
The Impact of Cancer Stem Cells in Colorectal Cancer
 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Cancer Stem Cells
Colorectal Cancer Stem Cells
3. Isolation and Identification of CCSCs
3.1. Utilizing Surface Markers
3.2. Isolation within In Vitro Cultures
3.3. CCSC Isolation via Biophysical Characteristics
4. CCSC Signaling Pathways
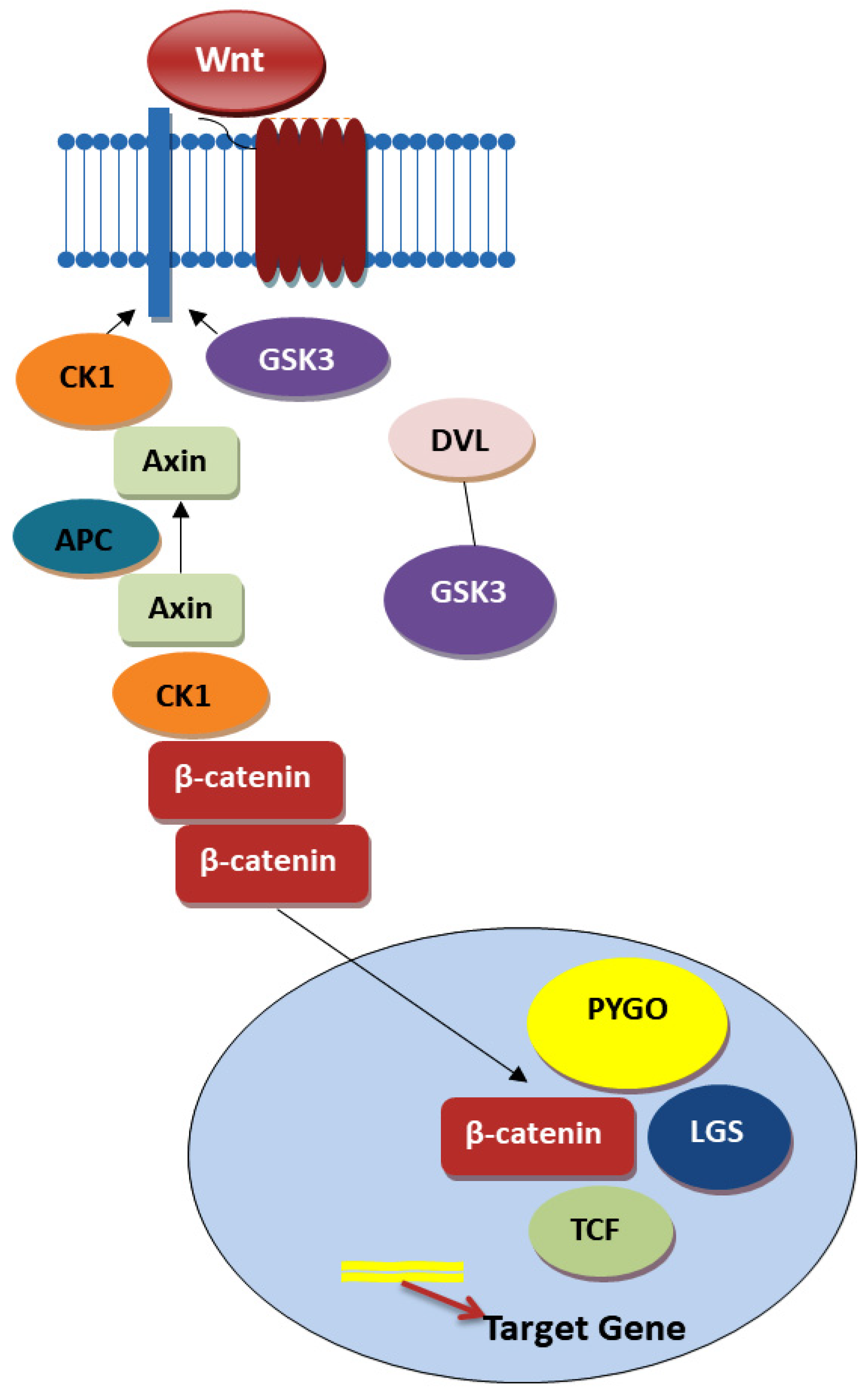
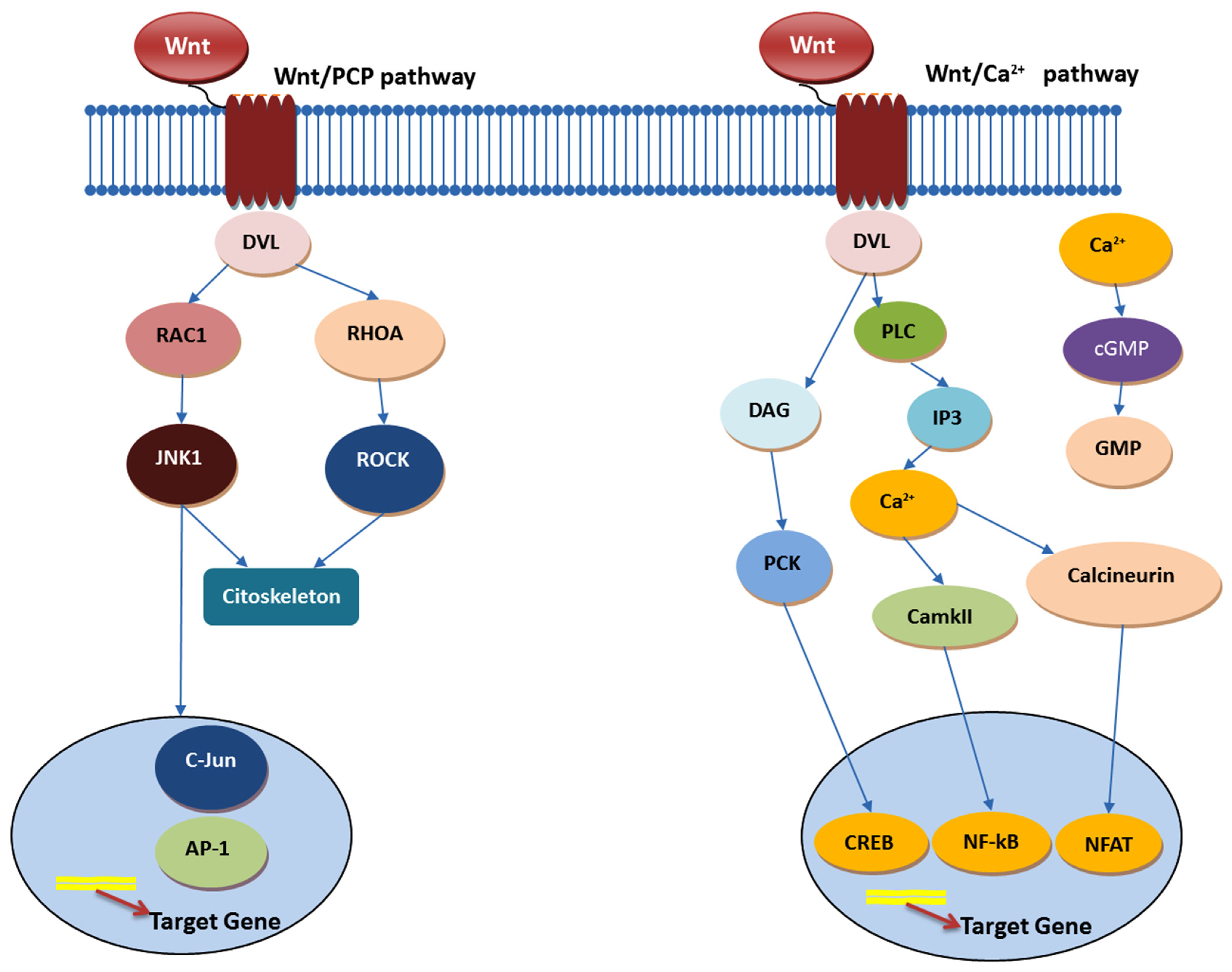
4.1. Wnt Signaling Pathway
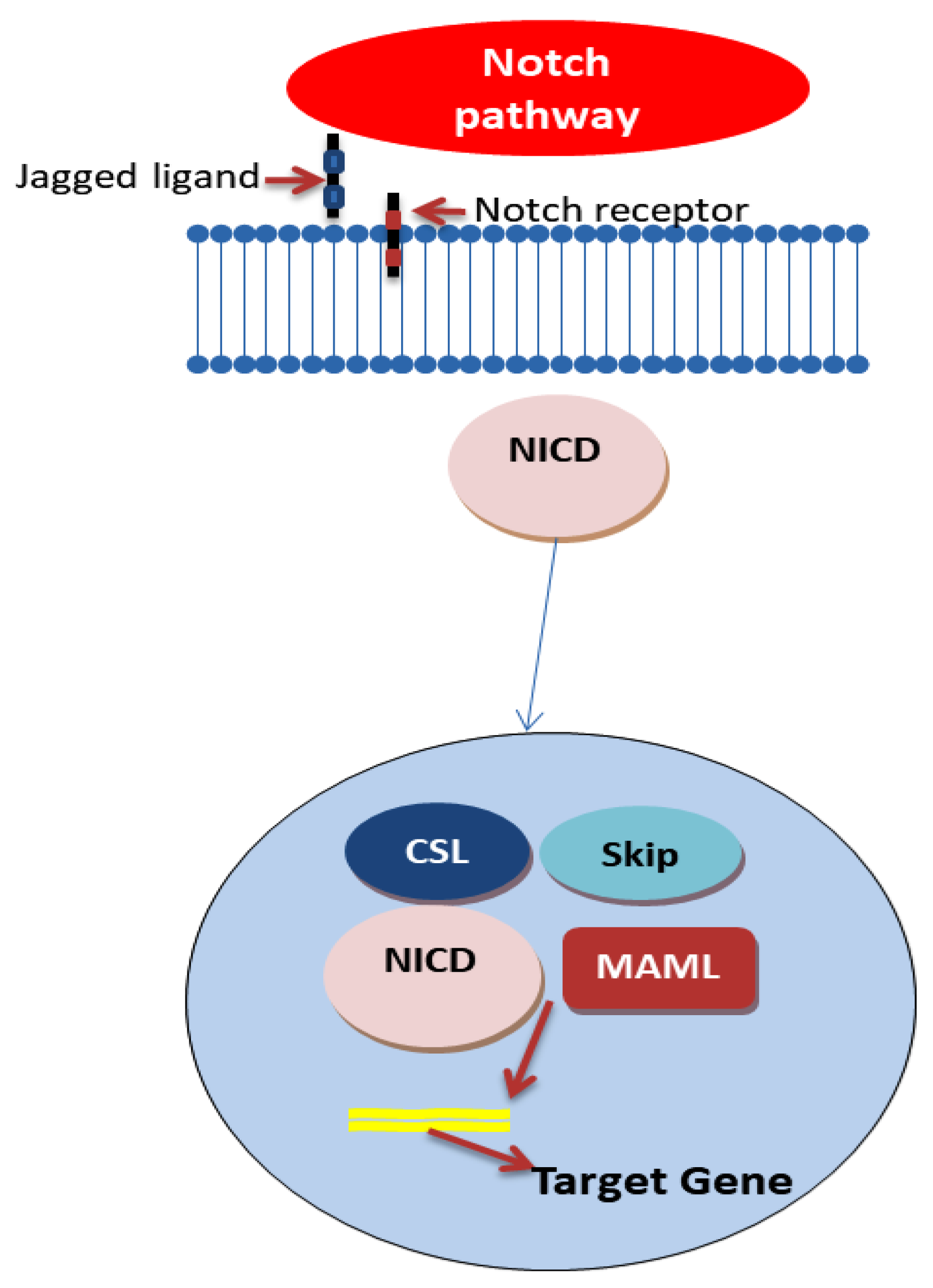
4.2. Notch Signaling Pathway

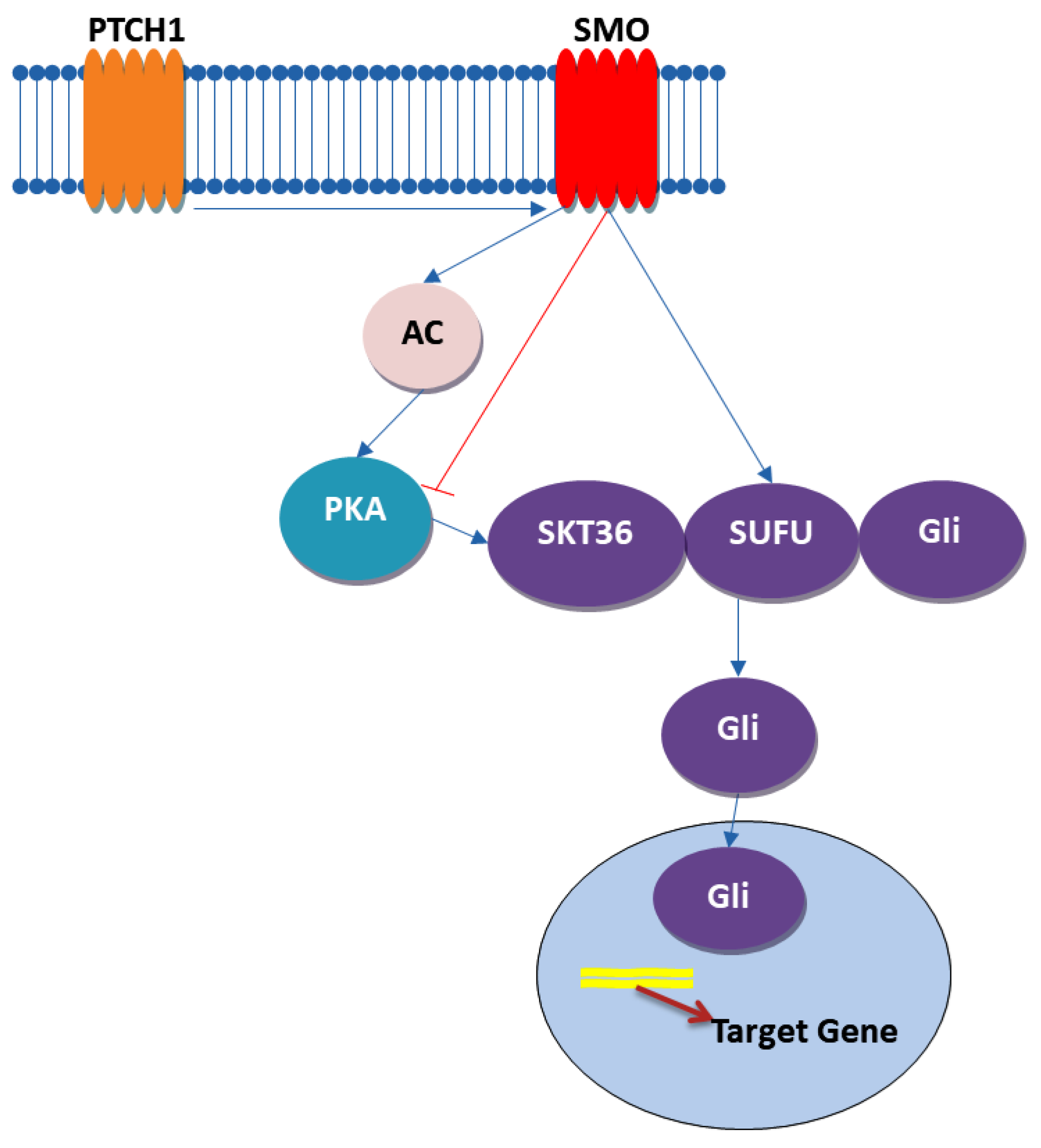
4.3. Hedgehog Signaling Pathway

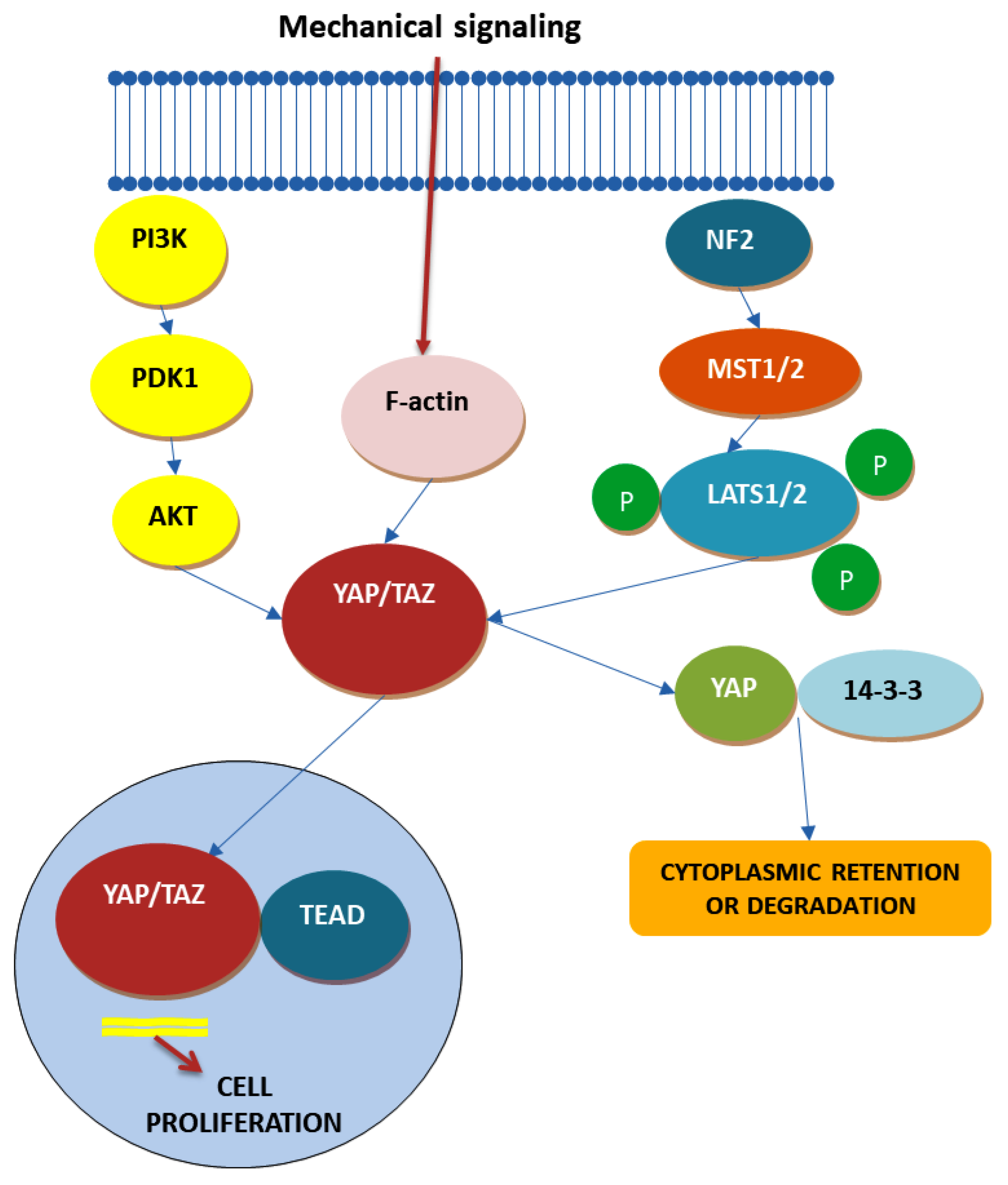
4.4. Hippo Signaling Pathway

5. CCSC and Tumor Microenvironment
6. CCSC and Drug Resistance
7. Therapeutic Strategies against CCSCs to Overcome Therapy Resistance
8. Conclusions
9. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K. Colorectal Cancers: An Update on Their Molecular Pathology. Cancers 2018, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Punt, C.J.; Koopman, M.; Vermeulen, L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Suyama, K.; Baba, H. Recent Advances in Targeting the EGFR Signaling Pathway for the Treatment of Metastatic Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 752. [Google Scholar] [CrossRef] [PubMed]
- Hervieu, C.; Christou, N.; Battu, S.; Mathonnet, M. The Role of Cancer Stem Cells in Colorectal Cancer: From the Basics to Novel Clinical Trials. Cancers 2021, 13, 92. [Google Scholar] [CrossRef]
- Lea, D.; Haland, S.; Hagland, H.R.; Soreide, K. Accuracy of TNM staging in colorectal cancer: A review of current culprits, the modern role of morphology and stepping-stones for improvements in the molecular era. Scand. J. Gastroenterol. 2014, 49, 1153–1163. [Google Scholar] [CrossRef]
- Printz, C. New AJCC cancer staging manual reflects changes in cancer knowledge. Cancer 2010, 116, 2–3. [Google Scholar] [CrossRef]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Wu, C.; Goldberg, R.M. Recent therapeutic advances in the treatment of colorectal cancer. Annu. Rev. Med. 2015, 66, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Shinji, S.; Yamada, T.; Matsuda, A.; Sonoda, H.; Ohta, R.; Iwai, T.; Takeda, K.; Yonaga, K.; Masuda, Y.; Yoshida, H. Recent Advances in the Treatment of Colorectal Cancer: A Review. J. Nippon Med. Sch. 2022, 89, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Marincas, M.; Cirimbei, C.; Prunoiu, V.; Eliescu, A.L.; Buzatu, R.; Stefan, I.; Bratucu, E.; Murarasu, D.; Puiu, L.; Mihalcea, C. Postradiotherapy regression—A prognostic factor in rectal neoplasm. Chirurgia 2011, 106, 753–758. [Google Scholar] [PubMed]
- Douaiher, J.; Ravipati, A.; Grams, B.; Chowdhury, S.; Alatise, O.; Are, C. Colorectal cancer-global burden, trends, and geographical variations. J. Surg. Oncol. 2017, 115, 619–630. [Google Scholar] [CrossRef]
- Sawicki, T.; Ruszkowska, M.; Danielewicz, A.; Niedzwiedzka, E.; Arlukowicz, T.; Przybylowicz, K.E. A Review of Colorectal Cancer in Terms of Epidemiology, Risk Factors, Development, Symptoms and Diagnosis. Cancers 2021, 13, 2025. [Google Scholar] [CrossRef] [PubMed]
- Baidoun, F.; Elshiwy, K.; Elkeraie, Y.; Merjaneh, Z.; Khoudari, G.; Sarmini, M.T.; Gad, M.; Al-Husseini, M.; Saad, A. Colorectal Cancer Epidemiology: Recent Trends and Impact on Outcomes. Curr. Drug Targets 2021, 22, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; Qiu, Y.H.; Cai, M.L.; Zhang, C.H.; Wang, X.W.; Liu, H.; Chen, Y.; Zhao, W.L.; Liu, J.B.; Shao, R.G. Role and molecular mechanism of stem cells in colorectal cancer initiation. J. Drug Target. 2020, 28, 1–10. [Google Scholar] [CrossRef]
- Munro, M.J.; Wickremesekera, S.K.; Peng, L.; Tan, S.T.; Itinteang, T. Cancer stem cells in colorectal cancer: A review. J. Clin. Pathol. 2018, 71, 110–116. [Google Scholar] [CrossRef]
- Relation, T.; Dominici, M.; Horwitz, E.M. Concise Review: An (Im)Penetrable Shield: How the Tumor Microenvironment Protects Cancer Stem Cells. Stem Cells 2017, 35, 1123–1130. [Google Scholar] [CrossRef]
