
The Golgi Apparatus: A Key Player in Innate Immunity

 ,
, {kind=link}
{kind=link}
Abstract
1. Introduction
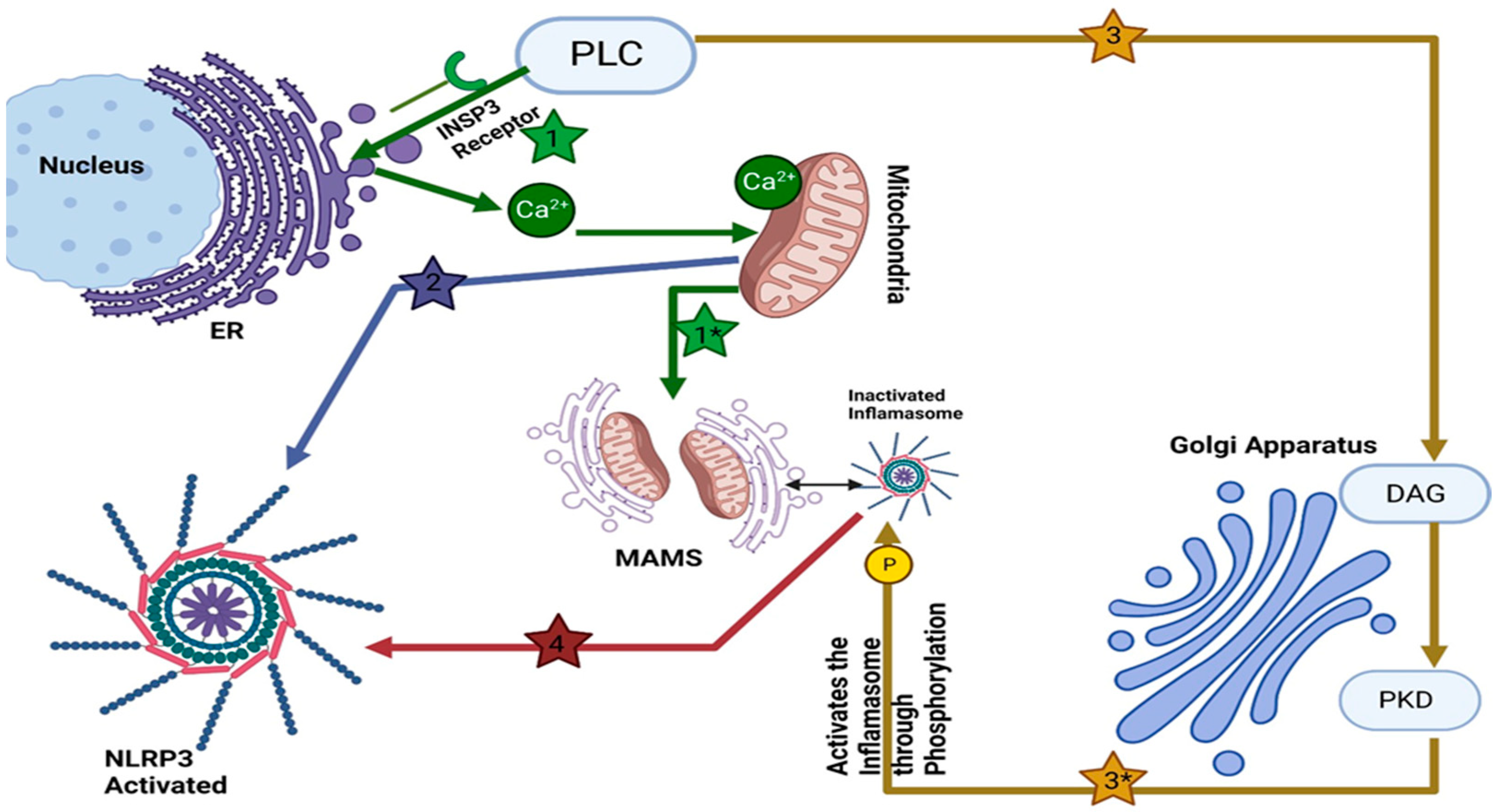
2. Golgi Apparatus and NLRP3 Inflammasome
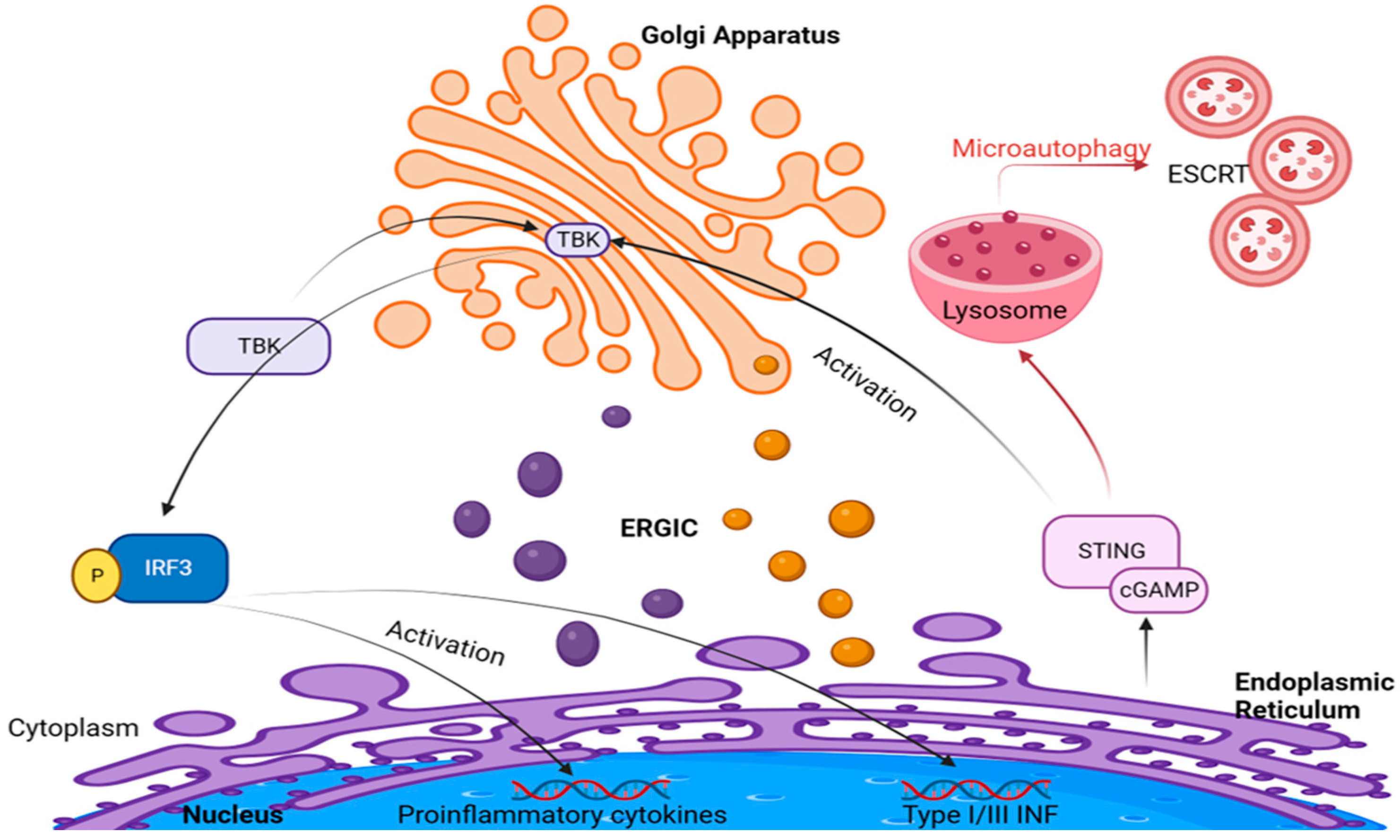
3. Golgi Apparatus and cGAS–STING Signaling
4. Golgi Apparatus and TLR/RLR Signaling
5. Golgi Apparatus and Microbial Infection
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Saceleanu, V.M.; Covache-Busuioc, R.-A.; Costin, H.-P.; Glavan, L.-A.; Ciurea, A.V. An Important Step in Neuroscience: Camillo Golgi and His Discoveries. Cells 2022, 11, 4112. [Google Scholar] [CrossRef]
- Shorter, J.; Warren, G. Golgi Architecture and Inheritance. Annu. Rev. Cell Dev. Biol. 2002, 18, 379–420. [Google Scholar] [CrossRef]
- Tang, D.; Wang, Y. Cell Cycle Regulation of Golgi Membrane Dynamics. Trends Cell Biol. 2013, 23, 296–304. [Google Scholar] [CrossRef]
- Boncompain, G.; Weigel, A.V. Transport and Sorting in the Golgi Complex: Multiple Mechanisms Sort Diverse Cargo. Curr. Opin. Cell Biol. 2018, 50, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, P.B.; Lee, J.E. Protein Trafficking Dysfunctions: Role in the Pathogenesis of Pulmonary Arterial Hypertension. Pulm. Circ. 2011, 1, 17–32. [Google Scholar] [CrossRef]
- Sehgal, P.B.; Mukhopadhyay, S.; Xu, F.; Patel, K.; Shah, M. Dysfunction of Golgi Tethers, SNAREs, and SNAPs in Monocrotaline-Induced Pulmonary Hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1526–L1542. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-M.; Lane, K.B.; Sehgal, P.B. Subcellular Mechanisms in Pulmonary Arterial Hypertension: Combinatorial Modalities That Inhibit Anterograde Trafficking and Cause Bone Morphogenetic Protein Receptor Type 2 Mislocalization. Pulm. Circ. 2013, 3, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Patel, K.; Almodóvar, S.; Tuder, R.M.; Flores, S.C.; Sehgal, P.B. Dependence of Golgi Apparatus Integrity on Nitric Oxide in Vascular Cells: Implications in Pulmonary Arterial Hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1141–H1158. [Google Scholar] [CrossRef]
- Lee, J.; Reich, R.; Xu, F.; Sehgal, P.B. Golgi, Trafficking, and Mitosis Dysfunctions in Pulmonary Arterial Endothelial Cells Exposed to Monocrotaline Pyrrole and NO Scavenging. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L715–L728. [Google Scholar] [CrossRef]
- Lee, J.E.; Yuan, H.; Liang, F.-X.; Sehgal, P.B. Nitric Oxide Scavenging Causes Remodeling of the Endoplasmic Reticulum, Golgi Apparatus and Mitochondria in Pulmonary Arterial Endothelial Cells. Nitric Oxide 2013, 33, 64–73. [Google Scholar] [CrossRef]
