
Design of New Schiff Bases and Their Heavy Metal Ion Complexes for Environmental Applications: A Molecular Dynamics and Density Function Theory Study
 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
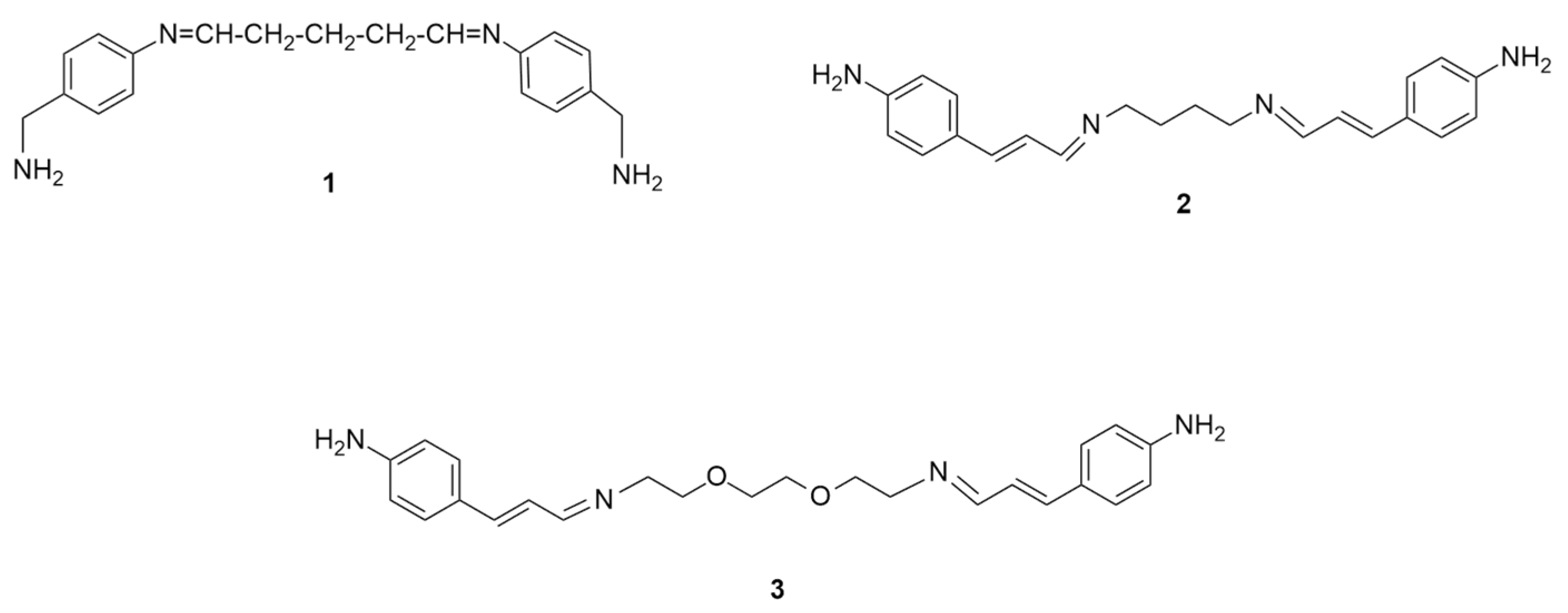
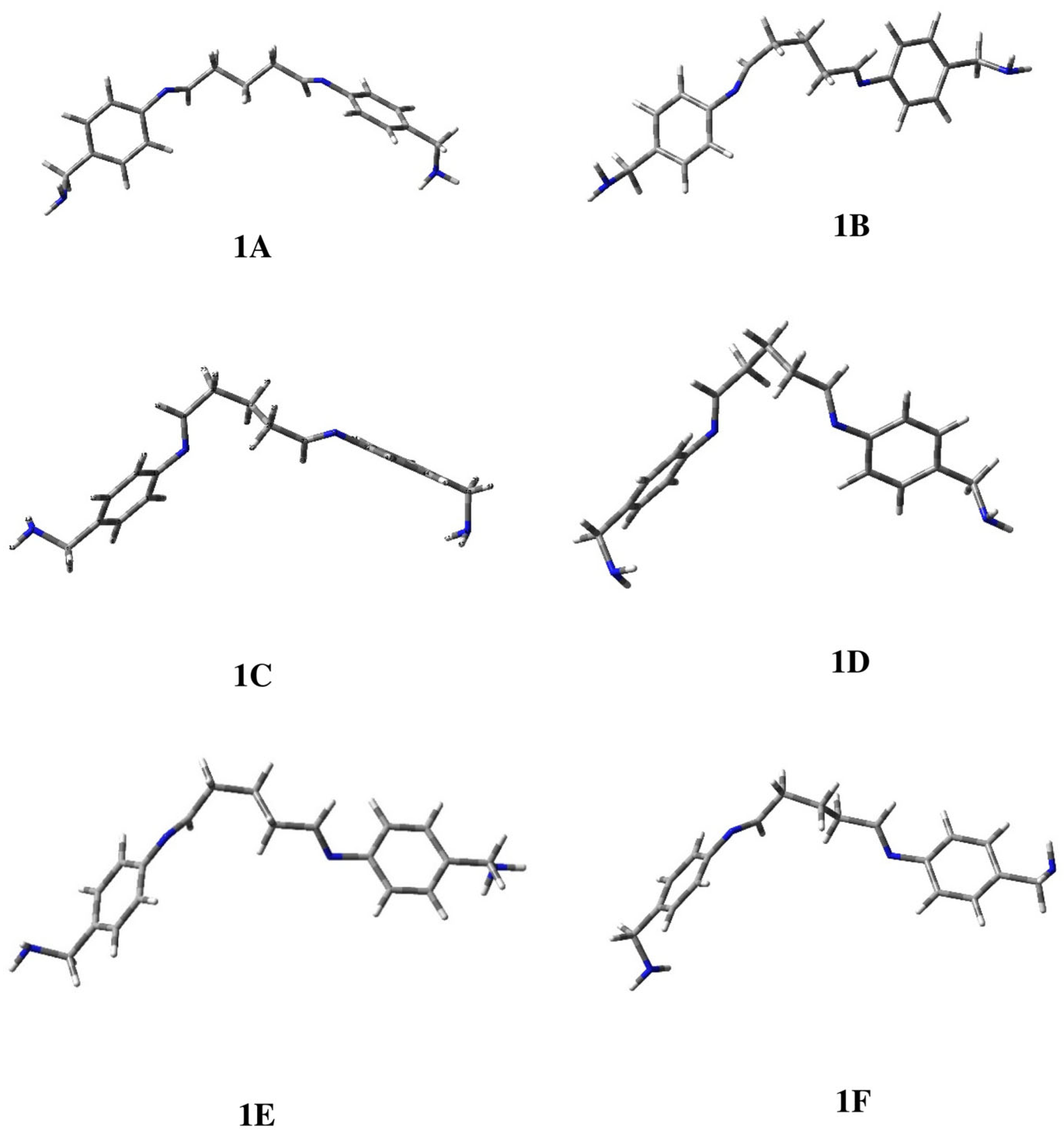
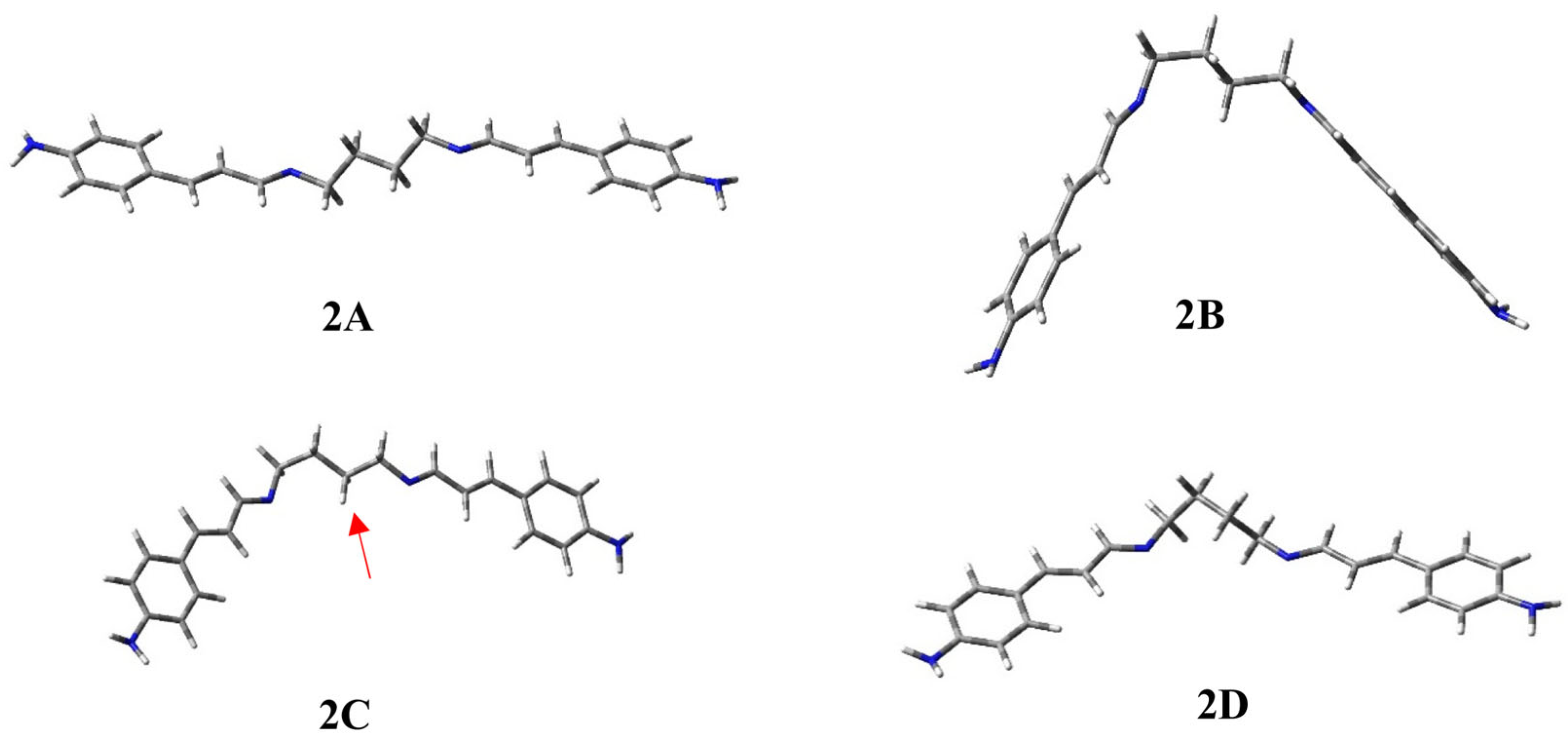
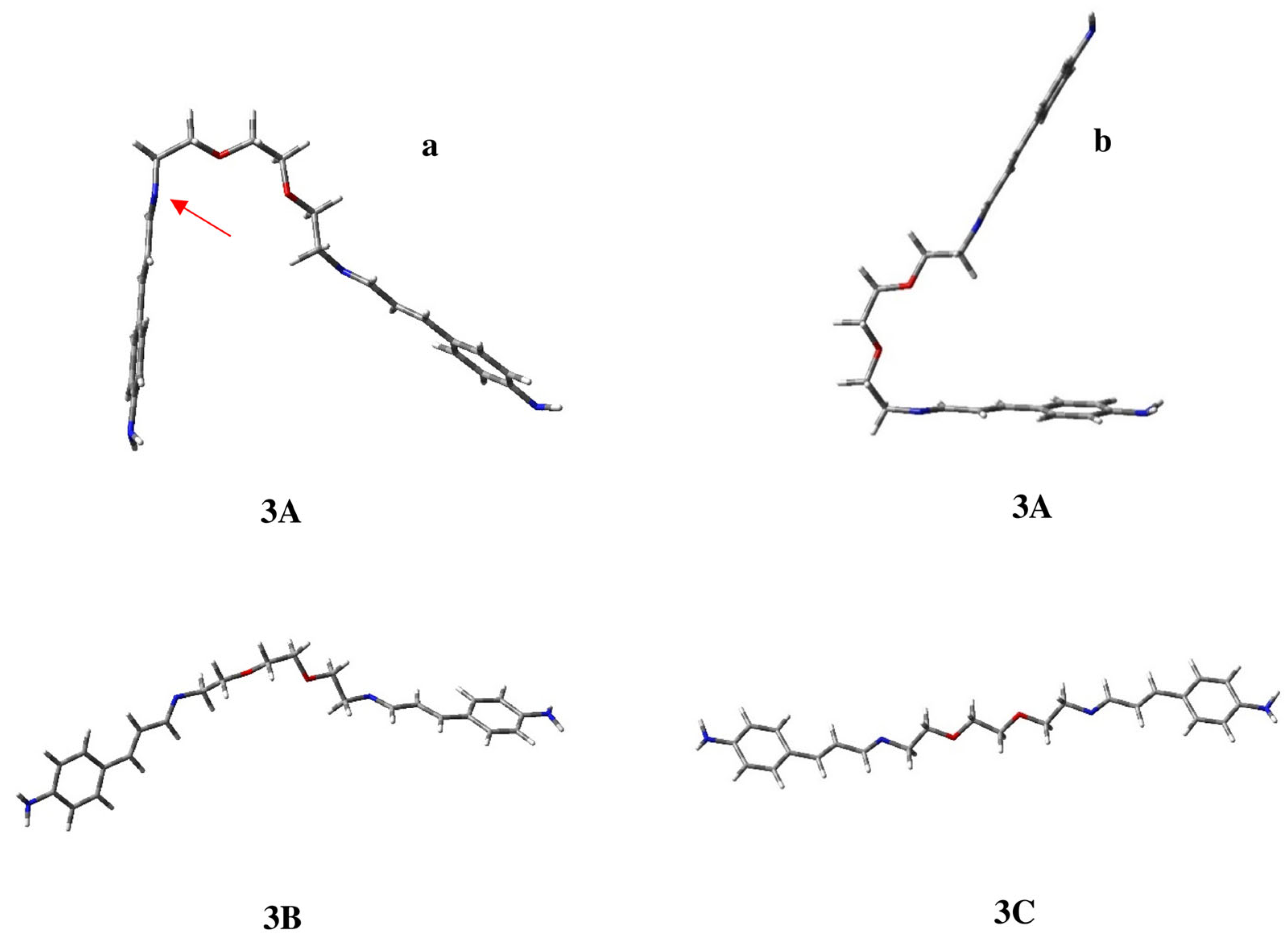
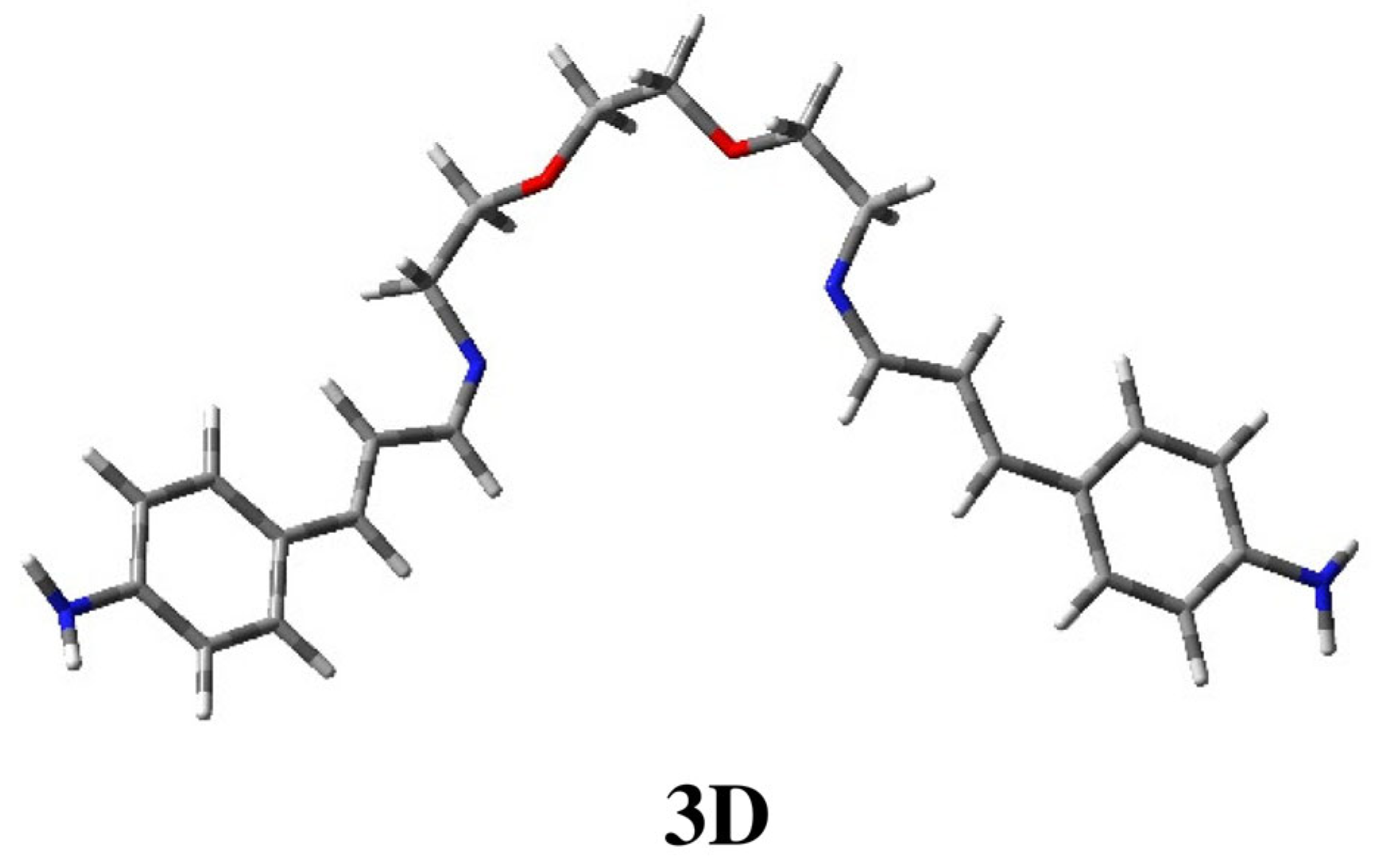
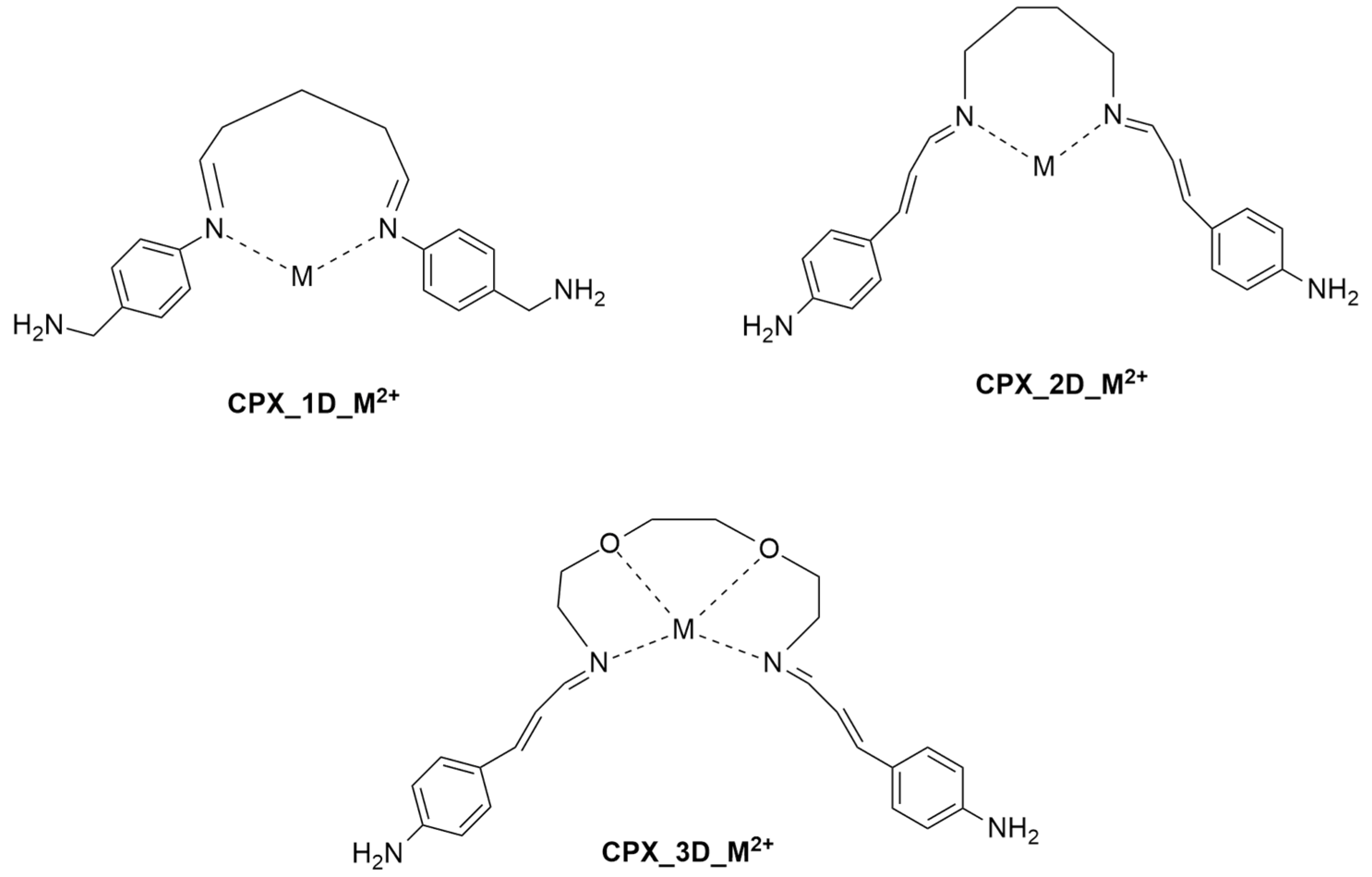
2.1. Conformational Study of Ligands 1–3
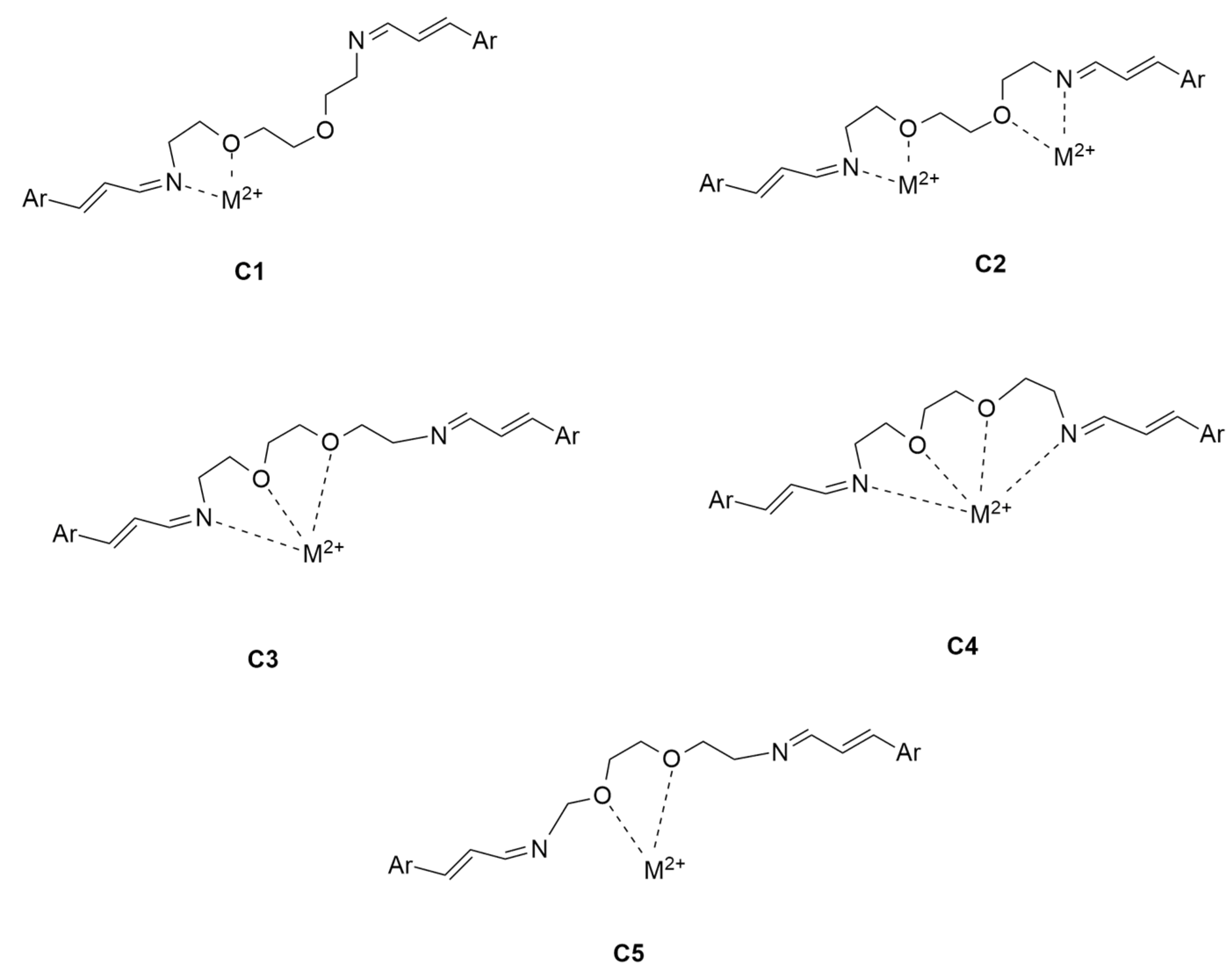
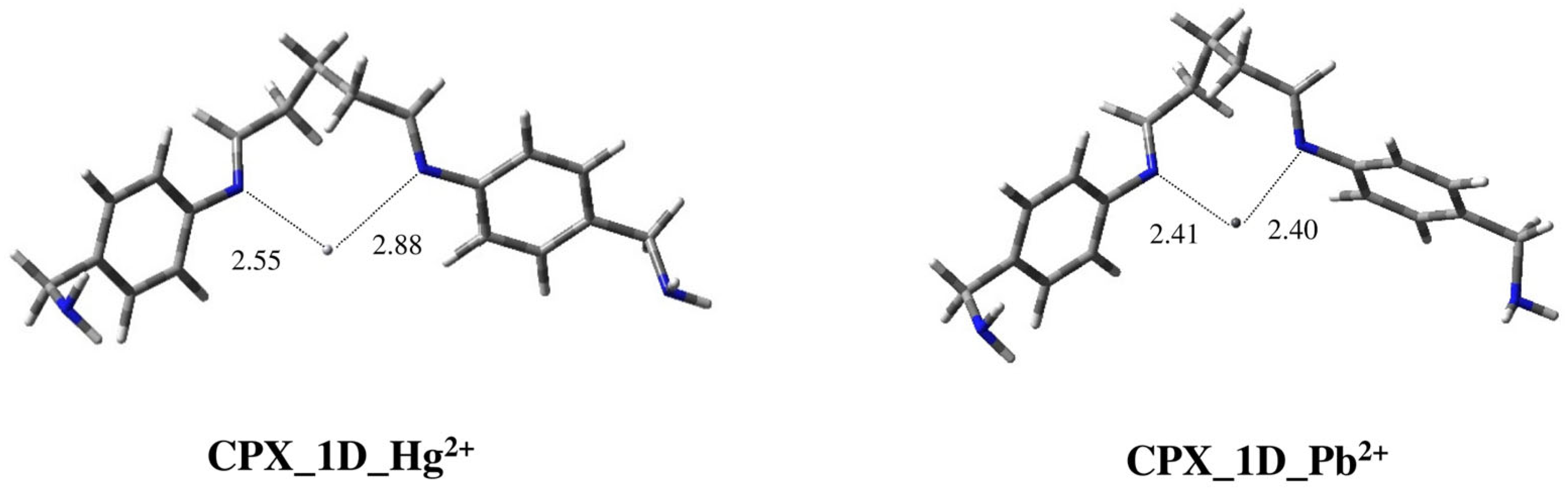
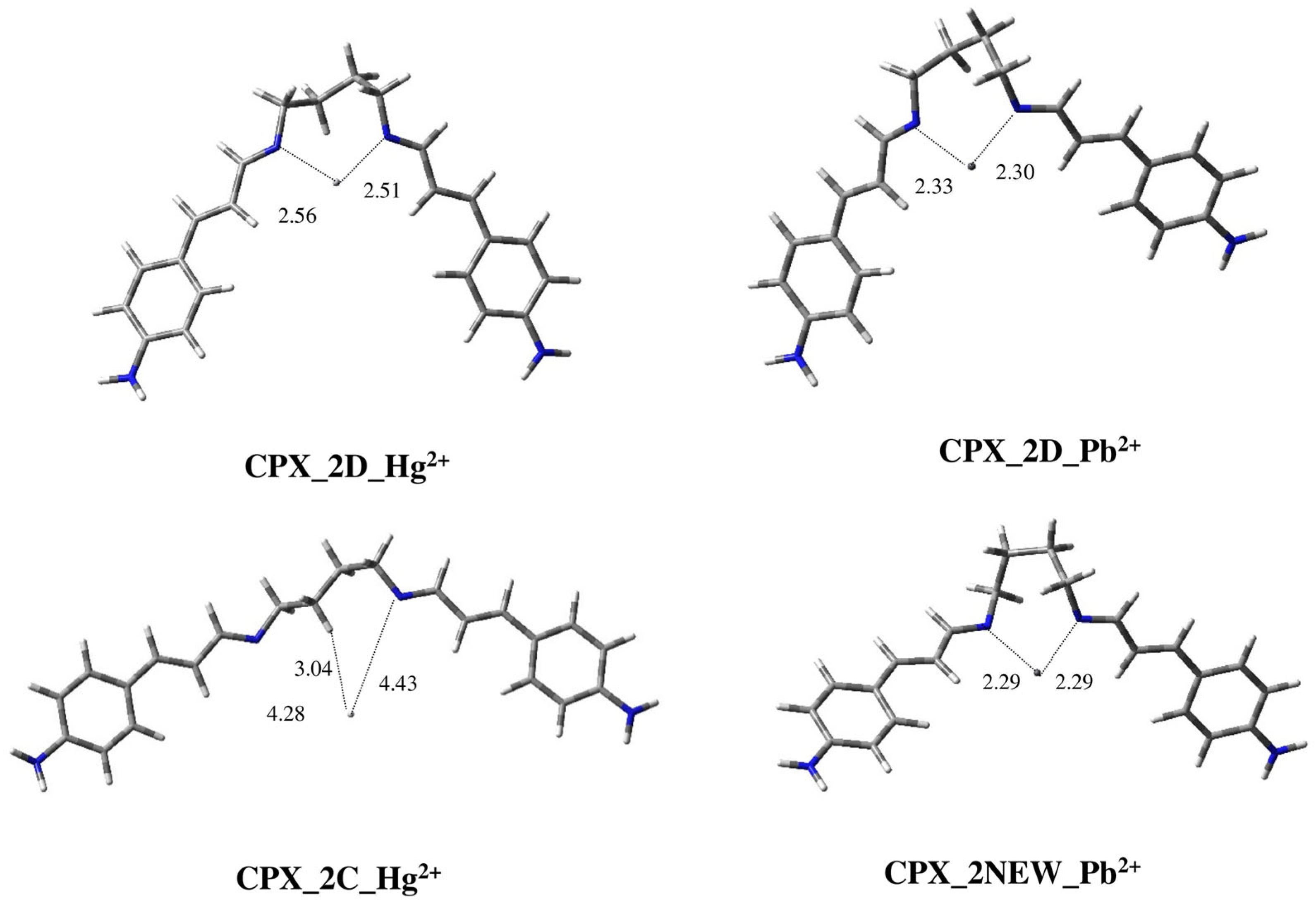
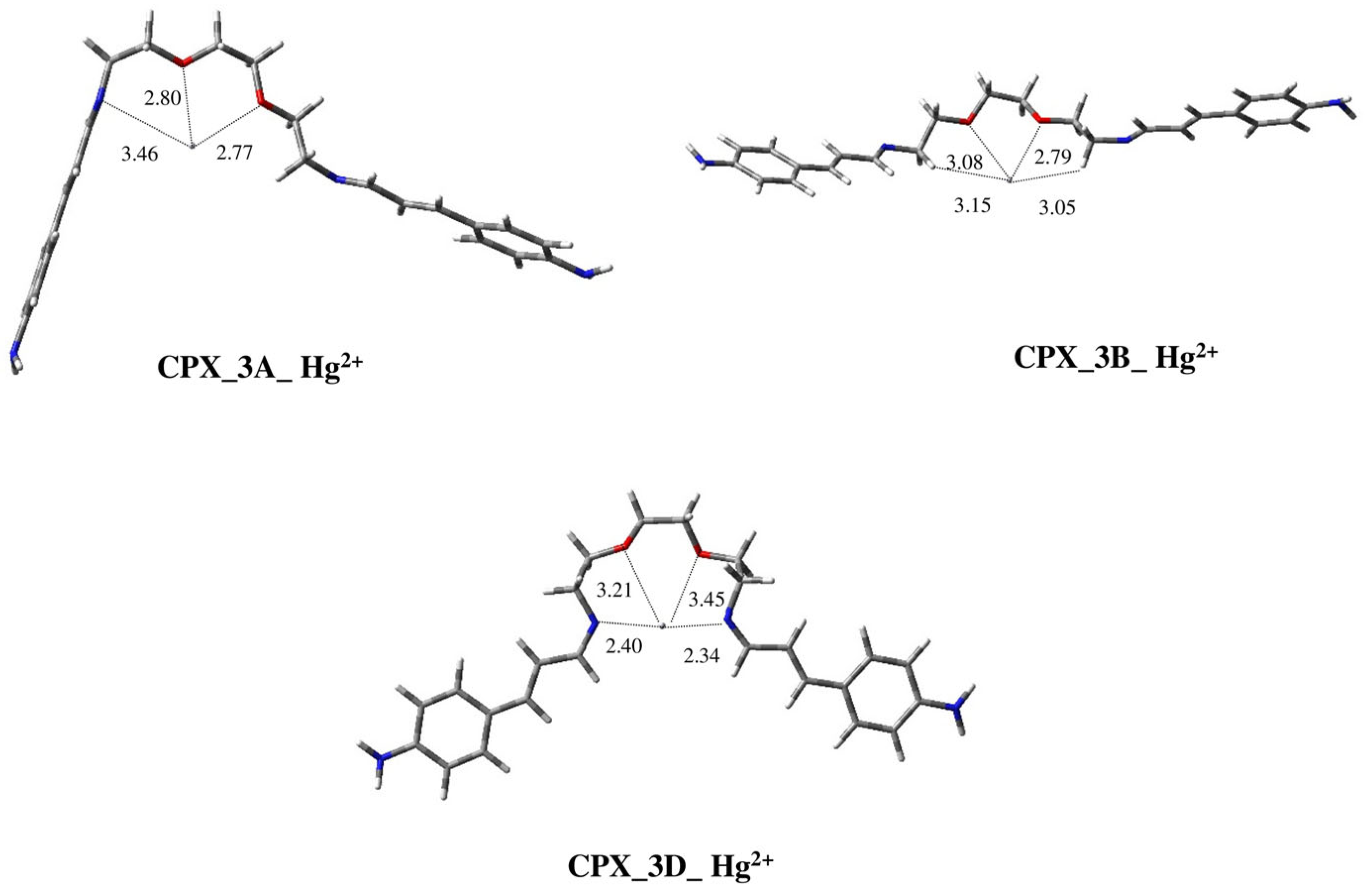
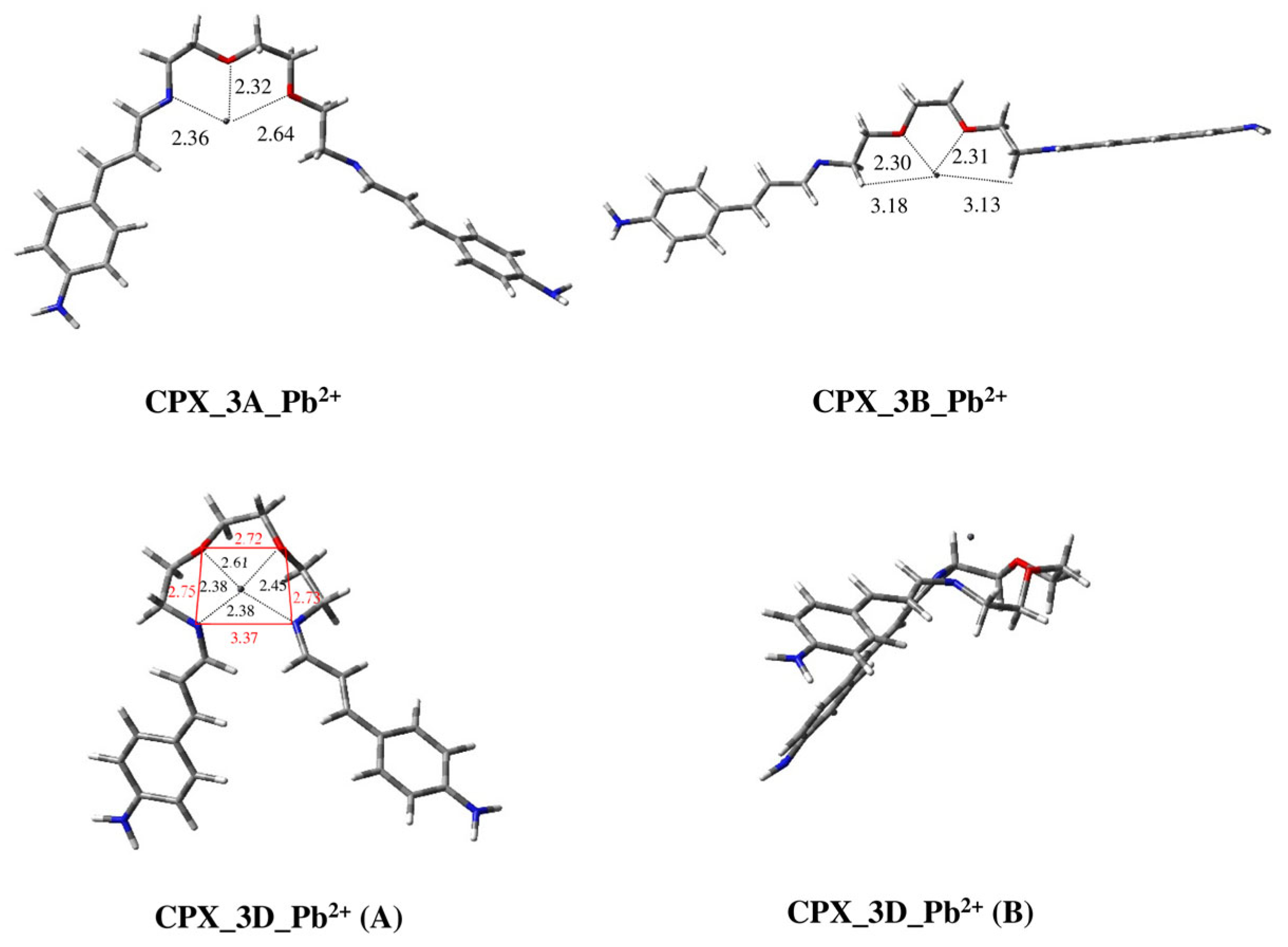
2.2. Modeling Study of Complexes of Ligands 1–3 with Hg2+ and Pb2+ Metal Ions
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Briffa, J.; Sinagra, E.; Blundell, R. Heavy metal pollution in the environment and their toxicological effects on humans. Heliyon 2020, 6, e04691. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, J.; Huang, X.; Zhang, L.; Yang, C.; Li, E.; Wang, Z. Heavy metal distribution and bioaccumulation combined with ecological and human health risk evaluation in a typical urban plateau lake, southwest China. Front. Environ. Sci. 2022, 10, 814678. [Google Scholar] [CrossRef]
