
DNA Damage and Parkinson’s Disease


Abstract
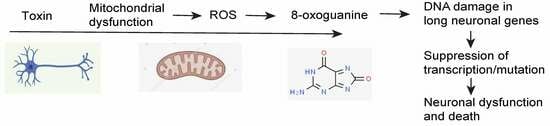
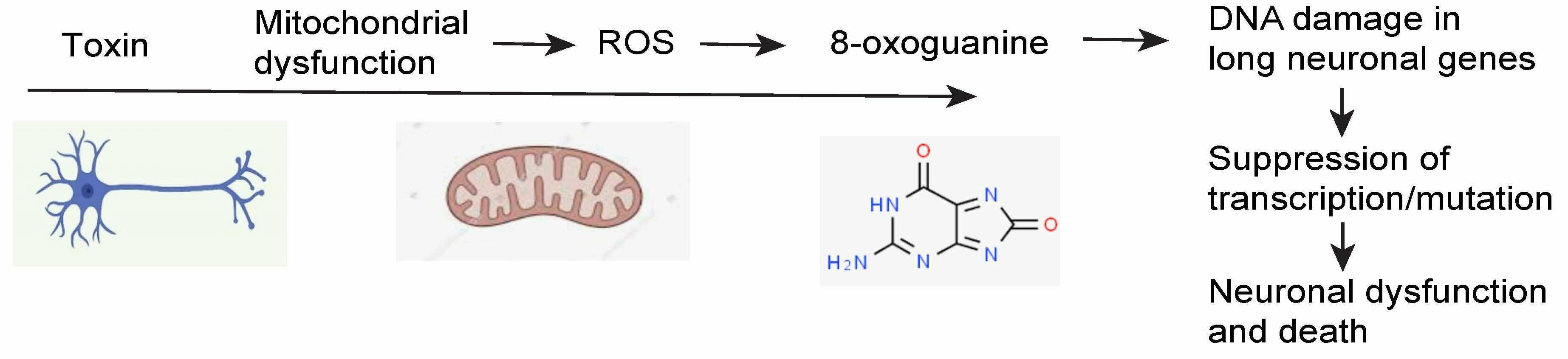
1. Introduction
2. DNA Damage and Parkinson’s Disease—The Intriguing Case of Trichloroethylene
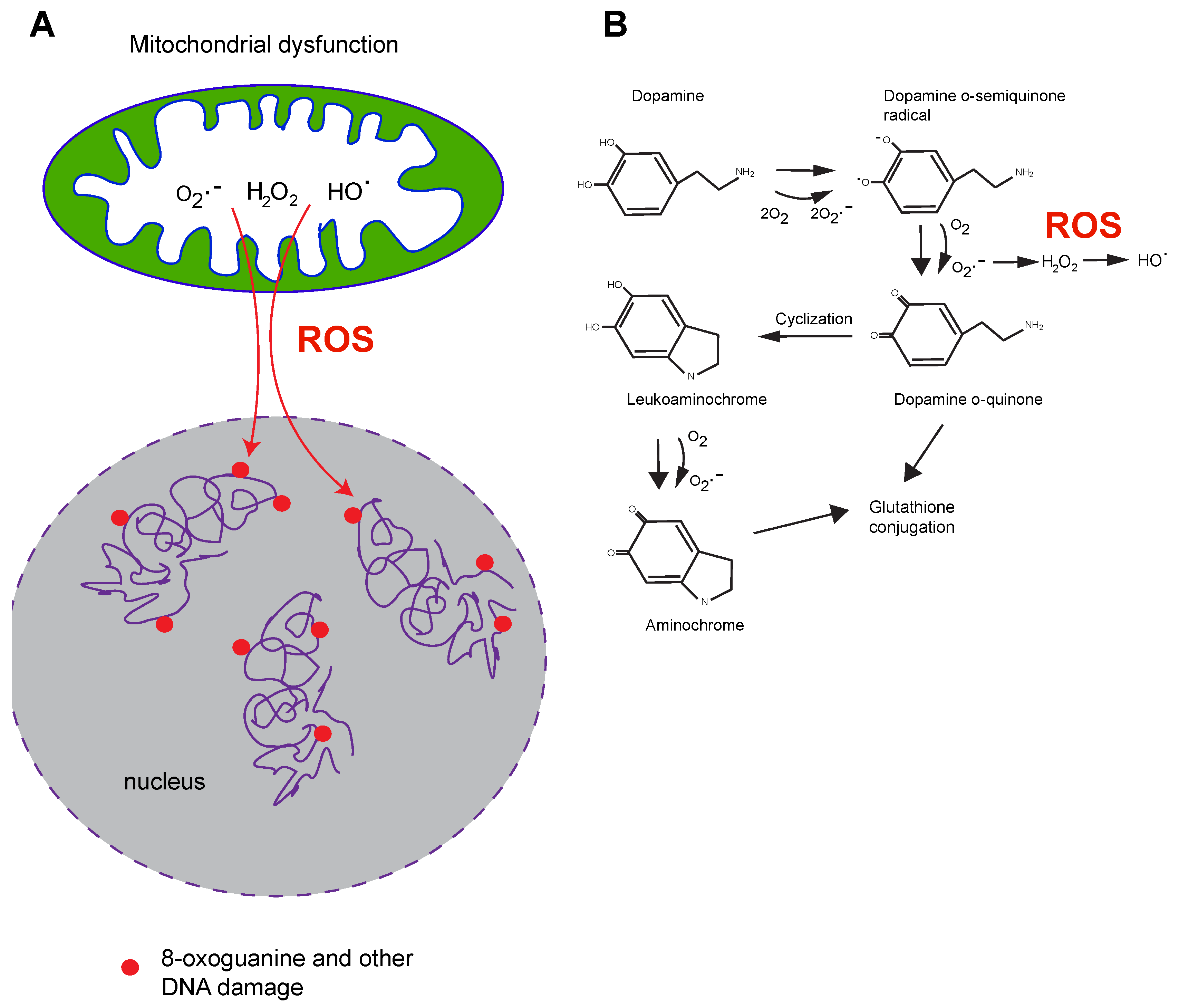
3. Mitochondrial Dysfunction, Mitochondrial Toxins, and Parkinson’s Disease
4. Defects of Mitochondrial Pathways in Familial and Sporadic PD
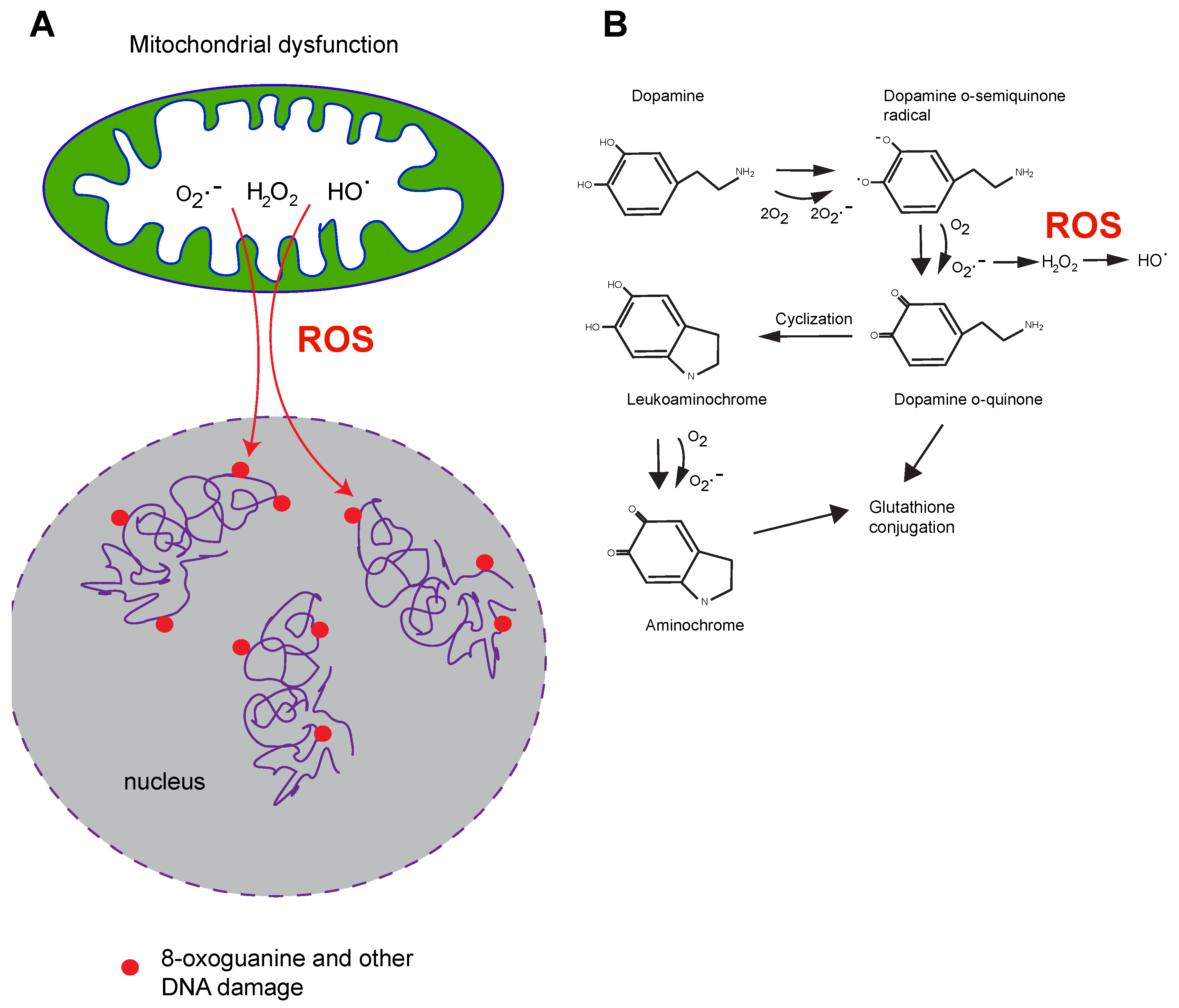
5. DNA Damage in PD
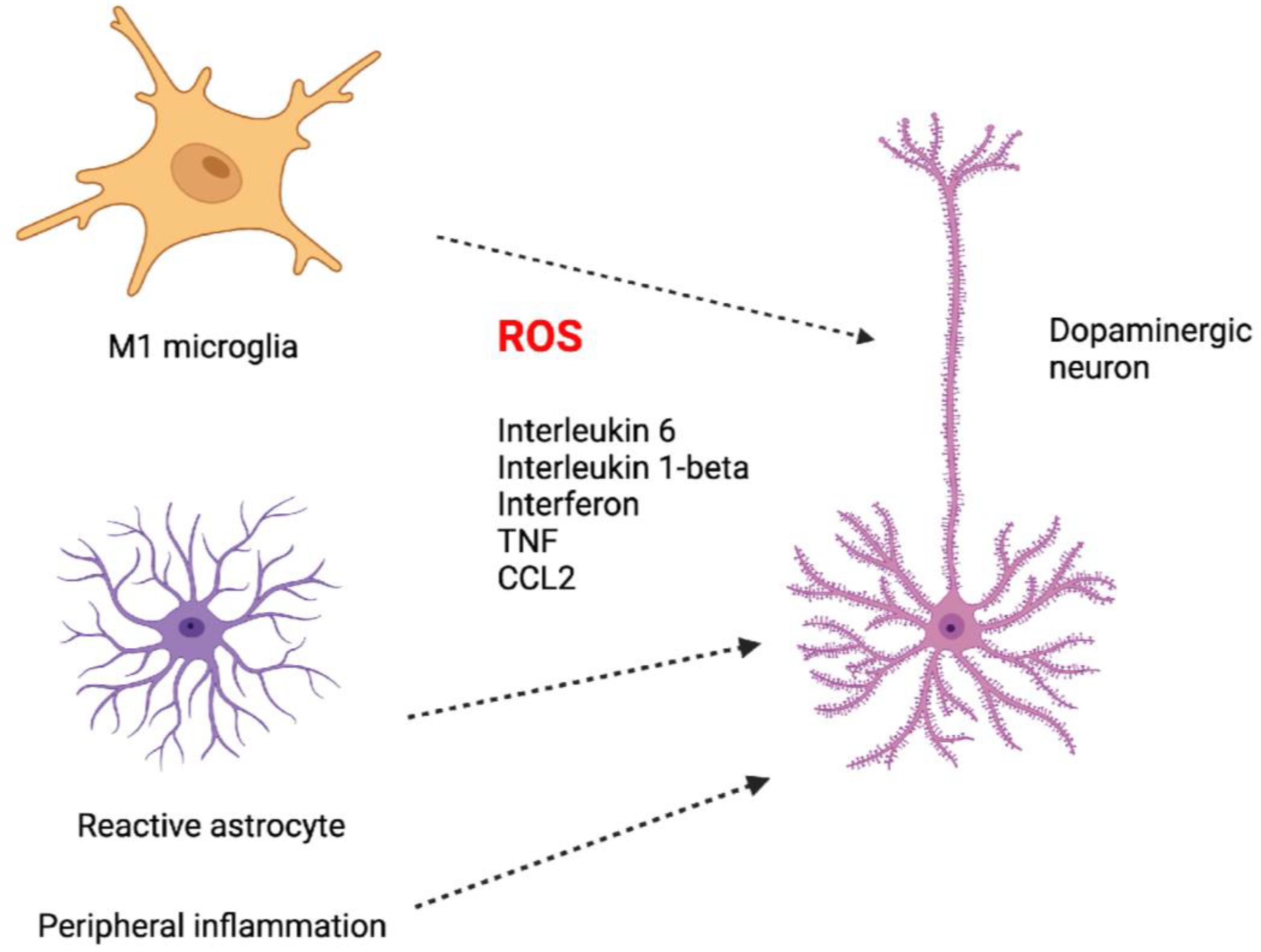
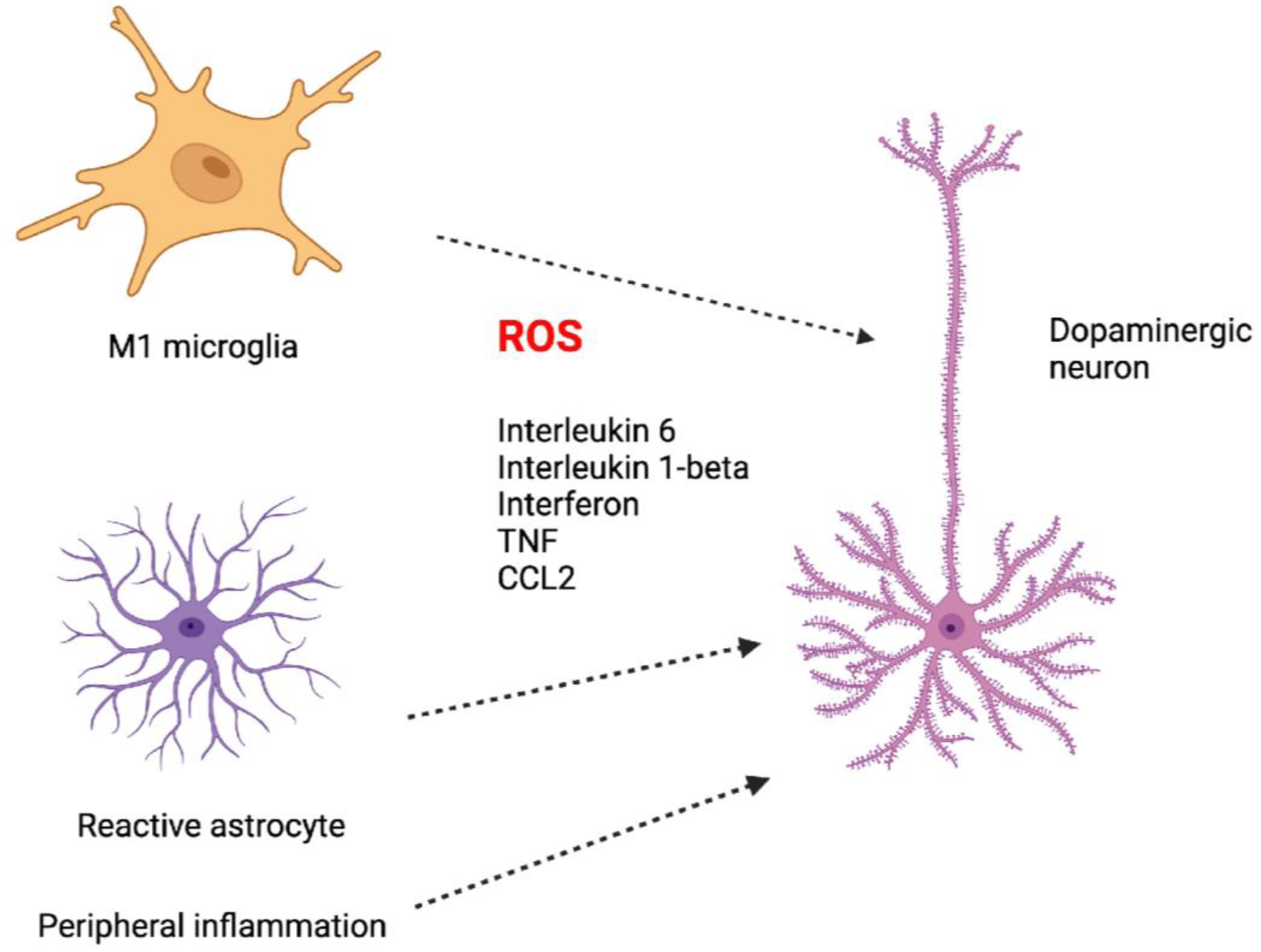
6. Inflammation and PD
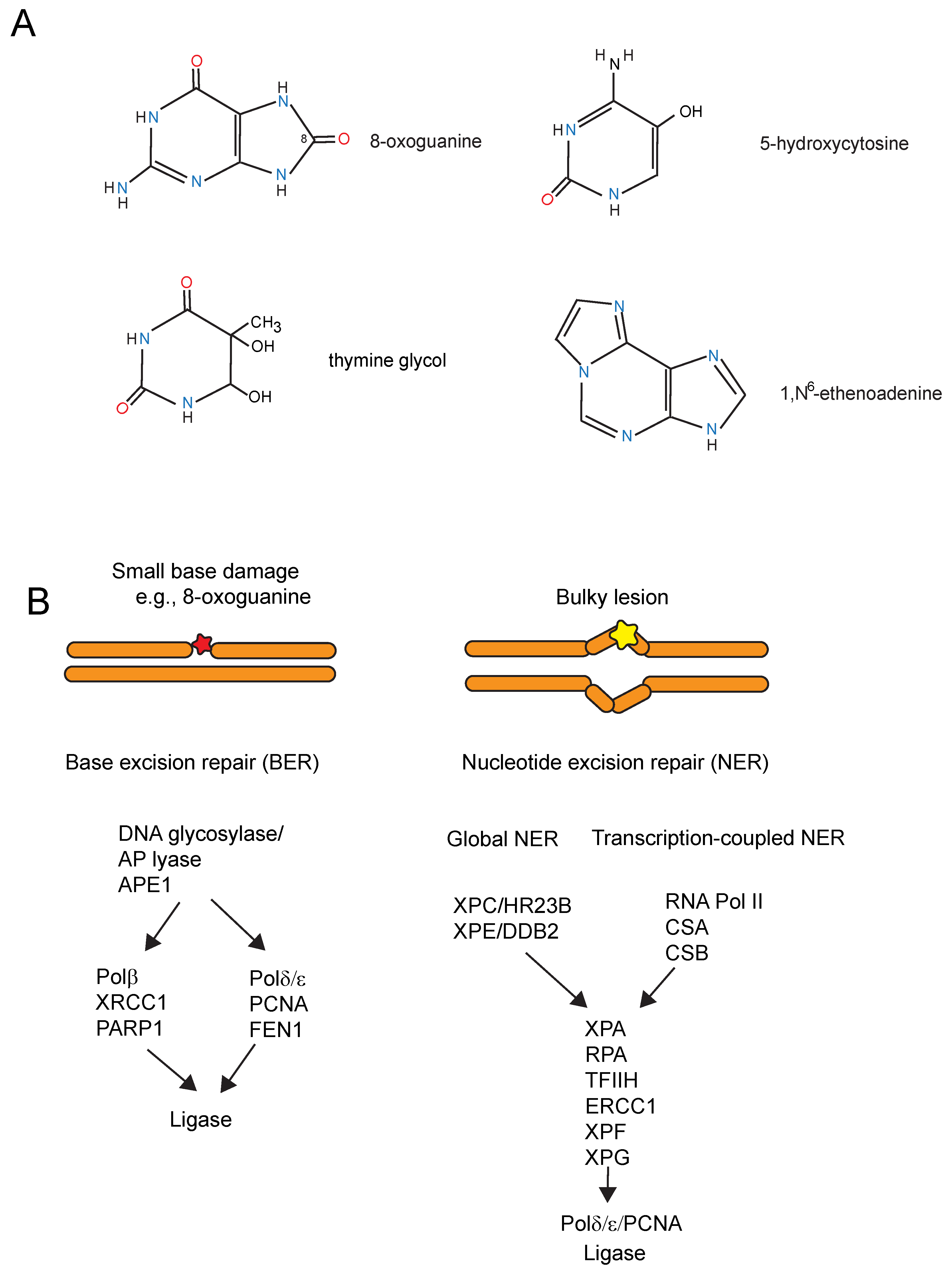
7. DNA Repair Deficiencies and Neurodegeneration
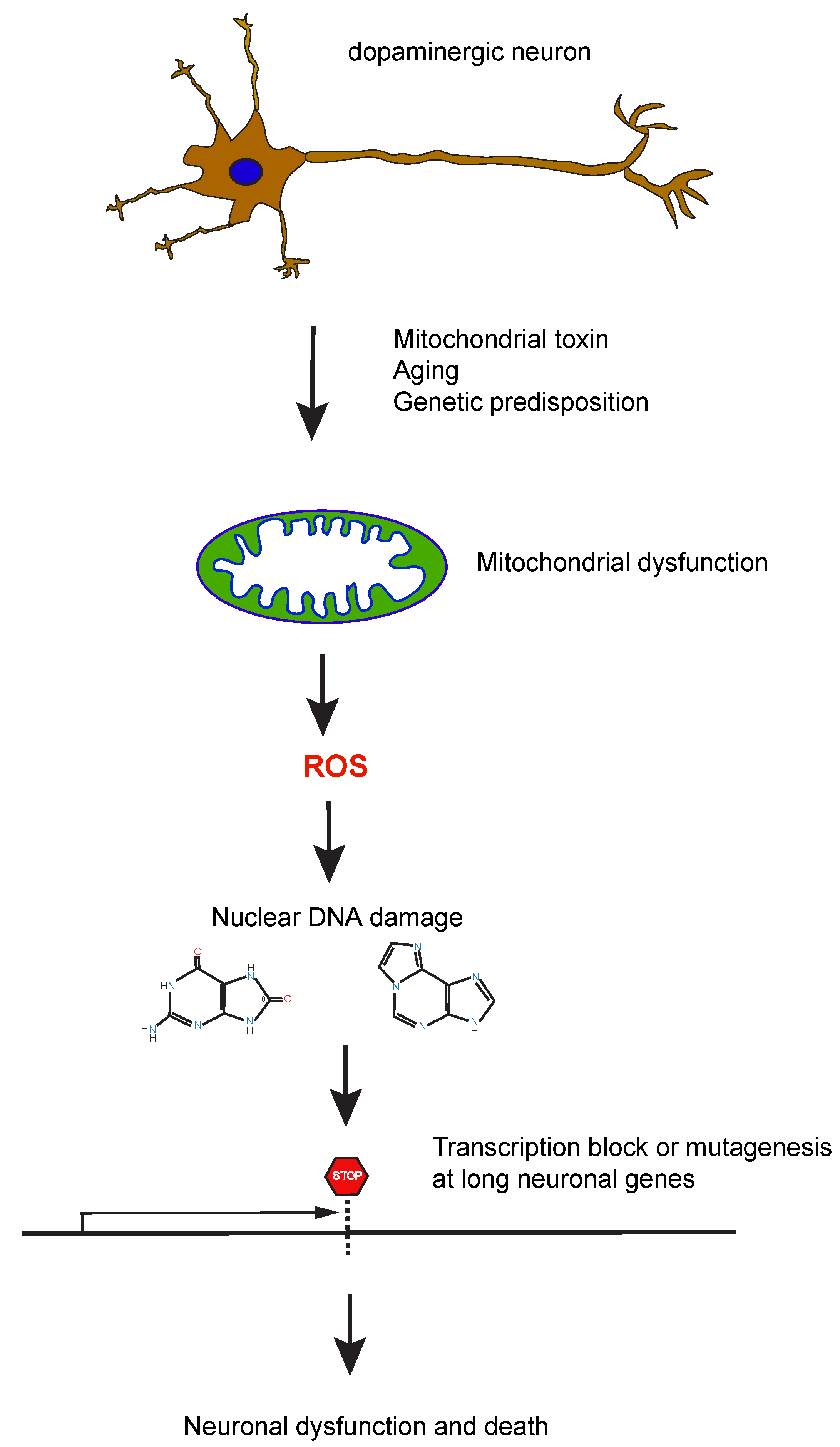
8. Potential Mechanisms of How DNA Damage or Repair Deficiency May Contribute to Parkinson’s Disease
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cherian, A.; Divya, K.P.; Vijayaraghavan, A. Parkinson’s disease—Genetic cause. Curr. Opin. Neurol. 2023, 36, 292–301. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.M.; Diederich, N.J.; Kruger, R.; Balling, R. The hallmarks of Parkinson’s disease. FEBS J. 2013, 280, 5981–5993. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M., 3rd; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Pavlou, M.A.S.; Outeiro, T.F. Epigenetics in Parkinson’s Disease. Adv. Exp. Med. Biol. 2017, 978, 363–390. [Google Scholar] [PubMed]
- van Heesbeen, H.J.; Smidt, M.P. Entanglement of genetics and epigenetics in Parkinson’s disease. Front. Neurosci. 2019, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shlomo, Y.; Darweesh, S.; Llibre-Guerra, J.; Marras, C.; San Luciano, M.; Tanner, C. The epidemiology of Parkinson’s disease. Lancet 2024, 403, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Ball, N.; Teo, W.P.; Chandra, S.; Chapman, J. Parkinson’s Disease and the environment. Front. Neurol. 2019, 10, 218. [Google Scholar] [CrossRef]
- Goldman, S.M.; Weaver, F.M.; Stroupe, K.T.; Cao, L.; Gonzalez, B.; Colletta, K.; Brown, E.G.; Tanner, C.M. Risk of Parkinson disease among service members at marine corps base Camp Lejeune. JAMA Neurol. 2023, 80, 673–681. [Google Scholar] [CrossRef]
- Wadman, M. Solvent exposure strongly linked to Parkinson’s. Science 2023, 380, 683. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Zafar, M.; Lettenberger, S.E.; Pawlik, M.E.; Kinel, D.; Frissen, M.; Schneider, R.B.; Kieburtz, K.; Tanner, C.M.; De Miranda, B.R.; et al. Trichloroethylene: An invisible cause of Parkinson’s disease? J. Parkinson’s Dis. 2023, 13, 203–218. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, B.R.; Greenamyre, J.T. Trichloroethylene, a ubiquitous environmental contaminant in the risk for Parkinson’s disease. Environ. Sci. Process. Impacts 2020, 22, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Choi, D.Y.; Hunter, R.L.; Pandya, J.D.; Cass, W.A.; Sullivan, P.G.; Kim, H.C.; Gash, D.M.; Bing, G. Trichloroethylene induces dopaminergic neurodegeneration in Fisher 344 rats. J. Neurochem. 2010, 112, 773–783. [Google Scholar] [CrossRef]
- De Miranda, B.R.; Castro, S.L.; Rocha, E.M.; Bodle, C.R.; Johnson, K.E.; Greenamyre, J.T. The industrial solvent trichloroethylene induces LRRK2 kinase activity and dopaminergic neurodegeneration in a rat model of Parkinson’s disease. Neurobiol. Dis. 2021, 153, 105312. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Shin, E.J.; Dang, D.K.; Jin, C.H.; Lee, P.H.; Jeong, J.H.; Park, S.J.; Kim, Y.S.; Xing, B.; Xin, T.; et al. Trichloroethylene and Parkinson’s disease: Risk assessment. Mol. Neurobiol. 2018, 55, 6201–6214. [Google Scholar] [CrossRef]