- Sehgal, P.B.; Mukhopadhyay, S. Dysfunctional Intracellular Trafficking in the Pathobiology of Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2007, 37, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, G.; D’Osualdo, A.; Schubert, D.A.; Weber, A.; Bruscia, E.M.; Hartl, D. Cellular Innate Immunity: An Old Game with New Players. J. Innate Immun. 2017, 9, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Chen, Y.; Wang, H.Y.; Wang, R.-F. Mechanisms and Pathways of Innate Immune Activation and Regulation in Health and Cancer. Hum. Vaccines Immunother. 2014, 10, 3270–3285. [Google Scholar] [CrossRef] [PubMed]
- Wicherska-Pawłowska, K.; Wróbel, T.; Rybka, J. Toll-Like Receptors (TLRs), NOD-Like Receptors (NLRs), and RIG-I-Like Receptors (RLRs) in Innate Immunity. TLRs, NLRs, and RLRs Ligands as Immunotherapeutic Agents for Hematopoietic Diseases. Int. J. Mol. Sci. 2021, 22, 13397. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.P.; Secord, E. Innate Immunity. Pediatr. Clin. N. Am. 2019, 66, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.J.; Xavier, R.J. Trained Immunity: A Program of Innate Immune Memory in Health and Disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining Trained Immunity and Its Role in Health and Disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Peignier, A.; Parker, D. Trained Immunity and Host-Pathogen Interactions. Cell. Microbiol. 2020, 22, e13261. [Google Scholar] [CrossRef]
- Murray, R.Z.; Stow, J.L. Cytokine Secretion in Macrophages: SNAREs, Rabs, and Membrane Trafficking. Front. Immunol. 2014, 5, 538. [Google Scholar] [CrossRef]
- Lieu, Z.Z.; Lock, J.G.; Hammond, L.A.; La Gruta, N.L.; Stow, J.L.; Gleeson, P.A. A trans-Golgi Network Golgin Is Required for the Regulated Secretion of TNF in Activated Macrophages In Vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 3351–3356. [Google Scholar] [CrossRef]
- Micaroni, M.; Stanley, A.C.; Khromykh, T.; Venturato, J.; Wong, C.X.F.; Lim, J.P.; Marsh, B.J.; Storrie, B.; Gleeson, P.A.; Stow, J.L. Rab6a/a’ Are Important Golgi Regulators of Pro-Inflammatory TNF Secretion in Macrophages. PLoS ONE 2013, 8, e57034. [Google Scholar] [CrossRef]
- Blank, U.; Madera-Salcedo, I.K.; Danelli, L.; Claver, J.; Tiwari, N.; Sánchez-Miranda, E.; Vázquez-Victorio, G.; Ramírez-Valadez, K.A.; Macias-Silva, M.; González-Espinosa, C. Vesicular Trafficking and Signaling for Cytokine and Chemokine Secretion in Mast Cells. Front. Immunol. 2014, 5, 453. [Google Scholar] [CrossRef]
- Low, P.C.; Misaki, R.; Schroder, K.; Stanley, A.C.; Sweet, M.J.; Teasdale, R.D.; Vanhaesebroeck, B.; Meunier, F.A.; Taguchi, T.; Stow, J.L. Phosphoinositide 3-Kinase δ Regulates Membrane Fission of Golgi Carriers for Selective Cytokine Secretion. J. Cell Biol. 2010, 190, 1053–1065. [Google Scholar] [CrossRef]