- Gall, J.E.; Boyd, R.S.; Rajakaruna, N. Transfer of heavy metals through terrestrial food webs: A review. Environ. Monit. Assess. 2015, 187, 201. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Chen, J.; Zhu, F.; Chai, L.; Lin, Z.; Zhang, K.; Shi, S. Biological toxicity of heavy metal(loid)s in natural environments: From microbes to humans. Front. Environ. Sci. 2022, 10, 920957. [Google Scholar] [CrossRef]
- Andreoli, V.; Sprovieri, F. Genetic aspects of susceptibility to mercury toxicity: An overview. Int. J. Environ. Res. Public Health 2017, 14, 93. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, M.; Kaleem, M.; Siddiqui, J. Environmental lead toxicity and nutritional factors. Clin. Nutr. 2007, 26, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Monger, A.; Wangdi, K. Lead and mercury exposure and related health problems in metal artisan workplaces and high-risk household contacts in Thimphu, Bhutan. Sci. World J. 2020, 2020, 9267181. [Google Scholar] [CrossRef] [PubMed]
- Nyamato, G.S. Perspectives and prospects of chelation extraction of heavy metals from wastewater: A review. Water Sci. Technol. 2023, 88, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Sayin, S.; Engin, M.S.; Eymur, S.; Çay, S. Synthesis and characterization of 1-(2-furoyl) piperazine calix[4]arene for the preconcentration of metal ions. Anal. Lett. 2018, 51, 111–118. [Google Scholar] [CrossRef]