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Gash, D.M.; Rutland, K.; Hudson, N.L.; Sullivan, P.G.; Bing, G.; Cass, W.A.; Pandya, J.D.; Liu, M.; Choi, D.Y.; Hunter, R.L.; et al. Trichloroethylene: Parkinsonism and complex 1 mitochondrial neurotoxicity. Ann. Neurol. 2008, 63, 184–192. [Google Scholar] [CrossRef]
- Ibarra-Gutierrez, M.T.; Serrano-Garcia, N.; Orozco-Ibarra, M. Rotenone-induced model of Parkinson’s disease: Beyond mitochondrial complex I inhibition. Mol. Neurobiol. 2023, 60, 1929–1948. [Google Scholar] [CrossRef]
- Paul, K.C.; Krolewski, R.C.; Lucumi Moreno, E.; Blank, J.; Holton, K.M.; Ahfeldt, T.; Furlong, M.; Yu, Y.; Cockburn, M.; Thompson, L.K.; et al. A pesticide and iPSC dopaminergic neuron screen identifies and classifies Parkinson-relevant pesticides. Nat. Commun. 2023, 14, 2803. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Huo, Q.; Tabassum, S.; Jiang, J.; Ahmed, A.; Chen, X.; Zhou, J.; Zhang, J.; Liu, S.; et al. Mitochondrial Deficits With Neural and Social Damage in Early-Stage Alzheimer’s Disease Model Mice. Front. Aging Neurosci. 2021, 13, 748388. [Google Scholar] [CrossRef] [PubMed]
- Caboni, P.; Sherer, T.B.; Zhang, N.; Taylor, G.; Na, H.M.; Greenamyre, J.T.; Casida, J.E. Rotenone, deguelin, their metabolites, and the rat model of Parkinson’s disease. Chem. Res. Toxicol. 2004, 17, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect 2011, 119, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Liu, B.; Hong, J.S. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J. Neurosci. 2003, 23, 6181–6187. [Google Scholar] [CrossRef] [PubMed]
- Barbeau, A.; Roy, M.; Bernier, G.; Campanella, G.; Paris, S. Ecogenetics of Parkinson’s disease: Prevalence and environmental aspects in rural areas. Can. J. Neurol. Sci. 1987, 14, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Tangamornsuksan, W.; Lohitnavy, O.; Sruamsiri, R.; Chaiyakunapruk, N.; Norman Scholfield, C.; Reisfeld, B.; Lohitnavy, M. Paraquat exposure and Parkinson’s disease: A systematic review and meta-analysis. Arch. Environ. Occup. Health 2019, 74, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.C.; Cockburn, M.; Gong, Y.; Bronstein, J.; Ritz, B. Agricultural paraquat dichloride use and Parkinson’s disease in California’s Central Valley. Int. J. Epidemiol. 2024, 53, dyae004. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Mittal, P. Paraquat (herbicide) as a cause of Parkinson’s Disease. Parkinsonism. Relat. Disord. 2024, 119, 105932. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular senescence is Induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef]
- Cristovao, A.C.; Campos, F.L.; Je, G.; Esteves, M.; Guhathakurta, S.; Yang, L.; Beal, M.F.; Fonseca, B.M.; Salgado, A.J.; Queiroz, J.; et al. Characterization of a Parkinson’s disease rat model using an upgraded paraquat exposure paradigm. Eur. J. Neurosci. 2020, 52, 3242–3255. [Google Scholar] [CrossRef] [PubMed]
- Burns, R.S.; Chiueh, C.C.; Markey, S.P.; Ebert, M.H.; Jacobowitz, D.M.; Kopin, I.J. A primate model of parkinsonism: Selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc. Natl. Acad. Sci. USA 1983, 80, 4546–4550. [Google Scholar] [CrossRef] [PubMed]
