-Renewal of Normal and Malignant Human Mammary Stem Cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef]
- Santini, R.; Vinci, M.C.; Pandolfi, S.; Penachioni, J.Y.; Montagnani, V.; Olivito, B.; Gattai, R.; Pimpinelli, N.; Gerlini, G.; Borgognoni, L.; et al. HEDGEHOG-GLI Signaling Drives Self-Renewal and Tumorigenicity of Human Melanoma-Initiating Cells. Stem Cells 2012, 30, 1808–1818. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog Signaling Is Essential for Maintenance of Cancer Stem Cells in Myeloid Leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Matsubara, S.; Ding, Q.; Tsukasa, K.; Yoshimitsu, M.; Kosai, K.I.; Takao, S. Efficient Elimination of Pancreatic Cancer Stem Cells by Hedgehog/GLI Inhibitor GANT61 in Combination with MTOR Inhibition. Mol. Cancer 2016, 15, 49. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Ray, H. Efficacy and Safety of Sonic Hedgehog Pathway Inhibitors in Cancer. Drug Saf. 2019, 42, 263. [Google Scholar] [CrossRef]
- Meiss, F.; Andrlová, H.; Zeiser, R. Vismodegib. Recent. Results Cancer Res. 2018, 211, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Fu, J.; Srivastava, R.K.; Shankar, S. Hedgehog Signaling Antagonist GDC-0449 (Vismodegib) Inhibits Pancreatic Cancer Stem Cell Characteristics: Molecular Mechanisms. PLoS ONE 2011, 6, e27306. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Basset-Seguin, N.; Garbe, C.; Gesierich, A.; Lao, C.D.; Miller, C.; Mortier, L.; Murrell, D.F.; Hamid, O.; et al. Long-Term Safety and Efficacy of Vismodegib in Patients with Advanced Basal Cell Carcinoma: Final Update of the Pivotal ERIVANCE BCC Study. BMC Cancer 2017, 17, 332. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the Hedgehog Pathway in Advanced Basal-Cell Carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172. [Google Scholar] [CrossRef]
- Kim, R.; Ji, J.H.; Kim, J.H.; Hong, J.Y.; Lim, H.Y.; Kang, W.K.; Lee, J.; Kim, S.T. Safety and Anti-Tumor Effects of Vismodegib in Patients with Refractory Advanced Gastric Cancer: A Single-Arm, Phase-II Trial. J. Cancer 2022, 13, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B.; Fehrenbacher, L.; Holloway, R.; Amit, A.; Karlan, B.; Slomovitz, B.; Sabbatini, P.; Fu, L.; Yauch, R.L.; Chang, I.; et al. A Phase II, Randomized, Placebo-Controlled Study of Vismodegib as Maintenance Therapy in Patients with Ovarian Cancer in Second or Third Complete Remission. Clin. Cancer Res. 2012, 18, 6509–6518. [Google Scholar] [CrossRef]
- De Jesus-Acosta, A.; Sugar, E.A.; O’Dwyer, P.J.; Ramanathan, R.K.; Von Hoff, D.D.; Rasheed, Z.; Zheng, L.; Begum, A.; Anders, R.; Maitra, A.; et al. Phase 2 Study of Vismodegib, a Hedgehog Inhibitor, Combined with Gemcitabine and Nab-Paclitaxel in Patients with Untreated Metastatic Pancreatic Adenocarcinoma. Br. J. Cancer 2020, 122, 498–505. [Google Scholar] [CrossRef]
- Burness, C.B. Sonidegib: First Global Approval. Drugs 2015, 75, 1559–1566. [Google Scholar] [CrossRef]
- Irvine, D.A.; Zhang, B.; Kinstrie, R.; Tarafdar, A.; Morrison, H.; Campbell, V.L.; Moka, H.A.; Ho, Y.; Nixon, C.; Manley, P.W.; et al. Deregulated Hedgehog Pathway Signaling Is Inhibited by the Smoothened Antagonist LDE225 (Sonidegib) in Chronic Phase Chronic Myeloid Leukaemia. Sci. Rep. 2016, 6, 25476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Vagiannis, D.; Budagaga, Y.; Sabet, Z.; Hanke, I.; Rozkoš, T.; Hofman, J. Sonidegib Potentiates the Cancer Cells’ Sensitivity to Cytostatic Agents by Functional Inhibition of ABCB1 and ABCG2 in Vitro and Ex Vivo. Biochem. Pharmacol. 2022, 199, 115009. [Google Scholar] [CrossRef]