- Kaushik, V.; Kulkarni, Y.; Felix, K.; Azad, N.; Iyer, A.K.V.; Yakisich, J.S. Alternative models of cancer stem cells: The stemness phenotype model, 10 years later. World J. Stem Cells 2021, 13, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Hayat, H.; Hayat, H.; Dwan, B.F.; Gudi, M.; Bishop, J.O.; Wang, P. A Concise Review: The Role of Stem Cells in Cancer Progression and Therapy. OncoTargets Ther. 2021, 14, 2761–2772. [Google Scholar] [CrossRef] [PubMed]
- Erkisa, M.; Karakas, D.; Ulukaya, E. Cancer Stem Cells: Root of the Evil. Crit. Rev. Oncog. 2019, 24, 69–87. [Google Scholar] [CrossRef]
- Gurel, C.; Inetas, G.; Hortu, I.; Tunc, E.; Kuscu, G.C.; Dindaroglu, F.C.; Sahin, O.; Buhur, A.; Oktem, G. Cancer and Cancer Stem Cells: New Molecular Perspectives. Crit. Rev. Oncog. 2019, 24, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Atashzar, M.R.; Baharlou, R.; Karami, J.; Abdollahi, H.; Rezaei, R.; Pourramezan, F.; Zoljalali Moghaddam, S.H. Cancer stem cells: A review from origin to therapeutic implications. J. Cell. Physiol. 2020, 235, 790–803. [Google Scholar] [CrossRef]
- Fedyanin, M.; Anna, P.; Elizaveta, P.; Sergei, T. Role of Stem Cells in Colorectal Cancer Progression and Prognostic and Predictive Characteristics of Stem Cell Markers in Colorectal Cancer. Curr. Stem Cell Res. Ther. 2017, 12, 19–30. [Google Scholar] [CrossRef]
- Zhou, Y.; Xia, L.; Wang, H.; Oyang, L.; Su, M.; Liu, Q.; Lin, J.; Tan, S.; Tian, Y.; Liao, Q.; et al. Cancer stem cells in progression of colorectal cancer. Oncotarget 2018, 9, 33403–33415. [Google Scholar] [CrossRef]
- Jahanafrooz, Z.; Mosafer, J.; Akbari, M.; Hashemzaei, M.; Mokhtarzadeh, A.; Baradaran, B. Colon cancer therapy by focusing on colon cancer stem cells and their tumor microenvironment. J. Cell. Physiol. 2020, 235, 4153–4166. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell. Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef] [PubMed]
- Desbats, M.A.; Giacomini, I.; Prayer-Galetti, T.; Montopoli, M. Metabolic Plasticity in Chemotherapy Resistance. Front. Oncol. 2020, 10, 281. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Melin, C.; Perraud, A.; Akil, H.; Jauberteau, M.O.; Cardot, P.; Mathonnet, M.; Battu, S. Cancer stem cell sorting from colorectal cancer cell lines by sedimentation field flow fractionation. Anal. Chem. 2012, 84, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.L.; Francescangeli, F.; Zeuner, A.; Baiocchi, M. Colorectal Cancer Stem Cells: An Overview of Evolving Methods and Concepts. Cancers 2021, 13, 5910. [Google Scholar] [CrossRef] [PubMed]
- Conciatori, F.; Bazzichetto, C.; Falcone, I.; Ferretti, G.; Cognetti, F.; Milella, M.; Ciuffreda, L. Colorectal cancer stem cells properties and features: Evidence of interleukin-8 involvement. Cancer Drug Resist. 2019, 2, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Parizadeh, S.M.; Jafarzadeh-Esfehani, R.; Hassanian, S.M.; Parizadeh, S.M.R.; Vojdani, S.; Ghandehari, M.; Ghazaghi, A.; Khazaei, M.; Shahidsales, S.; Rezayi, M.; et al. Targeting cancer stem cells as therapeutic approach in the treatment of colorectal cancer. Int. J. Biochem. Cell Biol. 2019, 110, 75–83. [Google Scholar] [CrossRef]
- Zalewski, A.; Snook, A.E.; Waldman, S.A. Stem cells as therapeutic targets in colorectal cancer. Pers. Med. 2021, 18, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Zeuner, A.; Todaro, M.; Stassi, G.; De Maria, R. Colorectal cancer stem cells: From the crypt to the clinic. Cell Stem Cell 2014, 15, 692–705. [Google Scholar] [CrossRef]
- Akbarzadeh, M.; Maroufi, N.F.; Tazehkand, A.P.; Akbarzadeh, M.; Bastani, S.; Safdari, R.; Farzane, A.; Fattahi, A.; Nejabati, H.R.; Nouri, M.; et al. Current approaches in identification and isolation of cancer stem cells. J. Cell. Physiol. 2019, 234, 14759–14772. [Google Scholar] [CrossRef]
- Kondo, J.; Endo, H.; Okuyama, H.; Ishikawa, O.; Iishi, H.; Tsujii, M.; Ohue, M.; Inoue, M. Retaining cell-cell contact enables preparation and culture of spheroids composed of pure primary cancer cells from colorectal cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 6235–6240. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Todaro, M.; de Sousa Mello, F.; Sprick, M.R.; Kemper, K.; Perez Alea, M.; Richel, D.J.; Stassi, G.; Medema, J.P. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 13427–13432. [Google Scholar] [CrossRef]