- Stanley, A.C.; Lieu, Z.Z.; Wall, A.A.; Venturato, J.; Khromykh, T.; Hamilton, N.A.; Gleeson, P.A.; Stow, J.L. Recycling Endosome-Dependent and -Independent Mechanisms for IL-10 Secretion in LPS-Activated Macrophages. J. Leukoc. Biol. 2012, 92, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Manderson, A.P.; Kay, J.G.; Hammond, L.A.; Brown, D.L.; Stow, J.L. Subcompartments of the Macrophage Recycling Endosome Direct the Differential Secretion of IL-6 and TNFalpha. J. Cell Biol. 2007, 178, 57–69. [Google Scholar] [CrossRef]
- Lacy, P.; Stow, J.L. Cytokine Release from Innate Immune Cells: Association with Diverse Membrane Trafficking Pathways. Blood 2011, 118, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5, 117833. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, Z.J. PtdIns4P on Dispersed Trans-Golgi Network Mediates NLRP3 Inflammasome Activation. Nature 2018, 564, 71–76. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-Sensing Receptors in Sterile Inflammation and Inflammatory Diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Dobbs, N.; Burnaevskiy, N.; Chen, D.; Gonugunta, V.K.; Alto, N.M.; Yan, N. STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe 2015, 18, 157–168. [Google Scholar] [CrossRef]
- Mukai, K.; Konno, H.; Akiba, T.; Uemura, T.; Waguri, S.; Kobayashi, T.; Barber, G.N.; Arai, H.; Taguchi, T. Activation of STING Requires Palmitoylation at the Golgi. Nat. Commun. 2016, 7, 11932. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Brinkmann, M.M.; Spooner, E.; Hoebe, K.; Beutler, B.; Ploegh, H.L.; Kim, Y.-M. The Interaction between the ER Membrane Protein UNC93B and TLR3, 7, and 9 Is Crucial for TLR Signaling. J. Cell Biol. 2007, 177, 265–275. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Brinkmann, M.M.; Paquet, M.-E.; Ploegh, H.L. UNC93B1 Delivers Nucleotide-Sensing Toll-like Receptors to Endolysosomes. Nature 2008, 452, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Meszaros, G.; He, W.; Xu, Y.; De Fatima Magliarelli, H.; Mailly, L.; Mihlan, M.; Liu, Y.; Puig Gámez, M.; Goginashvili, A.; et al. Protein Kinase D at the Golgi Controls NLRP3 Inflammasome Activation. J. Exp. Med. 2017, 214, 2671–2693. [Google Scholar] [CrossRef] [PubMed]
- Wakana, Y.; Campelo, F. The PKD-Dependent Biogenesis of TGN-to-Plasma Membrane Transport Carriers. Cells 2021, 10, 1618. [Google Scholar] [CrossRef]
- Malhotra, V.; Campelo, F. PKD Regulates Membrane Fission to Generate TGN to Cell Surface Transport Carriers. Cold Spring Harb. Perspect. Biol. 2011, 3, a005280. [Google Scholar] [CrossRef]
- Gutiérrez-Galindo, E.; Yilmaz, Z.H.; Hausser, A. Membrane Trafficking in Breast Cancer Progression: Protein Kinase D Comes into Play. Front. Cell Dev. Biol. 2023, 11, 1173387. [Google Scholar] [CrossRef]
- Guo, C.; Chi, Z.; Jiang, D.; Xu, T.; Yu, W.; Wang, Z.; Chen, S.; Zhang, L.; Liu, Q.; Guo, X.; et al. Cholesterol Homeostatic Regulator SCAP-SREBP2 Integrates NLRP3 Inflammasome Activation and Cholesterol Biosynthetic Signaling in Macrophages. Immunity 2018, 49, 842–856.e7. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory Caspases Are Innate Immune Receptors for Intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.-K.; Kim, J.K.; Shin, D.-M.; Sasakawa, C. Molecular Mechanisms Regulating NLRP3 Inflammasome Activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef]