- Dummer, R.; Guminksi, A.; Gutzmer, R.; Lear, J.T.; Lewis, K.D.; Chang, A.L.S.; Combemale, P.; Dirix, L.; Kaatz, M.; Kudchadkar, R.; et al. Long-term Efficacy and Safety of Sonidegib in Patients with Advanced Basal Cell Carcinoma: 42-month Analysis of the Phase II Randomized, Double-blind BOLT Study. Br. J. Dermatol. 2020, 182, 1369–1378. [Google Scholar] [CrossRef]
- Pietanza, M.C.; Litvak, A.M.; Varghese, A.M.; Krug, L.M.; Fleisher, M.; Teitcher, J.B.; Holodny, A.I.; Sima, C.S.; Woo, K.M.; Ng, K.K.; et al. A Phase I Trial of the Hedgehog Inhibitor, Sonidegib (LDE225), in Combination with Etoposide and Cisplatin for the Initial Treatment of Extensive Stage Small Cell Lung Cancer. Lung Cancer 2016, 99, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.L.; Wu, S.Z.; Roden, D.; Chan, C.L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting Stromal Remodeling and Cancer Stem Cell Plasticity Overcomes Chemoresistance in Triple Negative Breast Cancer. Nat. Commun. 2018, 9, 2897. [Google Scholar] [CrossRef]
- Munchhof, M.J.; Li, Q.; Shavnya, A.; Borzillo, G.V.; Boyden, T.L.; Jones, C.S.; Lagreca, S.D.; Martinez-Alsina, L.; Patel, N.; Pelletier, K.; et al. Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened. ACS Med. Chem. Lett. 2011, 3, 106–111. [Google Scholar] [CrossRef]
- Hoy, S.M. Glasdegib: First Global Approval. Drugs 2019, 79, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Heidel, F.H.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Candoni, A.; Leber, B.; Sekeres, M.A.; Pollyea, D.A.; et al. Survival Outcomes and Clinical Benefit in Patients with Acute Myeloid Leukemia Treated with Glasdegib and Low-Dose Cytarabine According to Response to Therapy. J. Hematol. Oncol. 2020, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.A.; Montesinos, P.; Novak, J.; Wang, J.; Jeyakumar, D.; Tomlinson, B.; Mayer, J.; Jou, E.; Robak, T.; Taussig, D.C.; et al. Glasdegib plus Intensive or Non-Intensive Chemotherapy for Untreated Acute Myeloid Leukemia: Results from the Randomized, Phase 3 BRIGHT AML 1019 Trial. Leukemia 2023, 37, 2017–2026. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog Signaling by Direct Binding of Cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743. [Google Scholar] [CrossRef]
- Che, J.; Zhang, F.Z.; Zhao, C.Q.; Hu, X.D.; Fan, S.J. Cyclopamine Is a Novel Hedgehog Signaling Inhibitor with Significant Anti-Proliferative, Anti-Invasive and Anti-Estrogenic Potency in Human Breast Cancer Cells. Oncol. Lett. 2013, 5, 1417. [Google Scholar] [CrossRef]
- Feldmann, G.; Fendrich, V.; McGovern, K.; Bedja, D.; Bisht, S.; Alvarez, H.; Koorstra, J.B.M.; Habbe, N.; Karikari, C.; Mullendore, M.; et al. An Orally Bioavailable Small-Molecule Inhibitor of Hedgehog Signaling Inhibits Tumor Initiation and Metastasis in Pancreatic Cancer. Mol. Cancer Ther. 2008, 7, 2725. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.R.; Lescarbeau, A.; Grogan, M.J.; Tan, E.; Lin, G.; Austad, B.C.; Yu, L.C.; Behnke, M.L.; Nair, S.J.; Hagel, M.; et al. Discovery of a Potent and Orally Active Hedgehog Pathway Antagonist (IPI-926). J. Med. Chem. 2009, 52, 4400–4418. [Google Scholar] [CrossRef]