- De Angelis, M.L.; Zeuner, A.; Policicchio, E.; Russo, G.; Bruselles, A.; Signore, M.; Vitale, S.; De Luca, G.; Pilozzi, E.; Boe, A.; et al. Cancer Stem Cell-Based Models of Colorectal Cancer Reveal Molecular Determinants of Therapy Resistance. Stem Cells Transl. Med. 2016, 5, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Kondo, J.; Ekawa, T.; Endo, H.; Yamazaki, K.; Tanaka, N.; Kukita, Y.; Okuyama, H.; Okami, J.; Imamura, F.; Ohue, M.; et al. High-throughput screening in colorectal cancer tissue-originated spheroids. Cancer Sci. 2019, 110, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Xu, X.; Yang, L.; Zhu, J.; Wan, J.; Shen, L.; Xia, F.; Fu, G.; Deng, Y.; Pan, M.; et al. Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell Stem Cell 2020, 26, 17–26.e6. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, V.; Wright, J.A.; Churchill, M.; Wang, T.; Rosati, R.; Lannagan, T.R.M.; Vrbanac, L.; Richardson, A.B.; Kobayashi, H.; Price, T.; et al. Medium-throughput Drug Screening of Patient-derived Organoids from Colorectal Peritoneal Metastases to Direct Personalized Therapy. Clin. Cancer Res. 2020, 26, 3662–3670. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef]
- Fumagalli, A.; Drost, J.; Suijkerbuijk, S.J.; van Boxtel, R.; de Ligt, J.; Offerhaus, G.J.; Begthel, H.; Beerling, E.; Tan, E.H.; Sansom, O.J.; et al. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc. Natl. Acad. Sci. USA 2017, 114, E2357–E2364. [Google Scholar] [CrossRef]
- O’Rourke, K.P.; Loizou, E.; Livshits, G.; Schatoff, E.M.; Baslan, T.; Manchado, E.; Simon, J.; Romesser, P.B.; Leach, B.; Han, T.; et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat. Biotechnol. 2017, 35, 577–582. [Google Scholar] [CrossRef]
- Sakai, E.; Nakayama, M.; Oshima, H.; Kouyama, Y.; Niida, A.; Fujii, S.; Ochiai, A.; Nakayama, K.I.; Mimori, K.; Suzuki, Y.; et al. Combined Mutation of Apc, Kras, and Tgfbr2 Effectively Drives Metastasis of Intestinal Cancer. Cancer Res. 2018, 78, 1334–1346. [Google Scholar] [CrossRef]
- Melin, C.; Perraud, A.; Bounaix Morand du Puch, C.; Loum, E.; Giraud, S.; Cardot, P.; Jauberteau, M.O.; Lautrette, C.; Battu, S.; Mathonnet, M. Sedimentation field flow fractionation monitoring of in vitro enrichment in cancer stem cells by specific serum-free culture medium. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 963, 40–46. [Google Scholar] [CrossRef]
- Melin, C.; Perraud, A.; Christou, N.; Bibes, R.; Cardot, P.; Jauberteau, M.O.; Battu, S.; Mathonnet, M. New ex-ovo colorectal-cancer models from different SdFFF-sorted tumor-initiating cells. Anal. Bioanal. Chem. 2015, 407, 8433–8443. [Google Scholar] [CrossRef]
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine 2016, 95 (Suppl. 1), S8–S19. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, X. Advances of Wnt Signalling Pathway in Colorectal Cancer. Cells 2023, 12, 447. [Google Scholar] [CrossRef] [PubMed]
- Miyako, S.; Matsuda, T.; Koma, Y.I.; Koide, T.; Sawada, R.; Hasegawa, H.; Yamashita, K.; Harada, H.; Urakawa, N.; Goto, H.; et al. Significance of Wnt/beta-Catenin Signal Activation for Resistance to Neoadjuvant Chemoradiotherapy in Rectal Cancer. Biomedicines 2023, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, M.; Deng, K. Blocking the Wnt/beta-catenin signaling pathway to treat colorectal cancer: Strategies to improve current therapies (Review). Int. J. Oncol. 2023, 62, 24. [Google Scholar] [CrossRef]
- He, K.; Gan, W.J. Wnt/β-Catenin Signaling Pathway in the Development and Progression of Colorectal Cancer. Cancer Manag. Res. 2023, 15, 435–448. [Google Scholar] [CrossRef]
- Zhao, H.; Ming, T.; Tang, S.; Ren, S.; Yang, H.; Liu, M.; Tao, Q.; Xu, H. Wnt signaling in colorectal cancer: Pathogenic role and therapeutic target. Mol. Cancer 2022, 21, 144. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.X.; Gao, D.; Shao, Z.Z.; Chen, L.; Ding, W.J.; Yu, Q.F. Wnt/beta-catenin signaling: Causes and treatment targets of drug resistance in colorectal cancer (Review). Mol. Med. Rep. 2021, 23, 105. [Google Scholar] [CrossRef] [PubMed]