- Blue, L.Y.; Jana, P.; Atwood, D.A. Aqueous mercury precipitation with the synthetic dithiolate, BDTH2. Fuel 2010, 89, 1326–1330. [Google Scholar] [CrossRef]
- Ganjali, M.R.; Tavakoli, M.; Faridbod, F.; Riahi, S.; Norouzi, P.; Salavati-Niassari, M. Interaction Study of a new Bis-Bidentate Schiff’s Base with some Metal Ions and its Application in Fabrication of Sm(III) Potentiometric Membrane Sensor. Int. J. Electrochem. Sci. 2008, 3, 1559–1573. [Google Scholar] [CrossRef]
- Puterová, Z.; Valentová, J.; Bojková, Z.; Kožíšek, J.; Devínsky, F. Synthesis, crystal structure and antiradical effect of copper(II) Schiff base complexes containing five-, six- and unusual seven-membered rings. Dalton Trans. 2011, 40, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Abu-Dief, A.M.; Mohamed, I.M.A. A review on versatile applications of transition metal complexes incorporating Schiff bases. Beni-Suef Univ. J. Basic Appl. Sci. 2015, 4, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Oiye, É.N.; Muzetti Ribeiro, M.F.; Katayama, J.M.T.; Tadini, M.C.; Balbino, M.A.; Eleotério, I.C.; Magalhães, J.; Soares Castro, A.; Mota Silva, R.S.; da Cruz Júnior, J.W.; et al. Electrochemical Sensors Containing Schiff Bases and their Transition Metal Complexes to Detect Analytes of Forensic, Pharmaceutical and Environmental Interest. A Review. Crit. Rev. Anal. Chem. 2019, 49, 488–509. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhong, H.; Huang, Y.; Zhao, R. Recent Advances in AIEgens for Metal Ion Biosensing and Bioimaging. Molecules 2019, 24, 4593. [Google Scholar] [CrossRef] [PubMed]