- Markey, S.P.; Johannessen, J.N.; Chiueh, C.C.; Burns, R.S.; Herkenham, M.A. Intraneuronal generation of a pyridinium metabolite may cause drug-induced parkinsonism. Nature 1984, 311, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Pantic, I.; Cumic, J.; Skodric, S.R.; Dugalic, S.; Brodski, C. Oxidopamine and oxidative stress: Recent advances in experimental physiology and pharmacology. Chem. Biol. Interact. 2021, 336, 109380. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kim-Han, J.S.; Harmon, S.; Sakiyama-Elbert, S.E.; O’Malley, K.L. The Parkinsonian mimetic, 6-OHDA, impairs axonal transport in dopaminergic axons. Mol. Neurodegener. 2014, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Glinka, Y.; Tipton, K.F.; Youdim, M.B. Nature of inhibition of mitochondrial respiratory complex I by 6-Hydroxydopamine. J. Neurochem. 1996, 66, 2004–2010. [Google Scholar] [CrossRef] [PubMed]
- Dolle, C.; Flones, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef]
- Sian-Hülsmann, J.; Mandel, S.; Youdim, M.B.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. J. Neurochem. 2011, 118, 939–957. [Google Scholar] [CrossRef]
- Pyatha, S.; Kim, H.; Lee, D.; Kim, K. Association between heavy metal exposure and Parkinson’s disease: A review of the mechanisms related to oxidative stress. Antioxidants 2022, 11, 2467. [Google Scholar] [CrossRef] [PubMed]
- Trempe, J.F.; Gehring, K. Structural mechanisms of mitochondrial quality control mediated by PINK1 and Parkin. J. Mol. Biol. 2023, 435, 168090. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, R.; Cookson, M.R. Pathways to Parkinsonism Redux: Convergent pathobiological mechanisms in genetics of Parkinson’s disease. Hum. Mol. Genet. 2015, 24, R32–R44. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, K.J.; Barbosa, I.A.; Bandres-Ciga, S.; Quinn, J.P.; Bubb, V.J.; Deshpande, C.; Botia, J.A.; Reynolds, R.H.; Zhang, D.; Simpson, M.A.; et al. Mitochondria function associated genes contribute to Parkinson’s Disease risk and later age at onset. NPJ Parkinsons Dis. 2019, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.E.; Zheng, Q. The tale of DJ-1 (PARK7): A Swiss army knife in biomedical and psychological research. Int. J. Mol. Sci. 2023, 24, 7409. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, T.; Mirzaei-Behbahani, B.; Zadali, R.; Pirhaghi, M.; Morozova-Roche, L.A.; Meratan, A.A. Common mechanisms underlying alpha-synuclein-induced mitochondrial dysfunction in Parkinson’s disease. J. Mol. Biol. 2023, 435, 167992. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.L.; Grossmann, D.; Delcambre, S.; Hermann, A.; Grunewald, A. Novel insights into Parkin-mediated mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Curr. Opin. Neurobiol. 2023, 80, 102720. [Google Scholar] [CrossRef] [PubMed]
- Skou, L.D.; Johansen, S.K.; Okarmus, J.; Meyer, M. Pathogenesis of DJ-1/PARK7-mediated Parkinson’s disease. Cells 2024, 13, 296. [Google Scholar] [CrossRef]
- Singh, F.; Ganley, I.G. Parkinson’s disease and mitophagy: An emerging role for LRRK2. Biochem. Soc. Trans. 2021, 49, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.T.; Chen, X.; Otero, P.A.; Moore, D.J. Understanding the contributions of VPS35 and the retromer in neurodegenerative disease. Neurobiol. Dis. 2022, 170, 105768. [Google Scholar] [CrossRef]
- Dang, T.; Cao, W.J.; Zhao, R.; Lu, M.; Hu, G.; Qiao, C. ATP13A2 protects dopaminergic neurons in Parkinson’s disease: From biology to pathology. J. Biomed. Res. 2022, 36, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Schreglmann, S.R.; Houlden, H. VPS13C-another hint at mitochondrial dysfunction in familial Parkinson’s disease. Mov. Disord. 2016, 31, 1340. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.L.; Jiang, Z.; Wang, J.Y.; He, Q.; Li, S.X.; Gu, X.J.; Qi, Y.R.; Zhang, M.; Yang, W.J.; Cao, B.; et al. Loss of CHCHD2 stability coordinates with C1QBP/CHCHD2/CHCHD10 complex impairment to mediate PD-linked mitochondrial dysfunction. Mol. Neurobiol. 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef]