- Brisset, M.; Mehlen, P.; Meurette, O.; Hollande, F. Notch receptor/ligand diversity: Contribution to colorectal cancer stem cell heterogeneity. Front. Cell Dev. Biol. 2023, 11, 1231416. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, N.; Afshinpour, M.; Fakhr, S.S.; Kalkhoran, P.G.; Shadman-Manesh, V.; Adelian, S.; Beiranvand, S.; Rezaei-Tazangi, F.; Khorram, R.; Hamblin, M.R.; et al. Cancer stem cells in colorectal cancer: Signaling pathways involved in stemness and therapy resistance. Crit. Rev. Oncol. Hematol. 2023, 182, 103920. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Das, P.K.; Islam, F.; Lam, A.K. The Roles of Cancer Stem Cells and Therapy Resistance in Colorectal Carcinoma. Cells 2020, 9, 1392. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Sharma, A.K.; Damodaran, C. A Review on Notch Signaling and Colorectal Cancer. Cells 2020, 9, 1549. [Google Scholar] [CrossRef]
- Lin, A.; Yao, J.; Cheng, Q.; Liu, Z.; Luo, P.; Zhang, J. Mutations Status of NOTCH Signaling Pathway Predict Prognosis of Immune Checkpoint Inhibitors in Colorectal Cancer. J. Inflamm. Res. 2023, 16, 1693–1709. [Google Scholar] [CrossRef] [PubMed]
- Negri, F.; Bottarelli, L.; Pedrazzi, G.; Maddalo, M.; Leo, L.; Milanese, G.; Sala, R.; Lecchini, M.; Campanini, N.; Bozzetti, C.; et al. Notch-Jagged1 signaling and response to bevacizumab therapy in advanced colorectal cancer: A glance to radiomics or back to physiopathology? Front. Oncol. 2023, 13, 1132564. [Google Scholar] [CrossRef]
- Yahyanejad, S.; Theys, J.; Vooijs, M. Targeting Notch to overcome radiation resistance. Oncotarget 2016, 7, 7610–7628. [Google Scholar] [CrossRef]
- Yuan, X.; Wu, H.; Xu, H.; Xiong, H.; Chu, Q.; Yu, S.; Wu, G.S.; Wu, K. Notch signaling: An emerging therapeutic target for cancer treatment. Cancer Lett. 2015, 369, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Geyer, N.; Gerling, M. Hedgehog Signaling in Colorectal Cancer: All in the Stroma? Int. J. Mol. Sci. 2021, 22, 1025. [Google Scholar] [CrossRef] [PubMed]
- Sigafoos, A.N.; Paradise, B.D.; Fernandez-Zapico, M.E. Hedgehog/GLI Signaling Pathway: Transduction, Regulation, and Implications for Disease. Cancers 2021, 13, 3410. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Sakurai, M.; Umata, K.; Elbadawy, M.; Ohama, T.; Yamawaki, H.; Hazama, S.; Takenouchi, H.; Nakajima, M.; Tsunedomi, R.; et al. Hedgehog Signals Mediate Anti-Cancer Drug Resistance in Three-Dimensional Primary Colorectal Cancer Organoid Culture. Int. J. Mol. Sci. 2018, 19, 1098. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Bhatt, L.K.; Johnston, T.P.; Prabhavalkar, K.S. Colon cancer stem cells: Potential target for the treatment of colorectal cancer. Cancer Biol. Ther. 2019, 20, 1068–1082. [Google Scholar] [CrossRef] [PubMed]
- Giammona, A.; Crivaro, E.; Stecca, B. Emerging Roles of Hedgehog Signaling in Cancer Immunity. Int. J. Mol. Sci. 2023, 24, 1321. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Saraf, S.; Verma, A.; Panda, P.K.; Jain, S.K. Novel targeting approaches and signaling pathways of colorectal cancer: An insight. World J. Gastroenterol. 2018, 24, 4428–4435. [Google Scholar] [CrossRef] [PubMed]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Mo, J.S.; Park, H.W.; Guan, K.L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef]
- Park, J.H.; Shin, J.E.; Park, H.W. The Role of Hippo Pathway in Cancer Stem Cell Biology. Mol. Cells 2018, 41, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Maugeri-Sacca, M.; De Maria, R. The Hippo pathway in normal development and cancer. Pharmacol. Ther. 2018, 186, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Mohajan, S.; Jaiswal, P.K.; Vatanmakarian, M.; Yousefi, H.; Sankaralingam, S.; Alahari, S.K.; Koul, S.; Koul, H.K. Hippo pathway: Regulation, deregulation and potential therapeutic targets in cancer. Cancer Lett. 2021, 507, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Zinatizadeh, M.R.; Miri, S.R.; Zarandi, P.K.; Chalbatani, G.M.; Raposo, C.; Mirzaei, H.R.; Akbari, M.E.; Mahmoodzadeh, H. The Hippo Tumor Suppressor Pathway (YAP/TAZ/TEAD/MST/LATS) and EGFR-RAS-RAF-MEK in cancer metastasis. Genes Dis. 2021, 8, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Dong, J. The Hippo Signaling Pathway in Drug Resistance in Cancer. Cancers 2021, 13, 318. [Google Scholar] [CrossRef] [PubMed]