- Boulechfar, C.; Ferkous, H.; Delimi, A.; Djedouani, A.; Kahlouche, A.; Boublia, A.; Darwish, A.S.; Lemaoui, T.; Verma, R.; Benguerba, Y. Schiff bases and their metal Complexes: A review on the history, synthesis, and applications. Inorg. Chem. Commun. 2023, 150, 110451. [Google Scholar] [CrossRef]
- More, M.S.; Joshi, P.G.; Mishra, Y.K.; Khanna, P.K. Metal complexes driven from Schiff bases and semicarbazones for biomedical and allied applications: A review. Mater. Today Chem. 2019, 14, 100195. [Google Scholar] [CrossRef]
- Kumar, R.; Singh, B.; Gahlyan, P.; Kumar, R.; Pani, B. Recent developments on the colorimetric and fluorometric detection of 3d block metal ions using Schiff base probes: A review. J. Mol. Struct. 2023, 1289, 135859. [Google Scholar] [CrossRef]
- Wang, X.; Xu, T.; Duan, H. Schiff base fluorescence probes for Cu2+ based on imidazole and benzimidazole. Sens. Actuators B Chem. 2015, 214, 138–143. [Google Scholar] [CrossRef]
- Sherif, O.E.; Abdel-Kader, N.S. DFT calculations, spectroscopic studies, thermal analysis and biological activity of supramolecular Schiff base complexes. Arab. J. Chem. 2018, 11, 700–713. [Google Scholar] [CrossRef]