- Liu, J.; Tan, J.; Tang, B.; Guo, J. Unveiling the role of iPLA(2)beta in neurodegeneration: From molecular mechanisms to advanced therapies. Pharmacol. Res. 2024, 202, 107114. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Choi, W.S.; Sorscher, N.; Park, H.J.; Tronche, F.; Palmiter, R.D.; Xia, Z. Genetic reduction of mitochondrial complex I function does not lead to loss of dopamine neurons in vivo. Neurobiol. Aging 2015, 36, 2617–2627. [Google Scholar] [CrossRef] [PubMed]
- Subrahmanian, N.; LaVoie, M.J. Is there a special relationship between complex I activity and nigral neuronal loss in Parkinson’s disease? A critical reappraisal. Brain Res. 2021, 1767, 147434. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Harvey, L.M.R.; Mitchell, E.; Lawson, A.R.J.; Lensing, S.V.; Ellis, P.; Russell, A.J.C.; Alcantara, R.E.; Baez-Ortega, A.; Wang, Y.; et al. Somatic mutation landscapes at single-molecule resolution. Nature 2021, 593, 405–410. [Google Scholar] [CrossRef]
- Blokzijl, F.; de Ligt, J.; Jager, M.; Sasselli, V.; Roerink, S.; Sasaki, N.; Huch, M.; Boymans, S.; Kuijk, E.; Prins, P.; et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016, 538, 260–264. [Google Scholar] [CrossRef]
- Müller-Nedebock, A.C.; Brennan, R.R.; Venter, M.; Pienaar, I.S.; van der Westhuizen, F.H.; Elson, J.L.; Ross, O.A.; Bardien, S. The unresolved role of mitochondrial DNA in Parkinson’s disease: An overview of published studies, their limitations, and future prospects. Neurochem. Int. 2019, 129, 104495. [Google Scholar] [CrossRef]
- Müller, W.E.; Eckert, A.; Kurz, C.; Eckert, G.P.; Leuner, K. Mitochondrial dysfunction: Common final pathway in brain aging and Alzheimer’s disease--therapeutic aspects. Mol. Neurobiol. 2010, 41, 159–171. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Breitzig, M.; Bhimineni, C.; Lockey, R.; Kolliputi, N. 4-Hydroxy-2-nonenal: A critical target in oxidative stress? Am. J. Physiol. Cell Physiol. 2016, 311, C537–C543. [Google Scholar] [CrossRef] [PubMed]
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, C.D.; Jenner, P.; Halliwell, B. Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem. 1997, 69, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.G.; Meng, Y.; Johnson, J.; Szabo, P.E.; Pfeifer, G.P. Concordance of hydrogen peroxide-induced 8-oxo-guanine patterns with two cancer mutation signatures of upper GI tract tumors. Sci. Adv. 2022, 8, eabn3815. [Google Scholar] [CrossRef] [PubMed]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.D.; Zhao, X.; Li, Y.; Li, G.R.; Liu, X.L. Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease (Review). Int. J. Mol. Med. 2018, 41, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.B.; Huang, A.Y.; Kim, J.; Zhou, Z.; Kirkham, S.L.; Maury, E.A.; Ziegenfuss, J.S.; Reed, H.C.; Neil, J.E.; Rento, L.; et al. Somatic genomic changes in single Alzheimer’s disease neurons. Nature 2022, 604, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Harding, O.; Holzer, E.; Riley, J.F.; Martens, S.; Holzbaur, E.L.F. Damaged mitochondria recruit the effector NEMO to activate NF-kappaB signaling. Mol. Cell 2023, 83, 3188–3204.e7. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.I.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s disease: Mechanisms and therapeutic implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef]
- Bido, S.; Muggeo, S.; Massimino, L.; Marzi, M.J.; Giannelli, S.G.; Melacini, E.; Nannoni, M.; Gambare, D.; Bellini, E.; Ordazzo, G.; et al. Microglia-specific overexpression of alpha-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat. Commun. 2021, 12, 6237. [Google Scholar] [CrossRef]