- Zafari, N.; Khosravi, F.; Rezaee, Z.; Esfandyari, S.; Bahiraei, M.; Bahramy, A.; Ferns, G.A.; Avan, A. The role of the tumor microenvironment in colorectal cancer and the potential therapeutic approaches. J. Clin. Lab. Anal. 2022, 36, e24585. [Google Scholar] [CrossRef] [PubMed]
- Wozniakova, M.; Skarda, J.; Raska, M. The Role of Tumor Microenvironment and Immune Response in Colorectal Cancer Development and Prognosis. Pathol. Oncol. Res. 2022, 28, 1610502. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; De Sousa, E.M.F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Novoa Diaz, M.B.; Martin, M.J.; Gentili, C. Tumor microenvironment involvement in colorectal cancer progression via Wnt/beta-catenin pathway: Providing understanding of the complex mechanisms of chemoresistance. World J. Gastroenterol. 2022, 28, 3027–3046. [Google Scholar] [CrossRef]
- Nallasamy, P.; Nimmakayala, R.K.; Parte, S.; Are, A.C.; Batra, S.K.; Ponnusamy, M.P. Tumor microenvironment enriches the stemness features: The architectural event of therapy resistance and metastasis. Mol. Cancer 2022, 21, 225. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y. Role of Epigenetic Regulation in Plasticity of Tumor Immune Microenvironment. Front. Immunol. 2021, 12, 640369. [Google Scholar] [CrossRef]
- Li, J.; Chen, D.; Shen, M. Tumor Microenvironment Shapes Colorectal Cancer Progression, Metastasis, and Treatment Responses. Front. Med. 2022, 9, 869010. [Google Scholar] [CrossRef]
- Bregenzer, M.; Horst, E.; Mehta, P.; Snyder, C.; Repetto, T.; Mehta, G. The Role of the Tumor Microenvironment in CSC Enrichment and Chemoresistance: 3D Co-culture Methods. Methods Mol. Biol. 2022, 2424, 217–245. [Google Scholar] [CrossRef]
- Taeb, S.; Ashrafizadeh, M.; Zarrabi, A.; Rezapoor, S.; Musa, A.E.; Farhood, B.; Najafi, M. Role of Tumor Microenvironment in Cancer Stem Cells Resistance to Radiotherapy. Curr. Cancer Drug Targets 2022, 22, 18–30. [Google Scholar] [CrossRef]
- Rainho, M.A.; Siqueira, P.B.; de Amorim, I.S.S.; Mencalha, A.L.; Thole, A.A. Mitochondria in colorectal cancer stem cells—A target in drug resistance. Cancer Drug Resist. 2023, 6, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Wang, Z.; Chai, Q.; Li, Z.; Zhang, M.; Zhang, Y.; Zhang, L.; Tang, Q.; Zhu, H.; Sui, H. Exosomes derived from MDR cells induce cetuximab resistance in CRC via PI3K/AKT signaling-mediated Sox2 and PD-L1 expression. Exp. Ther. Med. 2023, 25, 86. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yuan, Y.; Yang, L.; Chen, H.; Zhang, X.; Wen, T.; Liao, W.; Zhao, M.; Zhao, Z.; Hu, Q. AT7867 Inhibits the Growth of Colorectal Cancer Stem-Like Cells and Stemness by Regulating the Stem Cell Maintenance Factor Ascl2 and Akt Signaling. Stem Cells Int. 2023, 2023, 4199052. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Choi, H.S.; Choi, J.H.; Kim, H.S.; Jang, Y.S.; Seo, J.W. 7S,15R-Dihydroxy-16S,17S-epoxy-docosapentaenoic Acid Overcomes Chemoresistance of 5-Fluorouracil by Suppressing the Infiltration of Tumor-Associated Macrophages and Inhibiting the Activation of Cancer Stem Cells in a Colorectal Cancer Xenograft Model. Mar. Drugs 2023, 21, 80. [Google Scholar] [CrossRef]
- Mangiapane, L.R.; Nicotra, A.; Turdo, A.; Gaggianesi, M.; Bianca, P.; Di Franco, S.; Sardina, D.S.; Veschi, V.; Signore, M.; Beyes, S.; et al. PI3K-driven HER2 expression is a potential therapeutic target in colorectal cancer stem cells. Gut 2022, 71, 119–128. [Google Scholar] [CrossRef]
- Roy, S.; Zhao, Y.; Yuan, Y.C.; Goel, A. Metformin and ICG-001 Act Synergistically to Abrogate Cancer Stem Cells-Mediated Chemoresistance in Colorectal Cancer by Promoting Apoptosis and Autophagy. Cancers 2022, 14, 1281. [Google Scholar] [CrossRef]
- Zhang, X.; MaY, Y.; Ma, J.; Yang, L.; Song, Q.; Wang, H.; Lv, G. Glutathione Peroxidase 4 as a Therapeutic Target for Anti-Colorectal Cancer Drug-Tolerant Persister Cells. Front. Oncol. 2022, 12, 913669. [Google Scholar] [CrossRef]