- Chai, L.-Q.; An, H.-L.; Chen, T.-T.; Cai, Y.-Y. Structural, spectroscopic, theoretical calculation and Hirshfeld surface analyses of 3-D supramolecular dinuclear zinc(II) and copper(II) complexes. J. Mol. Struct. 2024, 1299, 137151. [Google Scholar] [CrossRef]
- Bouzaheur, A.; Bouchoucha, A.; Larbi, K.S.; Zaater, S. Experimental and DFT studies of a novel Schiff base sulfonamide derivative ligand and its palladium (II) and platinum (IV) complexes: Antimicrobial activity, cytotoxicity, and molecular docking study. J. Mol. Struct. 2022, 1261, 132811. [Google Scholar] [CrossRef]
- Iannazzo, D.; Pistone, A.; Ziccarelli, I.; Espro, C.; Galvagno, S.; Giofré, S.V.; Romeo, R.; Cicero, N.; Bua, G.D.; Lanza, G.; et al. Removal of heavy metal ions from wastewaters using dendrimer-functionalized multi-walled carbon nanotubes. Environ. Sci. Pollut. Res. 2017, 24, 14735–14747. [Google Scholar] [CrossRef]
- Chiacchio, M.A.; Legnani, L. Density functional theory calculations: A useful tool to investigate mechanisms of 1,3-dipolar cycloaddition reactions. Int. J. Mol. Sci. 2024, 25, 1298. [Google Scholar] [CrossRef]
- Biagiotti, G.; Legnani, L.; Aresta, G.; Chiacchio, M.A.; Richichi, B. Benzo[c] [1,2]thiazine-based analogs in the inverse electron demand [4+2] hetero Diels-Alder reaction with glycals: Access to tetracyclic fused galactose and fucose derivatives. Eur. J. Org. Chem. 2022, 2022, e20220076. [Google Scholar] [CrossRef]