- Wang, H.; Lautrup, S.; Caponio, D.; Zhang, J.; Fang, E.F. DNA damage-induced neurodegeneration in accelerated ageing and Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 6748. [Google Scholar] [CrossRef] [PubMed]
- Jaarsma, D.; van der Pluijm, I.; de Waard, M.C.; Haasdijk, E.D.; Brandt, R.; Vermeij, M.; Rijksen, Y.; Maas, A.; van Steeg, H.; Hoeijmakers, J.H.; et al. Age-related neuronal degeneration: Complementary roles of nucleotide excision repair and transcription-coupled repair in preventing neuropathology. PLoS Genet. 2011, 7, e1002405. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Bohr, V.A.; Stevnsner, T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 2011, 94, 166–200. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, G.S.; Nascimento, L.L.S.; Neves, M.R.C.; Leandro, G.D.S.; Garcia, C.C.M.; Menck, C.F.M. Transcription blockage by DNA damage in nucleotide excision repair-related neurological dysfunctions. Semin. Cell Dev. Biol. 2021, 114, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Sepe, S.; Milanese, C.; Gabriels, S.; Derks, K.W.; Payan-Gomez, C.; van IJcken, W.F.; Rijksen, Y.M.; Nigg, A.L.; Moreno, S.; Cerri, S.; et al. Inefficient DNA repair Is an aging-related modifier of Parkinson’s disease. Cell Rep. 2016, 15, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Weidenheim, K.M.; Dickson, D.W.; Rapin, I. Neuropathology of Cockayne syndrome: Evidence for impaired development, premature aging, and neurodegeneration. Mech. Ageing Dev. 2009, 130, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Selby, C.P.; Lindsey-Boltz, L.A.; Li, W.; Sancar, A. Molecular mechanisms of transcription-coupled repair. Annu. Rev. Biochem. 2023, 92, 115–144. [Google Scholar] [CrossRef]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef]
- Sheng, Z.; Oka, S.; Tsuchimoto, D.; Abolhassani, N.; Nomaru, H.; Sakumi, K.; Yamada, H.; Nakabeppu, Y. 8-Oxoguanine causes neurodegeneration during MUTYH-mediated DNA base excision repair. J. Clin. Investig. 2012, 122, 4344–4361. [Google Scholar] [CrossRef]
- Cardozo-Pelaez, F.; Sanchez-Contreras, M.; Nevin, A.B. Ogg1 null mice exhibit age-associated loss of the nigrostriatal pathway and increased sensitivity to MPTP. Neurochem. Int. 2012, 61, 721–730. [Google Scholar] [CrossRef]
- Pao, P.C.; Patnaik, D.; Watson, L.A.; Gao, F.; Pan, L.; Wang, J.; Adaikkan, C.; Penney, J.; Cam, H.P.; Huang, W.C.; et al. HDAC1 modulates OGG1-initiated oxidative DNA damage repair in the aging brain and Alzheimer’s disease. Nat. Commun. 2020, 11, 2484. [Google Scholar] [CrossRef]
- Zylka, M.J.; Simon, J.M.; Philpot, B.D. Gene length matters in neurons. Neuron 2015, 86, 353–355. [Google Scholar] [CrossRef]
- Raychaudhuri, S.; Korn, J.M.; McCarroll, S.A.; International Schizophrenia, C.; Altshuler, D.; Sklar, P.; Purcell, S.; Daly, M.J. Accurately assessing the risk of schizophrenia conferred by rare copy-number variation affecting genes with brain function. PLoS Genet. 2010, 6, e1001097. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.I.; Zhu, Y.; McAvoy, S.; Kuhn, R. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 2006, 232, 48–57. [Google Scholar] [CrossRef] [PubMed]
- King, I.F.; Yandava, C.N.; Mabb, A.M.; Hsiao, J.S.; Huang, H.S.; Pearson, B.L.; Calabrese, J.M.; Starmer, J.; Parker, J.S.; Magnuson, T.; et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature 2013, 501, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Gabel, H.W.; Kinde, B.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015, 522, 89–93. [Google Scholar] [CrossRef]