- Li, Y.; Wu, M.; Xu, S.; Huang, H.; Yan, L.; Gu, Y. Colorectal cancer stem cell-derived exosomal long intergenic noncoding RNA 01315 (LINC01315) promotes proliferation, migration, and stemness of colorectal cancer cells. Bioengineered 2022, 13, 10827–10842. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef]
- de Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, A.; Oost, K.C.; Kester, L.; Morgner, J.; Bornes, L.; Bruens, L.; Spaargaren, L.; Azkanaz, M.; Schelfhorst, T.; Beerling, E.; et al. Plasticity of Lgr5-Negative Cancer Cells Drives Metastasis in Colorectal Cancer. Cell Stem Cell 2020, 26, 569–578. [Google Scholar] [CrossRef]
- Lenos, K.J.M.; Miedema, D.M.; Lodestijn, S.C.; Nijman, L.E.; van den Bosch, T.; Romero Ros, X.; Lourenço, F.C.; Lecca, M.C.; van der Heijden, M.; van Neerven, S.M.; et al. Stem Cell Functionality Is Microenvironmentally Defined during Tumour Expansion and Therapy Response in Colon Cancer. Nat. Cell Biol. 2018, 20, 1193–1202. [Google Scholar] [CrossRef]
- de Sousa, E.M.F.; de Sauvage, F.J. Cellular Plasticity in Intestinal Homeostasis and Disease. Cell Stem Cell 2019, 24, 54–64. [Google Scholar] [CrossRef]
- Damane, B.P.; Marima, R.; Mulaudzi, T.V.; Dlamini, Z. Targeting Stem Cells in the Colorectal Cancer Microenvironment to Avert Drug Resistance in Pursuit of Novel Oncotherapies. J. Biol. Regul. Homeost. Agents 2023, 37, 4519–4530. [Google Scholar] [CrossRef]
- Zhao, H.; Han, R.; Wang, Z.; Xian, J.; Bai, X. Colorectal Cancer Stem Cells and Targeted Agents. Pharmaceutics 2023, 15, 2763. [Google Scholar] [CrossRef]
- Citarella, A.; Catanzaro, G.; Besharat, Z.M.; Trocchianesi, S.; Barbagallo, F.; Gosti, G.; Leonetti, M.; Di Fiore, A.; Coppola, L.; Autilio, T.M.; et al. Hedgehog-GLI and Notch Pathways Sustain Chemoresistance and Invasiveness in Colorectal Cancer and Their Inhibition Restores Chemotherapy Efficacy. Cancers 2023, 15, 1471. [Google Scholar] [CrossRef]
- Shao, Z.; Wang, H.; Ren, H.; Sun, Y.; Chen, X. The Anticancer Effect of Napabucasin (BBI608), a Natural Naphthoquinone. Molecules 2023, 28, 5678. [Google Scholar] [CrossRef]
- Jing, B.; Guo, F.; An, R.; Gao, Y.; Li, Y.; Xie, Y.; Wang, J.; Chen, Y.; Li, H.; Gao, T.; et al. Apoptotic tumor cell-derived microparticles loading Napabucasin inhibit CSCs and synergistic immune therapy. J. Nanobiotechnol. 2023, 21, 37. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, Z.; Liu, W.; Zhu, W.; Yin, L.; Han, Z.; Xian, Y.; Wen, J.; Tang, H.; Lin, X.; et al. Disheveled3 enhanced EMT and cancer stem-like cells properties via Wnt/beta-catenin/c-Myc/SOX2 pathway in colorectal cancer. J. Transl. Med. 2023, 21, 302. [Google Scholar] [CrossRef] [PubMed]
- Tsochantaridis, I.; Roupas, A.; Mohlin, S.; Pappa, A.; Voulgaridou, G.P. The Concept of Cancer Stem Cells: Elaborating on ALDH1B1 as an Emerging Marker of Cancer Progression. Life 2023, 13, 197. [Google Scholar] [CrossRef] [PubMed]
- Sonbol, M.B.; Ahn, D.H.; Bekaii-Saab, T. Therapeutic Targeting Strategies of Cancer Stem Cells in Gastrointestinal Malignancies. Biomedicines 2019, 7, 17. [Google Scholar] [CrossRef]
- Atreya, C.E.; Yaeger, R.; Chu, E. Systemic Therapy for Metastatic Colorectal Cancer: From Current Standards to Future Molecular Targeted Approaches. Am. Soc. Clin. Oncol. Educ. Book. 2017, 37, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Mahmood, F.; Akingboye, A. Biomarkers in Colorectal Cancer: Current Research and Future Prospects. Int. J. Mol. Sci. 2020, 21, 5311. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCSC Marker | Biological Function | Prognostic Significance |
|---|---|---|
| CD24 | Facilitates cell adhesion and serves as a P-selectin ligand | High cytoplasmic CD24 expression is closely tied to decreased patient survival. |
| CD29 | Plays a role in cell adhesion processes | Increased expression of CD29 correlates with a poorer prognosis and increased aggressiveness |
| CD44 | Regulates cell interactions, adhesion, and migration | Elevated CD44 levels linked to lymph node metastasis, distant metastases, and worse prognosis |