- Di, A.; Xiong, S.; Ye, Z.; Malireddi, R.K.S.; Kometani, S.; Zhong, M.; Mittal, M.; Hong, Z.; Kanneganti, T.-D.; Rehman, J.; et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity 2018, 49, 56–65.e4. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The Adaptor MAVS Promotes NLRP3 Mitochondrial Localization and Inflammasome Activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.-W.; Alnemri, E.S. The Mitochondrial Antiviral Protein MAVS Associates with NLRP3 and Regulates Its Inflammasome Activity. J. Immunol. 2013, 191, 4358–4366. [Google Scholar] [CrossRef]
- Liu, J.; Huang, Y.; Li, T.; Jiang, Z.; Zeng, L.; Hu, Z. The Role of the Golgi Apparatus in Disease (Review). Int. J. Mol. Med. 2021, 47, 38. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Taguchi, T.; Mukai, K.; Takaya, E.; Shindo, R. STING Operation at the ER/Golgi Interface. Front. Immunol. 2021, 12, 646304. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.; Jiang, W.; Hao, J. Research Advances in How the cGAS-STING Pathway Controls the Cellular Inflammatory Response. Front. Immunol. 2020, 11, 615. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal Roles of cGAS-cGAMP Signaling in Antiviral Defense and Immune Adjuvant Effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, F.; Cao, Y.; Dang, Y.; Ge, B. The Multifaceted Functions of cGAS. J. Mol. Cell Biol. 2022, 14, mjac031. [Google Scholar] [CrossRef] [PubMed]
- Volkman, H.E.; Cambier, S.; Gray, E.E.; Stetson, D.B. Tight Nuclear Tethering of cGAS Is Essential for Preventing Autoreactivity. eLife 2019, 8, e47491. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Liu, P. Cytosolic DNA Sensing by cGAS: Regulation, Function, and Human Diseases. Signal Transduct. Target Ther. 2021, 6, 170. [Google Scholar] [CrossRef] [PubMed]
- Civril, F.; Deimling, T.; de Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.-P. Structural Mechanism of Cytosolic DNA Sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef]
- Kranzusch, P.J.; Vance, R.E. cGAS Dimerization Entangles DNA Recognition. Immunity 2013, 39, 992–994. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Chen, Z.J. DNA-Induced Liquid Phase Condensation of cGAS Activates Innate Immune Signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef]
- Luecke, S.; Holleufer, A.; Christensen, M.H.; Jønsson, K.L.; Boni, G.A.; Sørensen, L.K.; Johannsen, M.; Jakobsen, M.R.; Hartmann, R.; Paludan, S.R. cGAS Is Activated by DNA in a Length-Dependent Manner. EMBO Rep. 2017, 18, 1707–1715. [Google Scholar] [CrossRef]