- Chiacchio, M.A.; Legnani, L.; Campisi, A.; Bottino, P.; Lanza, G.; Iannazzo, D.; Veltri, L.; Giofrè, S.V.; Romeo, R. 1,2,4-Oxadiazole-5-ones as analogues of tamoxifen: Synthesis and biological evaluation. Org. Biomol. Chem. 2019, 17, 4892–4905. [Google Scholar] [CrossRef]
- Chiacchio, M.A.; Iannazzo, D.; Giofrè, S.V.; Romeo, R.; Legnani, L. Ruthenium tetroxide oxidation of N-methyl-isoxazolidine: Computational mechanistic study. Arab. J. Chem. 2022, 15, 104063. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Case, D.A.; Walker, R.C.; Cheatham, T.E., III; Simmerling, C.; Roitberg, A.; Merz, K.M.; Luo, R.; Li, P.; Darden, T.; Sagui, C.; et al. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comp. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K.V. MD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J.; Cammi, R.; Cheeseman, J.R.; Frisch, M.J.; Devlin, F.J.; Gabriel, S.; Stephens, P.J. Polarizable continuum model (PCM) calculations of solvent effects on optical rotations of chiral molecules. J. Phys. Chem. A 2002, 106, 6102–6113. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations—Potentials for the transition-metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔE [kcal mol−1] | % | d [Å] [a] | τ1 [°] [b] | τ2 [°] [c] | τ3 [°] [d] | τ4 [°] [e] | τ5 [°] [f] | τ6 [°] [g] | |
|---|---|---|---|---|---|---|---|---|---|
| 1A | 0.00 | 27.5 | 6.81 | −178 | −119 | −177 | 178 | 120 | −177 |