| CD44v6 | Binds hepatocyte growth factor, facilitating migration and metastases | High levels of CD44v6 negatively impact overall survival |
| CD133 | Governs self-renewal and contributes to tumor angiogenesis | CD133 expression correlates with decreased survival in CRC patients |
| CD166 | Mediates homophilic interactions among cells | Increased expression of CD166 associated with shortened patient survival |
| EpCAM | Controls cell adhesion, proliferation, and migration | Loss of EpCAM expression linked to advanced tumor stage, lymph node and distant metastases, and poor prognosis |
| Lgr5 | Serves as a downstream target of the Wnt pathway involved in self-renewal | Lgr5 expression linked to lymph node and distant metastases, and overexpression related to reduced overall survival |
| Bmi-1 | Functions as a self-renewal regulator | High Bmi-1 expression is associated with poor survival rates |
| CD26 | Promotes invasion and metastases | Elevated CD26 expression associated with advanced tumor staging and worse survival |
| Cells | Impact on Cancer Progression | Influence on CSC Dynamics | Pathways |
|---|---|---|---|
| Cancer-Associated Fibroblasts | Facilitates cancer advancement, invasion, and orchestrates morphological transformations through the release of growth-promoting substances such as HGF, CCL12, fibroblast growth factors, and stanniocalcin. | Enhances the characteristics of CSCs and their capacity for invasive metastasis. | Triggers Wnt/β-catenin pathway activation through the release of HGF, matrix metalloproteinases, and cytokines such as TNF-α. |
| Adipocytes | Produces signaling molecules, including leptin, adiponectin, IL-6, MCP-1, and TNF-α. | The leptin receptor sustains an autocatalytic signaling loop that amplifies the CSC population and accelerates tumor expansion. | - |
| Tumor-Associated Macrophages | Fosters tumor growth by triggering T cell inactivity, influencing ECM dynamics, tissue repair, and new blood vessel formation. | Facilitates CSC development through the involvement of Milk-fat globule-EGF factor 8 (MFG-E8). | Initiates STAT3 and Hedgehog signaling, promoting cancer cell growth and resistance to treatment in CSCs. |
| T-regulatory cells | Inhibits immune responses by releasing cytokines such as IL-10, IL-35, and TGF-β, and dampens the activity of cytotoxic T cells and NK cells. | In low-oxygen conditions, FOXP3+ Treg cells produce IL-17, leading to the growth of the CCSC population, as shown by elevated levels of CD133, CD44s, and EpCAM. Additionally, these cells modulate CSC characteristics by emitting prostaglandin (PGE2) via the NF-κB pathway. | IL-17 triggers the activation of the Akt and MAPK signaling pathways |
| Myeloid-Derived Suppressor Cells | They release arginase 1, reactive oxygen species (ROS), and inducible nitric oxide synthase (iNOS) to suppress the tumor-fighting abilities of NK cells and T cells, aiding in immune evasion and facilitating the onset and advancement of tumors. | Their presence in the intestinal mucosa is tied to the activation of CXCR2 on endothelial and immune cells within colorectal cancer regions. | - |
| HIF | HIF-1α and HIF-2α interact with hypoxia response elements (HRE), exacerbating tumor development, infiltration, and spread. | HIF-1α enhances the gene expression driven by β-catenin within the traditional Wnt pathway. Hypoxia’s role in keeping CSCs dormant further aids in their resistance to therapeutic drugs. | Wnt/β-catenin |
| Trial Number | Clinical Study | Interventions | Aim |
|---|---|---|---|
| NCT00004087 | Radiolabeled monoclonal antibody therapy plus peripheral stem cell transplantation in treating patients with metastatic or recurrent colorectal cancer or pancreatic cancer | Biological: filgrastim Procedure: autologous bone marrow transplantation Procedure: peripheral blood stem cell transplantation Radiation: indium In 111 monoclonal antibody MN-14 Radiation: yttrium Y 90 monoclonal antibody MN-14 | Effectiveness of combined treatment involving radiolabeled monoclonal antibodies and peripheral stem cell transplantation in treating metastatic or recurrent colorectal cancer unresponsive to prior therapies. |