- Campbell, V.T.; Nadesan, P.; Ali, S.A.; Wang, C.Y.Y.; Whetstone, H.; Poon, R.; Wei, Q.; Keilty, J.; Proctor, J.; Wang, L.W.; et al. Hedgehog Pathway Inhibition in Chondrosarcoma Using the Smoothened Inhibitor IPI-926 Directly Inhibits Sarcoma Cell Growth. Mol. Cancer Ther. 2014, 13, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.W.; Keysar, S.B.; Eagles, J.R.; Wang, G.; Glogowska, M.J.; McDermott, J.D.; Le, P.N.; Gao, D.; Ray, C.E.; Rochon, P.J.; et al. A Pilot Study of Cetuximab and the Hedgehog Inhibitor IPI-926 in Recurrent/Metastatic Head and Neck Squamous Cell Carcinoma. Oral. Oncol. 2016, 53, 74. [Google Scholar] [CrossRef]
- Jimeno, A.; Weiss, G.J.; Miller, W.H.; Gettinger, S.; Eigl, B.J.C.; Chang, A.L.S.; Dunbar, J.; Devens, S.; Faia, K.; Skliris, G.; et al. Phase I Study of the Hedgehog Pathway Inhibitor IPI-926 in Adult Patients with Solid Tumors. Clin. Cancer Res. 2013, 19, 2766–2774. [Google Scholar] [CrossRef]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kelley, R.K.; Lewis, S.; Chang, W.C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370. [Google Scholar] [CrossRef] [PubMed]
- van Maldegem, A.M.; Bovée, J.V.; Gelderblom, H. Comprehensive Analysis of Published Studies Involving Systemic Treatment for Chondrosarcoma of Bone between 2000 and 2013. Clin. Sarcoma Res. 2014, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.H.; Komatsu, Y.; Kelly, L.A.; Malhotra, U.; Rotoloni, C.; Kosovec, J.E.; Zahoor, H.; Makielski, R.; Bhatt, A.; Hoppo, T.; et al. Smoothened Inhibition Leads to Decreased Proliferation and Induces Apoptosis in Esophageal Adenocarcinoma Cells. Cancer Investig. 2013, 31, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Rocconi, R.; Meng, E.; Shevde, L.; Finan, M.; Reed, E. A Novel Hedgehog Pathway Smoothened Inhibitor (BMS-833923) Demonstrates in Vitro Synergy with Carboplatin in Ovarian Cancer Cells. Gynecol. Oncol. 2011, 120, S41–S42. [Google Scholar] [CrossRef]
- Arenas, A.; Lainez, D.; Serrano del Castillo, C.; Salgado, R.N.; Blas, C.; Atance, M.; Cordoba, R.; Llamas, P.; Alonso Dominguez, J.M. Inhibition of the Hedgehog Pathway Decreases the Quiescent CD34+CD38- Population in Acute Myeloid Leukemia. Blood 2018, 132 (Suppl. S1), 1509. [Google Scholar] [CrossRef]
- Shah, N.P.; Cortes, J.E.; Martinelli, G.; Smith, B.D.; Clarke, E.; Copland, M.; Strauss, L.; Talpaz, M. Dasatinib Plus Smoothened (SMO) Inhibitor BMS-833923 in Chronic Myeloid Leukemia (CML) with Resistance or Suboptimal Response to a Prior Tyrosine Kinase Inhibitor (TKI): Phase I Study CA180323. Blood 2014, 124, 4539. [Google Scholar] [CrossRef]
- Bender, M.H.; Hipskind, P.A.; Capen, A.R.; Cockman, M.; Credille, K.M.; Gao, H.; Bastian, J.A.; Clay, J.M.; Lobb, K.L.; Sall, D.J.; et al. Abstract 2819: Identification and Characterization of a Novel Smoothened Antagonist for the Treatment of Cancer with Deregulated Hedgehog Signaling. Cancer Res. 2011, 71 (Suppl. S8), 2819. [Google Scholar] [CrossRef]
- Bendell, J.; Andre, V.; Ho, A.; Kudchadkar, R.; Migden, M.; Infante, J.; Tiu, R.V.; Pitou, C.; Tucker, T.; Brail, L.; et al. Phase i Study of Ly2940680, a Smo Antagonist, in Patients with Advanced Cancer Including Treatment-Naïve and Previously Treated Basal Cell Carcinoma. Clin. Cancer Res. 2018, 24, 2082–2091. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, H.; Wu, M.; Wang, Q.; Luo, L.; Ma, H.; Zhang, X.; He, S. Discovery of a Potent Hedgehog Pathway Inhibitor Capable of Activating Caspase8-Dependent Apoptosis. J. Pharmacol. Sci. 2018, 137, 256–264. [Google Scholar] [CrossRef]