- Xiao, Q.; McAtee, C.K.; Su, X. Phase Separation in Immune Signaling. Nat. Rev. Immunol. 2022, 22, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, E.-C.; He, Y.; Chai, Z.-L.; Ji, B.-Z.; Tu, Y.; Wang, H.-L.; Wu, W.-Q.; Liu, Y.; Zhang, X.-H.; et al. ZYG11B Potentiates the Antiviral Innate Immune Response by Enhancing cGAS-DNA Binding and Condensation. Cell Rep. 2023, 42, 112278. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING Pathway Connects DNA Damage to Inflammation, Senescence, and Cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Fu, Y.; Zhou, X.; Du, H.; Chen, Q. Cyclic Dinucleotides Mediate Bacterial Immunity by Dinucleotide Cyclase in Vibrio. Front. Microbiol. 2022, 13, 1065945. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, S.; Woo, J.S.; Wu, B.; El-Sherbiny, Y.M.; Leung, J.; Chupradit, K.; Rice, L.; Seo, G.J.; Calmettes, G.; Ramakrishna, C.; et al. The Ca2+ Sensor STIM1 Regulates the Type I Interferon Response by Retaining the Signaling Adaptor STING at the Endoplasmic Reticulum. Nat. Immunol. 2019, 20, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Son, A.; de Jesus, A.A.; Schwartz, D.M. STIM1 Holds a STING in Its (N-Terminal) Tail. Cell Calcium 2019, 80, 192–193. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS Produces a 2’-5’-Linked Cyclic Dinucleotide Second Messenger That Activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy Induction via STING Trafficking Is a Primordial Function of the cGAS Pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. Atg9a Controls dsDNA-Driven Dynamic Translocation of STING and the Innate Immune Response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846. [Google Scholar] [CrossRef]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with Covalent Small-Molecule Inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef]
- Fang, R.; Jiang, Q.; Guan, Y.; Gao, P.; Zhang, R.; Zhao, Z.; Jiang, Z. Golgi Apparatus-Synthesized Sulfated Glycosaminoglycans Mediate Polymerization and Activation of the cGAMP Sensor STING. Immunity 2021, 54, 962–975.e8. [Google Scholar] [CrossRef] [PubMed]
- Yum, S.; Li, M.; Fang, Y.; Chen, Z.J. TBK1 Recruitment to STING Activates Both IRF3 and NF-κB That Mediate Immune Defense against Tumors and Viral Infections. Proc. Natl. Acad. Sci. USA 2021, 118, e2100225118. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-κB Activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- Kuchitsu, Y.; Mukai, K.; Uematsu, R.; Takaada, Y.; Shinojima, A.; Shindo, R.; Shoji, T.; Hamano, S.; Ogawa, E.; Sato, R.; et al. STING Signalling Is Terminated through ESCRT-Dependent Microautophagy of Vesicles Originating from Recycling Endosomes. Nat. Cell Biol. 2023, 25, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Uhlorn, B.L.; Gamez, E.R.; Li, S.; Campos, S.K. Attenuation of cGAS/STING Activity during Mitosis. Life Sci. Alliance 2020, 3, e201900636. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Rong, M.; Lv, Y.; Zhu, D.; Xiang, Y. Regulation of cGAS Activity by RNA-Modulated Phase Separation. EMBO Rep. 2023, 24, e51800. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Ortiz Serrano, T.P.; Davis, J.; Prigge, A.D.; Ridge, K.M. The cGAS-STING Pathway: The Role of Self-DNA Sensing in Inflammatory Lung Disease. FASEB J. 2020, 34, 13156–13170. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, F.; Zhang, X. STING-Associated Vasculopathy with Onset in Infancy: A Familial Case Series Report and Literature Review. Ann. Transl. Med. 2021, 9, 176. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Q.; Wang, N.; Li, S.; Bian, W.; Sun, Z.; Wang, L.; Wang, L.; Liu, C.; Song, C.; et al. Oleic Acid Dissolves cGAS–DNA Phase Separation to Inhibit Immune Surveillance. Adv. Sci. 2023, 10, 2206820. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Cao, A.; Luo, Q.; Chen, D.; Zhao, W.; Xu, J.; Li, Q.; Bu, X.; Quan, J. Development of Cyclopeptide Inhibitors of cGAS Targeting Protein-DNA Interaction and Phase Separation. Nat. Commun. 2023, 14, 6132. [Google Scholar] [CrossRef]
- Gack, M.U. Mechanisms of RIG-I-Like Receptor Activation and Manipulation by Viral Pathogens. J. Virol. 2014, 88, 5213–5216. [Google Scholar] [CrossRef] [PubMed]
- Thoresen, D.; Wang, W.; Galls, D.; Guo, R.; Xu, L.; Pyle, A.M. The Molecular Mechanism of RIG-I Activation and Signaling. Immunol. Rev. 2021, 304, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Pourcelot, M.; Zemirli, N.; Silva Da Costa, L.; Loyant, R.; Garcin, D.; Vitour, D.; Munitic, I.; Vazquez, A.; Arnoult, D. The Golgi Apparatus Acts as a Platform for TBK1 Activation after Viral RNA Sensing. BMC Biol. 2016, 14, 69. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.T.; Gale, M.; Loo, Y.-M. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Chen, Z.J. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci. Signal. 2012, 5, ra20. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Wu, T.; Wang, Y.; Kang, Y.; Shan, Q.; Xu, L.; Jiang, Z.; Lin, X.; Ye, X.-Y.; Xie, T.; et al. 5-Hydroxymethylfurfural Enhances the Antiviral Immune Response in Macrophages through the Modulation of RIG-I-Mediated Interferon Production and the JAK/STAT Signaling Pathway. ACS Omega 2021, 6, 28019–28030. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, G.; Saha, S.K.; Guo, B.; He, J.Q.; Shahangian, A.; Zarnegar, B.; Perry, A.; Cheng, G. Critical Role of TRAF3 in the Toll-like Receptor-Dependent and -Independent Antiviral Response. Nature 2006, 439, 208–211. [Google Scholar] [CrossRef]
- van Zuylen, W.J.; Doyon, P.; Clément, J.-F.; Khan, K.A.; D’Ambrosio, L.M.; Dô, F.; St-Amant-Verret, M.; Wissanji, T.; Emery, G.; Gingras, A.-C.; et al. Proteomic Profiling of the TRAF3 Interactome Network Reveals a New Role for the ER-to-Golgi Transport Compartments in Innate Immunity. PLoS Pathog. 2012, 8, e1002747. [Google Scholar] [CrossRef]