| NCT01075893 | Changes in stem cells of the colon in response to increased risk of colorectal cancer | Not provided | Exploration of stem cell frequency and distribution among individuals at high and normal risk for colorectal cancer. Enhanced cell proliferation at the crypt apex in high-risk patients is attributed to alterations in the stem cell count at the crypt base. |
| NCT01286883 | Cancer stem cell markers and prognostic markers in circulating tumor cells | Not provided | Analyzing the genetic profiles of circulating and primary tumors to determine the prevalence of cancer cell genotypes in patients exhibiting elevated circulating tumor cell counts or experiencing early disease recurrence. |
| NCT01483001 | Feasibility study on stem cells sensitivity assay | Cancer Stem Cells Sensitivity Assay To test in vitro sensitivity of cancer stem cells to several antineoplastic drugs in order to personalize treatment | Isolation and identification of cancer stem cells within solid tumors, including colorectal cancer. |
| NCT01577511 | Invasiveness and chemoresistance of cancer stem cells in colon cancer | Biological: Samples and followup | Exploration of the traits related to the spread and drug resistance of colorectal cancer stem cells, along with their genetic characteristics. |
| NCT02176746 | A phase I/II study of active immunotherapy with cancer stem cells vaccine for CRC | Biological: cancer stem cell vaccine | The investigation evaluated the anticancer immune responses elicited by cytotoxic T-cells and B-cell antibodies, both activated through exposure to dendritic cells derived from colorectal cancer stem cells. |
| NCT03002727 | Role of CD133 and microsatellite status in evaluation of rectosigmoid cancer; young adults received neoadjuvant treatment | Not provided | The study aims to explore the potential link between microsatellite status and the prevalence of colorectal cancer stem cells, examining how this relationship could influence the outcomes of the disease and the effectiveness of treatment strategies. |
| NCT03803241 | CD133+ cell infusion in patients with colorectal liver metastases | Drug: CD133+ infusion Other: portal vein embolization | Patients deemed unsuitable for surgery underwent treatment with CD133+ cell infusion and portal vein embolization to potentially qualify them for surgical intervention. |
| NCT02753127 | A Study of Napabucasin (BBI-608) in Combination With FOLFIRI in Adult Patients With Previously Treated Metastatic Colorectal Cancer (CanStem303C) | Drug: Napabucasin | To evaluate the impact of combining napabucasin with biweekly FOLFIRI, compared to biweekly FOLFIRI alone, with or without the addition of bevacizumab, on the overall survival of individuals with metastatic colorectal cancer who have undergone previous treatments. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radu, P.; Zurzu, M.; Tigora, A.; Paic, V.; Bratucu, M.; Garofil, D.; Surlin, V.; Munteanu, A.C.; Coman, I.S.; Popa, F.; et al. The Impact of Cancer Stem Cells in Colorectal Cancer. Int. J. Mol. Sci. 2024, 25, 4140. https://doi.org/10.3390/ijms25084140
Radu P, Zurzu M, Tigora A, Paic V, Bratucu M, Garofil D, Surlin V, Munteanu AC, Coman IS, Popa F, et al. The Impact of Cancer Stem Cells in Colorectal Cancer. International Journal of Molecular Sciences. 2024; 25(8):4140. https://doi.org/10.3390/ijms25084140
Chicago/Turabian StyleRadu, Petru, Mihai Zurzu, Anca Tigora, Vlad Paic, Mircea Bratucu, Dragos Garofil, Valeriu Surlin, Alexandru Claudiu Munteanu, Ionut Simion Coman, Florian Popa, and et al. 2024. "The Impact of Cancer Stem Cells in Colorectal Cancer" International Journal of Molecular Sciences 25, no. 8: 4140. https://doi.org/10.3390/ijms25084140
APA StyleRadu, P., Zurzu, M., Tigora, A., Paic, V., Bratucu, M., Garofil, D., Surlin, V., Munteanu, A. C., Coman, I. S., Popa, F., Strambu, V., & Ramboiu, S. (2024). The Impact of Cancer Stem Cells in Colorectal Cancer. International Journal of Molecular Sciences, 25(8), 4140. https://doi.org/10.3390/ijms25084140