- Li, C.L.; Fang, Z.X.; Wu, Z.; Hou, Y.Y.; Wu, H.T.; Liu, J. Repurposed Itraconazole for Use in the Treatment of Malignancies as a Promising Therapeutic Strategy. Biomed. Pharmacother. 2022, 154, 113616. [Google Scholar] [CrossRef]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a Commonly Used Anti-Fungal That Inhibits Hedgehog Pathway Activity and Cancer Growth. Cancer Cell 2010, 17, 388. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Huang, L.; Liao, Z.; Liu, M.; Li, Q.; Xu, R. Itraconazole Inhibits the Hedgehog Signaling Pathway Thereby Inducing Autophagy-Mediated Apoptosis of Colon Cancer Cells. Cell Death Dis. 2020, 11, 539. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wei, S.; Zhao, Y.; Shi, C.; Liu, P.; Zhang, C.; Lei, Y.; Zhang, B.; Bai, B.; Huang, Y.; et al. Anti-Proliferation of Breast Cancer Cells with Itraconazole: Hedgehog Pathway Inhibition Induces Apoptosis and Autophagic Cell Death. Cancer Lett. 2017, 385, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Tsubamoto, H.; Sonoda, T.; Inoue, K. Impact of Itraconazole on the Survival of Heavily Pre-Treated Patients with Triple-Negative Breast Cancer. Anticancer Res. 2014, 34, 3839–3844. [Google Scholar]
- Inoue, K.; Tsubamoto, H.; Sonoda, T.; Ikuta, S.; Tani, S.; Yamanaka, N. Efficacy of Combination Chemotherapy with Itraconazole for Treating Metastatic Pancreatic Cancer in the Second-Line Setting. J. Clin. Oncol. 2015, 33 (Suppl. S15), e15255. [Google Scholar] [CrossRef]
- Tsubamoto, H.; Sonoda, T.; Inoue, K.; Ikuta, S.; Tani, S.; Yamanaka, N. Impact of Itraconazole after First-Line Chemotherapy on the Survival of Patients with Distant Metastatic Biliary Tract Cancer. J. Clin. Oncol. 2015, 33 (Suppl. S15), e15145. [Google Scholar] [CrossRef]
- Rudin, C.M.; Brahmer, J.R.; Juergens, R.A.; Hann, C.L.; Ettinger, D.S.; Sebree, R.; Smith, R.; Aftab, B.T.; Huang, P.; Liu, J.O. Phase 2 Study of Pemetrexed and Itraconazole as Second-Line Therapy for Metastatic Nonsquamous Non–Small-Cell Lung Cancer. J. Thorac. Oncol. 2013, 8, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Heath, E.I.; Smith, D.C.; Rathkopf, D.; Blackford, A.L.; Danila, D.C.; King, S.; Frost, A.; Ajiboye, A.S.; Zhao, M.; et al. Repurposing Itraconazole as a Treatment for Advanced Prostate Cancer: A Noncomparative Randomized Phase II Trial in Men with Metastatic Castration-Resistant Prostate Cancer. Oncologist 2013, 18, 163–173. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic Antagonizes the Hedgehog Pathway by Preventing Ciliary Accumulation and Reducing Stability of the Gli2 Transcriptional Effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef]
- Yang, D.; Cao, F.; Ye, X.; Zhao, H.; Liu, X.; Li, Y.; Shi, C.; Wang, H.; Zhou, J. Arsenic Trioxide Inhibits the Hedgehog Pathway Which Is Aberrantly Activated in Acute Promyelocytic Leukemia. Acta Haematol. 2013, 130, 260–267. [Google Scholar] [CrossRef]
- Chang, K.J.; Yin, J.Z.; Huang, H.; Li, B.; Yang, M.H. Arsenic Trioxide Inhibits the Growth of Cancer Stem Cells Derived from Small Cell Lung Cancer by Downregulating Stem Cell- Maintenance Factors and Inducing Apoptosis via the Hedgehog Signaling Blockade. Transl. Lung Cancer Res. 2020, 9, 1379–1396. [Google Scholar] [CrossRef]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Özdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic Trioxide Inhibits Human Cancer Cell Growth and Tumor Development in Mice by Blocking Hedgehog/GLI Pathway. J. Clin. Investig. 2011, 121, 148. [Google Scholar] [CrossRef] [PubMed]
- Bin Han, J.; Sang, F.; Chang, J.J.; Hua, Y.Q.; Shi, W.D.; Tang, L.H.; Liu, L. ming. Arsenic Trioxide Inhibits Viability of Pancreatic Cancer Stem Cells in Culture and in a Xenograft Model via Binding to SHH-Gli. Onco Targets Ther. 2013, 6, 1129. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergström, Å.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-Mediated Transcription and Tumor Cell Growth by Small-Molecule Antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed]