- Wei, P.C.; Chang, A.N.; Kao, J.; Du, Z.; Meyers, R.M.; Alt, F.W.; Schwer, B. Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell 2016, 164, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.J.; Li, W.X.; Liu, J.Q.; Guo, Y.C.; Wang, Q.; Li, G.H.; Dai, S.X.; Huang, J.F. Low expression of aging-related NRXN3 is associated with Alzheimer disease: A systematic review and meta-analysis. Medicine 2018, 97, e11343. [Google Scholar] [CrossRef]
- Oliver, P.L.; Finelli, M.J.; Edwards, B.; Bitoun, E.; Butts, D.L.; Becker, E.B.; Cheeseman, M.T.; Davies, B.; Davies, K.E. Oxr1 is essential for protection against oxidative stress-induced neurodegeneration. PLoS Genet. 2011, 7, e1002338. [Google Scholar] [CrossRef]
- Kearney, P.J.; Zhang, Y.; Tan, Y.; Kahuno, E.; Conklin, T.L.; Fagan, R.R.; Pavchinskiy, R.G.; Shafer, S.A.; Yue, Z.; Melikian, H.E. Rit2 silencing in dopamine neurons drives a progressive Parkinsonian phenotype. bioRxiv 2023, Preprint. [Google Scholar] [CrossRef]
- Griesius, S.; O’Donnell, C.; Waldron, S.; Thomas, K.L.; Dwyer, D.M.; Wilkinson, L.S.; Hall, J.; Robinson, E.S.J.; Mellor, J.R. Reduced expression of the psychiatric risk gene DLG2 (PSD93) impairs hippocampal synaptic integration and plasticity. Neuropsychopharmacology 2022, 47, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Jagomae, T.; Singh, K.; Philips, M.A.; Jayaram, M.; Seppa, K.; Tekko, T.; Gilbert, S.F.; Vasar, E.; Lillevali, K. Alternative promoter use governs the expression of IgLON cell adhesion molecules in histogenetic fields of the embryonic mouse brain. Int. J. Mol. Sci. 2021, 22, 6955. [Google Scholar] [CrossRef] [PubMed]
- Prashad, S.; Gopal, P.P. RNA-binding proteins in neurological development and disease. RNA Biol. 2021, 18, 972–987. [Google Scholar] [CrossRef]
- Montillot, C.; Skutunova, E.; Ayushma; Dubied, M.; Lahmar, A.; Nguyen, S.; Peerally, B.; Prin, F.; Duffourd, Y.; Thauvin-Robinet, C.; et al. Characterization of Vps13b-mutant mice reveals neuroanatomical and behavioral phenotypes with females less affected. Neurobiol. Dis. 2023, 185, 106259. [Google Scholar] [CrossRef] [PubMed]
- Aldaz, C.M.; Hussain, T. WWOX loss of function in neurodevelopmental and neurodegenerative disorders. Int. J. Mol. Sci. 2020, 21, 8922. [Google Scholar] [CrossRef] [PubMed]
- Sagner, A.; Zhang, I.; Watson, T.; Lazaro, J.; Melchionda, M.; Briscoe, J. A shared transcriptional code orchestrates temporal patterning of the central nervous system. PLoS Biol. 2021, 19, e3001450. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Medina-Menendez, C.; Garcia-Corzo, L.; Cordoba-Beldad, C.M.; Quiroga, A.C.; Calleja Barca, E.; Zinchuk, V.; Munoz-Lopez, S.; Rodriguez-Martin, P.; Ciorraga, M.; et al. SoxD genes are required for adult neural stem cell activation. Cell Rep. 2022, 38, 110313. [Google Scholar] [CrossRef] [PubMed]
- Gcwensa, N.Z.; Russell, D.L.; Cowell, R.M.; Volpicelli-Daley, L.A. Molecular mechanisms underlying synaptic and axon degeneration in Parkinson’s Ddsease. Front. Cell. Neurosci. 2021, 15, 626128. [Google Scholar] [CrossRef]
- Kuraoka, I.; Endou, M.; Yamaguchi, Y.; Wada, T.; Handa, H.; Tanaka, K. Effects of endogenous DNA base lesions on transcription elongation by mammalian RNA polymerase II. Implications for transcription-coupled DNA repair and transcriptional mutagenesis. J. Biol. Chem. 2003, 278, 7294–7299. [Google Scholar] [CrossRef]