- Saha, S.K.; Pietras, E.M.; He, J.Q.; Kang, J.R.; Liu, S.-Y.; Oganesyan, G.; Shahangian, A.; Zarnegar, B.; Shiba, T.L.; Wang, Y.; et al. Regulation of Antiviral Responses by a Direct and Specific Interaction between TRAF3 and Cardif. EMBO J. 2006, 25, 3257–3263. [Google Scholar] [CrossRef]
- Michallet, M.-C.; Meylan, E.; Ermolaeva, M.A.; Vazquez, J.; Rebsamen, M.; Curran, J.; Poeck, H.; Bscheider, M.; Hartmann, G.; König, M.; et al. TRADD Protein Is an Essential Component of the RIG-like Helicase Antiviral Pathway. Immunity 2008, 28, 651–661. [Google Scholar] [CrossRef]
- Pietras, E.M.; Cheng, G. A New TRADDition in Intracellular Antiviral Signaling. Sci. Signal. 2008, 1, pe36. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, D.; Glick, B.S. Two Mammalian Sec16 Homologues Have Nonredundant Functions in Endoplasmic Reticulum (ER) Export and Transitional ER Organization. Mol. Biol. Cell 2007, 18, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Iinuma, T.; Shiga, A.; Nakamoto, K.; O’Brien, M.B.; Aridor, M.; Arimitsu, N.; Tagaya, M.; Tani, K. Mammalian Sec16/P250 Plays a Role in Membrane Traffic from the Endoplasmic Reticulum. J. Biol. Chem. 2007, 282, 17632–17639. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.; Townley, A.K.; Koka, P.; Palmer, K.J.; Stephens, D.J. Sec16 Defines Endoplasmic Reticulum Exit Sites and Is Required for Secretory Cargo Export in Mammalian Cells. Traffic 2006, 7, 1678–1687. [Google Scholar] [CrossRef] [PubMed]
- Allan, B.B.; Moyer, B.D.; Balch, W.E. Rab1 Recruitment of P115 into a Cis-SNARE Complex: Programming Budding COPII Vesicles for Fusion. Science 2000, 289, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Lowe, M.; Levine, T.P.; Rabouille, C.; Warren, G. The Vesicle Docking Protein P115 Binds GM130, a Cis-Golgi Matrix Protein, in a Mitotically Regulated Manner. Cell 1997, 89, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.; VanScoy, S.; Cheng, T.-F.; Gomez, D.; Reich, N.C. IRF-3-Dependent and Augmented Target Genes during Viral Infection. Genes Immun. 2008, 9, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Wilkins, C.; Badil, S.; Iskarpatyoti, J.; Gale, M. Proteomic Analysis of Mitochondrial-Associated ER Membranes (MAM) during RNA Virus Infection Reveals Dynamic Changes in Protein and Organelle Trafficking. PLoS ONE 2015, 10, e0117963. [Google Scholar] [CrossRef] [PubMed]
- Beachboard, D.C.; Park, M.; Vijayan, M.; Snider, D.L.; Fernando, D.J.; Williams, G.D.; Stanley, S.; McFadden, M.J.; Horner, S.M. The Small GTPase RAB1B Promotes Antiviral Innate Immunity by Interacting with TNF Receptor–Associated Factor 3 (TRAF3). J. Biol. Chem. 2019, 294, 14231–14240. [Google Scholar] [CrossRef]