| 1B | 0.34 | 15.4 | 5.89 | 177 | −119 | −176 | −66 | 127 | 177 |
| 1C | 0.36 | 15.0 | 5.63 | 179 | 119 | 177 | −73 | −7 | −177 |
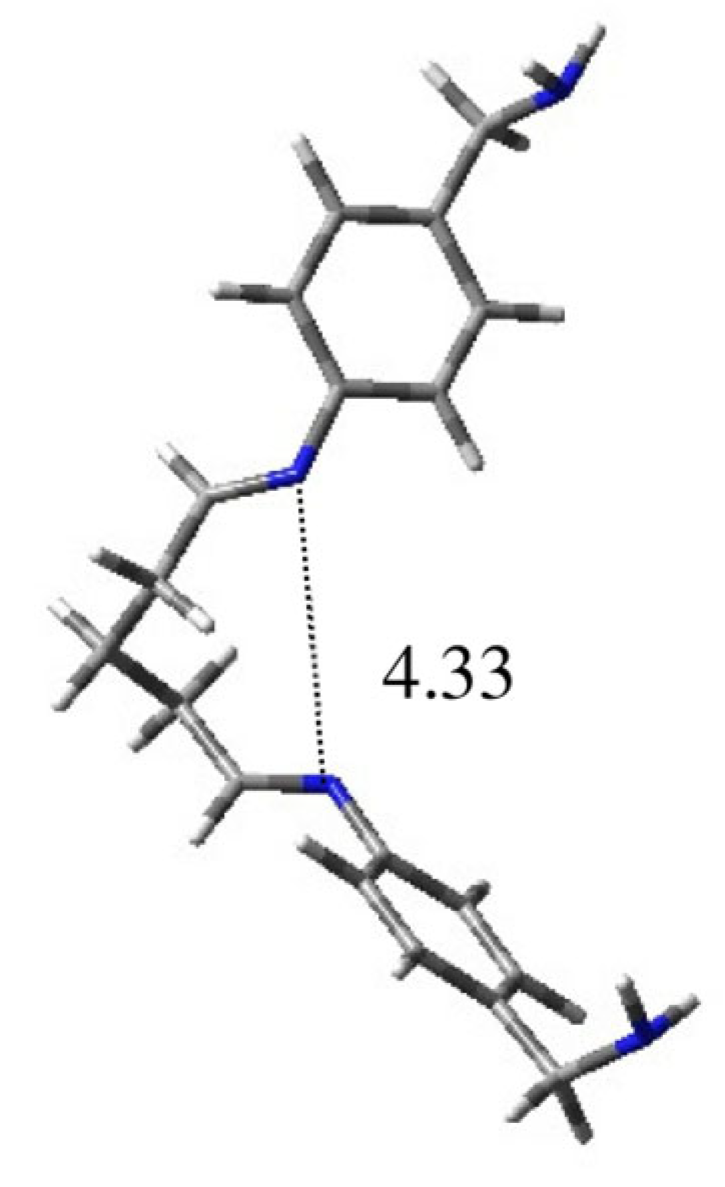
| 1D | 0.39 | 14.3 | 4.33 | 179 | −127 | 67 | 67 | −128 | −176 |
| 1E | 0.40 | 14.1 | 5.87 | 177 | 126 | −66 | −176 | −119 | 177 |
| 1F | 0.53 | 11.2 | 6.28 | 177 | 118 | 66 | 173 | 121 | 178 |
| others | / | 2.0 | / | / | / | / | / | / | / |
| ΔE [kcal mol−1] | % | d [Å] [a] | τ1 [°] [b] | τ2 [°] [c] | τ3 [°] [d] | τ4 [°] [e] | τ5 [°] [f] | τ6 [°] [g] | τ7 [°] [h] | |
|---|---|---|---|---|---|---|---|---|---|---|
| 2A | 0.00 | 45.4 | 6.24 | −180 | 121 | 176 | 179 | −177 | −122 | 180 |
| 2B | 0.23 | 30.5 | 5.44 | 180 | −121 | −176 | 179 | 67 | −126 | −179 |
| 2C | 0.65 | 15.1 | 5.40 | −180 | 121 | 177 | 178 | 64 | 123 | 180 |
| 2D | 1.53 | 3.4 | 4.80 | 180 | −122 | 179 | 65 | 61 | 123 | 180 |
| others | / | 6 | / | / | / | / | / | / | / | / |
| ΔE [kcal mol−1] | % | τ1 [°] [a] | τ2 [°] [b] | τ3 [°] [c] | τ4 [°] [d] | τ5 [°] [e] | τ6 [°] [f] | τ7 [°] [g] | τ8 [°] [h] | τ9 [°] [i] | τ10 [°] [l] | τ11 [°] [m] | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3A | 0.00 | 66.4 | 180 | −123 | 69 | 180 | 180 | −70 | 179 | 179 | 175 | 124 | 180 |
| 3B | 0.55 | 26.1 | 180 | −124 | −175 | −179 | 180 | −70 | 180 | 180 | 176 | 124 | 180 |
| 3C | 1.35 | 6.9 | 180 | −124 | −175 | −179 | 180 | 180 | 180 | 180 | 175 | 125 | 180 |