- Tornaletti, S.; Maeda, L.S.; Kolodner, R.D.; Hanawalt, P.C. Effect of 8-oxoguanine on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. DNA Repair 2004, 3, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W.; Ward, M.E.; Nussenzweig, A. The threat of programmed DNA damage to neuronal genome integrity and plasticity. Nat. Genet. 2022, 54, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P. DNA repair in neurons and its possible link to the epigenetic machinery at enhancers. Epigenomics 2021, 13, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.G.; Wu, X.; Li, A.X.; Pfeifer, G.P. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011, 39, 5015–5024. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.A.; Qiu, R.; Wu, X.; Li, A.X.; Zhang, H.; Wang, J.; Jui, J.; Jin, S.G.; Jiang, Y.; Pfeifer, G.P.; et al. Dynamics of 5-hydroxymethylcytosine and chromatin marks in mammalian neurogenesis. Cell Rep. 2013, 3, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Gyenis, A.; Chang, J.; Demmers, J.; Bruens, S.T.; Barnhoorn, S.; Brandt, R.M.C.; Baar, M.P.; Raseta, M.; Derks, K.W.J.; Hoeijmakers, J.H.J.; et al. Genome-wide RNA polymerase stalling shapes the transcriptome during aging. Nat. Genet. 2023, 55, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, A.; Gyenis, A.; Pothof, J.; Hoeijmakers, J.H.J.; Papantonis, A.; Beyer, A. Age-associated transcriptional stress due to accelerated elongation and increased stalling of RNAPII. Nat. Genet. 2023, 55, 2011–2012. [Google Scholar] [CrossRef] [PubMed]
- Ibanez-Sole, O.; Barrio, I.; Izeta, A. Age or lifestyle-induced accumulation of genotoxicity is associated with a length-dependent decrease in gene expression. iScience 2023, 26, 106368. [Google Scholar] [CrossRef] [PubMed]
- Soheili-Nezhad, S.; van der Linden, R.J.; Olde Rikkert, M.; Sprooten, E.; Poelmans, G. Long genes are more frequently affected by somatic mutations and show reduced expression in Alzheimer’s disease: Implications for disease etiology. Alzheimers Dement. 2021, 17, 489–499. [Google Scholar] [CrossRef]
- Stoeger, T.; Grant, R.A.; McQuattie-Pimentel, A.C.; Anekalla, K.R.; Liu, S.S.; Tejedor-Navarro, H.; Singer, B.D.; Abdala-Valencia, H.; Schwake, M.; Tetreault, M.P.; et al. Aging is associated with a systemic length-associated transcriptome imbalance. Nat. Aging 2022, 2, 1191–1206. [Google Scholar] [CrossRef]
- Vermeij, W.P.; Dolle, M.E.; Reiling, E.; Jaarsma, D.; Payan-Gomez, C.; Bombardieri, C.R.; Wu, H.; Roks, A.J.; Botter, S.M.; van der Eerden, B.C.; et al. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature 2016, 537, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Toomey, C.E.; Heywood, W.E.; Evans, J.R.; Lachica, J.; Pressey, S.N.; Foti, S.C.; Al Shahrani, M.; D’Sa, K.; Hargreaves, I.P.; Heales, S.; et al. Mitochondrial dysfunction is a key pathological driver of early stage Parkinson’s. Acta Neuropathol. Commun. 2022, 10, 134. [Google Scholar] [CrossRef] [PubMed]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply alpha-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Fu, Y.; Halliday, G.M.; Sue, C.M. PARK genes link mitochondrial dysfunction and alpha-synuclein pathology in sporadic Parkinson’s disease. Front. Cell. Dev. Biol. 2021, 9, 612476. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical | Use | Mode of Action | Structure |
|---|---|---|---|
| MPTP | Synthetic chemical | Converted to neurotoxic MPP+ in the brain. Inhibits mitochondrial complex 1. |  |
| 6-hydroxydopamine | Synthetic chemical used to induce Parkinsonism | Undergoes oxidation to quinones. Production of ROS. |  |
| Trichloroethylene | Cleaning and degreasing agent | Inhibits mitochondrial complex 1. Carcinogenic. |  |
| Rotenone | Insecticide, piscicide, and pesticide | Inhibits mitochondrial complex 1. | 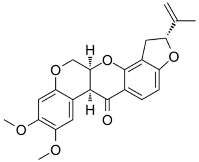 |