- Alvarez, C.; Garcia-Mata, R.; Brandon, E.; Sztul, E. COPI Recruitment Is Modulated by a Rab1b-Dependent Mechanism. Mol. Biol. Cell 2003, 14, 2116–2127. [Google Scholar] [CrossRef]
- Gao, J.; Gao, A.; Liu, W.; Chen, L. Golgi Stress Response: A Regulatory Mechanism of Golgi Function. Biofactors 2021, 47, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.K.; Choi, W.; Deshar, B.; Kang, S.; Kim, J. Golgi Stress Response: New Insights into the Pathogenesis and Therapeutic Targets of Human Diseases. Mol. Cells 2023, 46, 191. [Google Scholar] [CrossRef] [PubMed]
- Mousnier, A.; Swieboda, D.; Pinto, A.; Guedán, A.; Rogers, A.V.; Walton, R.; Johnston, S.L.; Solari, R. Human Rhinovirus 16 Causes Golgi Apparatus Fragmentation without Blocking Protein Secretion. J. Virol. 2014, 88, 11671–11685. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, M.; Matsuo, H.; Koito, T.; Murata, M.; Kubori, T.; Nagai, H.; Tagaya, M.; Arasaki, K. Legionella Hijacks the Host Golgi-to-ER Retrograde Pathway for the Association of Legionella-Containing Vacuole with the ER. PLoS Pathog. 2021, 17, e1009437. [Google Scholar] [CrossRef] [PubMed]
- Gatselis, N.K.; Tornai, T.; Shums, Z.; Zachou, K.; Saitis, A.; Gabeta, S.; Albesa, R.; Norman, G.L.; Papp, M.; Dalekos, G.N. Golgi Protein-73: A Biomarker for Assessing Cirrhosis and Prognosis of Liver Disease Patients. World J. Gastroenterol. 2020, 26, 5130–5145. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-Y.; Huang, L.; Wu, J.-F.; Zhang, H.-B.; Ai, W.-B.; Zhang, R.-T. Possible Roles of Golgi Protein-73 in Liver Diseases. Ann. Hepatol. 2022, 27, 100720. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, Y.; Shen, M.; Xu, H.; Li, Z.; He, N. Golgi Protein 73 and Its Diagnostic Value in Liver Diseases. Cell Prolif. 2018, 52, e12538. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mărunţelu, I.; Constantinescu, A.-E.; Covache-Busuioc, R.-A.; Constantinescu, I. The Golgi Apparatus: A Key Player in Innate Immunity. Int. J. Mol. Sci. 2024, 25, 4120. https://doi.org/10.3390/ijms25074120
Mărunţelu I, Constantinescu A-E, Covache-Busuioc R-A, Constantinescu I. The Golgi Apparatus: A Key Player in Innate Immunity. International Journal of Molecular Sciences. 2024; 25(7):4120. https://doi.org/10.3390/ijms25074120
Chicago/Turabian StyleMărunţelu, Ion, Alexandra-Elena Constantinescu, Razvan-Adrian Covache-Busuioc, and Ileana Constantinescu. 2024. "The Golgi Apparatus: A Key Player in Innate Immunity" International Journal of Molecular Sciences 25, no. 7: 4120. https://doi.org/10.3390/ijms25074120
APA StyleMărunţelu, I., Constantinescu, A.-E., Covache-Busuioc, R.-A., & Constantinescu, I. (2024). The Golgi Apparatus: A Key Player in Innate Immunity. International Journal of Molecular Sciences, 25(7), 4120. https://doi.org/10.3390/ijms25074120