- Kiesslich, T.; Mayr, C.; Wachter, J.; Bach, D.; Fuereder, J.; Wagner, A.; Alinger, B.; Pichler, M.; Di Fazio, P.; Ocker, M.; et al. Activated Hedgehog Pathway Is a Potential Target for Pharmacological Intervention in Biliary Tract Cancer. Mol. Cell Biochem. 2014, 396, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Ohashi, R.; Naito, K.; Kanki, K. Hedgehog Signal Inhibitor GANT61 Inhibits the Malignant Behavior of Undifferentiated Hepatocellular Carcinoma Cells by Targeting Non-Canonical GLI Signaling. Int. J. Mol. Sci. 2020, 21, 3126. [Google Scholar] [CrossRef] [PubMed]
- Michaud, N.R.; Wang, Y.; McEachern, K.A.; Jordan, J.J.; Mazzola, A.M.; Hernandez, A.; Jalla, S.; Chesebrough, J.W.; Hynes, M.J.; Belmonte, M.A.; et al. Novel Neutralizing Hedgehog Antibody Medi-5304 Exhibits Antitumor Activity by Inhibiting Paracrine Hedgehog Signaling. Mol. Cancer Ther. 2014, 13, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Bausch, D.; Fritz, S.; Bolm, L.; Wellner, U.F.; Fernandez-del-Castillo, C.; Warshaw, A.L.; Thayer, S.P.; Liss, A.S. Hedgehog Signaling Promotes Angiogenesis Directly and Indirectly in Pancreatic Cancer. Angiogenesis 2020, 23, 479–492. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, S.A.; Machalek, D.A.; Shearer, R.F.; Millar, E.K.A.; Nair, R.; Schofield, P.; McLeod, D.; Cooper, C.L.; McNeil, C.M.; McFarland, A.; et al. Hedgehog Overexpression Is Associated with Stromal Interactions and Predicts for Poor Outcome in Breast Cancer. Cancer Res. 2011, 71, 4002–4014. [Google Scholar] [CrossRef]
- Bissey, P.A.; Mathot, P.; Guix, C.; Jasmin, M.; Goddard, I.; Costechareyre, C.; Gadot, N.; Delcros, J.G.; Mali, S.M.; Fasan, R.; et al. Blocking SHH/Patched Interaction Triggers Tumor Growth Inhibition through Patched-Induced Apoptosis. Cancer Res. 2020, 80, 1970–1980. [Google Scholar] [CrossRef]
- Nakamura, M.; Kubo, M.; Yanai, K.; Mikami, Y.; Ikebe, M.; Nagai, S.; Yamaguchi, K.; Tanaka, M.; Katano, M. Anti-Patched-1 Antibodies Suppress Hedgehog Signaling Pathway and Pancreatic Cancer Proliferation. Anticancer Res. 2007, 27, 3743–3747. [Google Scholar] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MacLean, M.R.; Walker, O.L.; Arun, R.P.; Fernando, W.; Marcato, P. Informed by Cancer Stem Cells of Solid Tumors: Advances in Treatments Targeting Tumor-Promoting Factors and Pathways. Int. J. Mol. Sci. 2024, 25, 4102. https://doi.org/10.3390/ijms25074102
MacLean MR, Walker OL, Arun RP, Fernando W, Marcato P. Informed by Cancer Stem Cells of Solid Tumors: Advances in Treatments Targeting Tumor-Promoting Factors and Pathways. International Journal of Molecular Sciences. 2024; 25(7):4102. https://doi.org/10.3390/ijms25074102
Chicago/Turabian StyleMacLean, Maya R., Olivia L. Walker, Raj Pranap Arun, Wasundara Fernando, and Paola Marcato. 2024. "Informed by Cancer Stem Cells of Solid Tumors: Advances in Treatments Targeting Tumor-Promoting Factors and Pathways" International Journal of Molecular Sciences 25, no. 7: 4102. https://doi.org/10.3390/ijms25074102
APA StyleMacLean, M. R., Walker, O. L., Arun, R. P., Fernando, W., & Marcato, P. (2024). Informed by Cancer Stem Cells of Solid Tumors: Advances in Treatments Targeting Tumor-Promoting Factors and Pathways. International Journal of Molecular Sciences, 25(7), 4102. https://doi.org/10.3390/ijms25074102