| Paraquat | Herbicide | Production of ROS. Mitochondrial toxicity. | 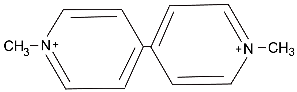 |
| Gene | Mutation Type | Mode of Action | Effect on Mitochondria | Reference |
|---|---|---|---|---|
| SNCA | Missense, amplification | Unknown. Disordered protein prone to aggregation | Deposition of aggregates inhibits mitochondrial function and produces ROS. | [46] |
| PRKN | Missense, copy number change, LOF | Ubiquitin ligase | Promotes mitochondrial quality control. | [47] |
| PINK1 | Missense, deletion, LOF | Serin/threonine protein kinase | Recruits PRKN to mitochondria. Controls respiratory chain function. | [47] |
| DJ-1 | Missense, LOF | Located at outer mitochondrial membrane. | Promotes mitochondrial function. Inhibits ROS formation. | [48] |
| LRRK2 | Missense, GOF | Kinase and GTPase | Promotes mitophagy. | [49] |
| VPS35 | Missense, D620N, GOF (?) | Vacuolar protein sorting | Loss of VPS35 causes mitochondrial dysfunction and fragmentation. | [50] |
| ATP13A2 | Missense, LOF | Lysosomal protein | Loss of ATP13A2 increases ROS and cell death. | [51] |
| VPS13C | Missense, LOF | Vacuolar protein sorting | Role in normal mitochondrial biogenesis and function. | [52] |
| CHCHD2 | Missense, T61I | CHCH domain containing protein | Maintains mitochondrial matrix structure. | [53] |
| FBXO7 | Missense, LOF | Adapter protein for ubiquitin E3 ligase | Recruits PRKN to damaged mitochondria. | [54] |
| PLA2G6 | Missense, LOF | Phospholipase | Maintains mitochondrial function. | [55] |
| Gene | Name | Length | Presumed Function | Human Disorders |
|---|---|---|---|---|
| PRKN | Parkin | 1379 kb | Ubiquitin ligase, regulates mitochondrial quality control | Familial PD gene |
| NRXN3 | Neurexin 3 | 1695 kb | Cell adhesion molecule | Autism |
| OXR1 | Oxidation resistance 1 | 484 kb | Critical for oxidative stress resistance of neurons | Cerebellar hypoplasia |
| RIT2 | RIC-like protein | 375 kb | Small GTPase Rit2 loss is causal for SNc cell death and motor dysfunction in mice | PD risk allele |
| DLG2 | Disks large homolog 2 | 2169 kb | Synaptic protein, membrane-associated guanylate kinase | Neurodevelop-mental disorders |
| LSAMP | Limbic system associated membrane protein | 644 kb | Cell adhesion molecule on axonal membranes | unknown |
| RBFOX1 | RNA binding FOX1 homologue | 1692 | RNA binding protein involved in splicing | Neurodevelop-mental disorders |
| VPS13B | Vacuolar protein sorting-associated 13B | 865 kb | Golgi associated protein | Autism, Cohen syndrome |
| WWOX | WW domain containing oxidoreductase | 1112 kb | Multifunctional protein | Spinocerebellar ataxia, epileptic encephalopathy |
| CNTNAP2 | Contactin-associated protein-like 2 | 2299 kb | Cell adhesion molecule | autism |
| DAB1 | Disabled 1 | 1255 kb | Reelin signaling, critical for neurodevelopment | Neurodevelop-mental disorders |
| SOX5 | SRY-related box 5 | 1033 kb | Transcription factor | Neurodevelop-mental disorder |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pfeifer, G.P. DNA Damage and Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 4187. https://doi.org/10.3390/ijms25084187
Pfeifer GP. DNA Damage and Parkinson’s Disease. International Journal of Molecular Sciences. 2024; 25(8):4187. https://doi.org/10.3390/ijms25084187
Chicago/Turabian StylePfeifer, Gerd P. 2024. "DNA Damage and Parkinson’s Disease" International Journal of Molecular Sciences 25, no. 8: 4187. https://doi.org/10.3390/ijms25084187
APA StylePfeifer, G. P. (2024). DNA Damage and Parkinson’s Disease. International Journal of Molecular Sciences, 25(8), 4187. https://doi.org/10.3390/ijms25084187