| others | / | 0.6 | / | / | / | / | / | / | / | / | / | / | / |
| Name | ΔE (kcal/mol) |
|---|---|
| CPX_1D_Hg2+ | 0.00 |
| CPX_1D_Pb2+ | 0.00 |
| CPX_2C_Hg2+ | 2.38 |
| CPX_2D_Hg2+ | 0.00 |
| CPX_2NEW_Pb2+ | 0.00 |
| CPX_2D_Pb2+ | 2.38 |
| CPX_3A_Hg2+ | 7.90 |
| CPX_3B_Hg2+ | 8.24 |
| CPX_3D_Hg2+ | 0.00 |
| CPX_3A_Pb2+ | >20 |
| CPX_3B_Pb2+ | >20 |
| CPX_3D_Pb2+ | 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiacchio, M.A.; Campisi, A.; Iannazzo, D.; Giofrè, S.V.; Legnani, L. Design of New Schiff Bases and Their Heavy Metal Ion Complexes for Environmental Applications: A Molecular Dynamics and Density Function Theory Study. Int. J. Mol. Sci. 2024, 25, 4159. https://doi.org/10.3390/ijms25084159
Chiacchio MA, Campisi A, Iannazzo D, Giofrè SV, Legnani L. Design of New Schiff Bases and Their Heavy Metal Ion Complexes for Environmental Applications: A Molecular Dynamics and Density Function Theory Study. International Journal of Molecular Sciences. 2024; 25(8):4159. https://doi.org/10.3390/ijms25084159
Chicago/Turabian StyleChiacchio, Maria Assunta, Agata Campisi, Daniela Iannazzo, Salvatore V. Giofrè, and Laura Legnani. 2024. "Design of New Schiff Bases and Their Heavy Metal Ion Complexes for Environmental Applications: A Molecular Dynamics and Density Function Theory Study" International Journal of Molecular Sciences 25, no. 8: 4159. https://doi.org/10.3390/ijms25084159