
Sterol O-Acyltransferase 1 (SOAT1): A Genetic Modifier of Niemann-Pick Disease, Type C1

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
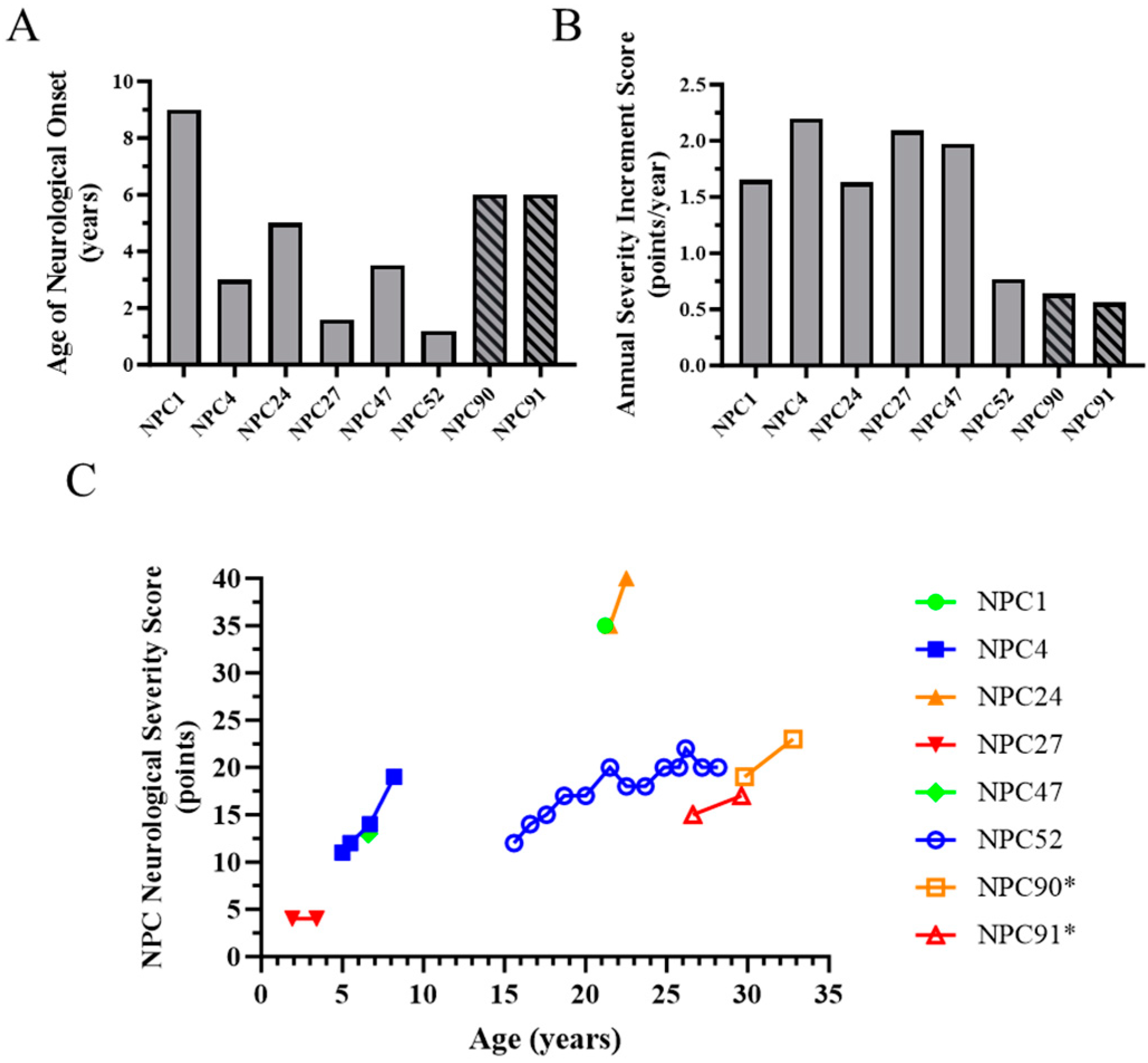
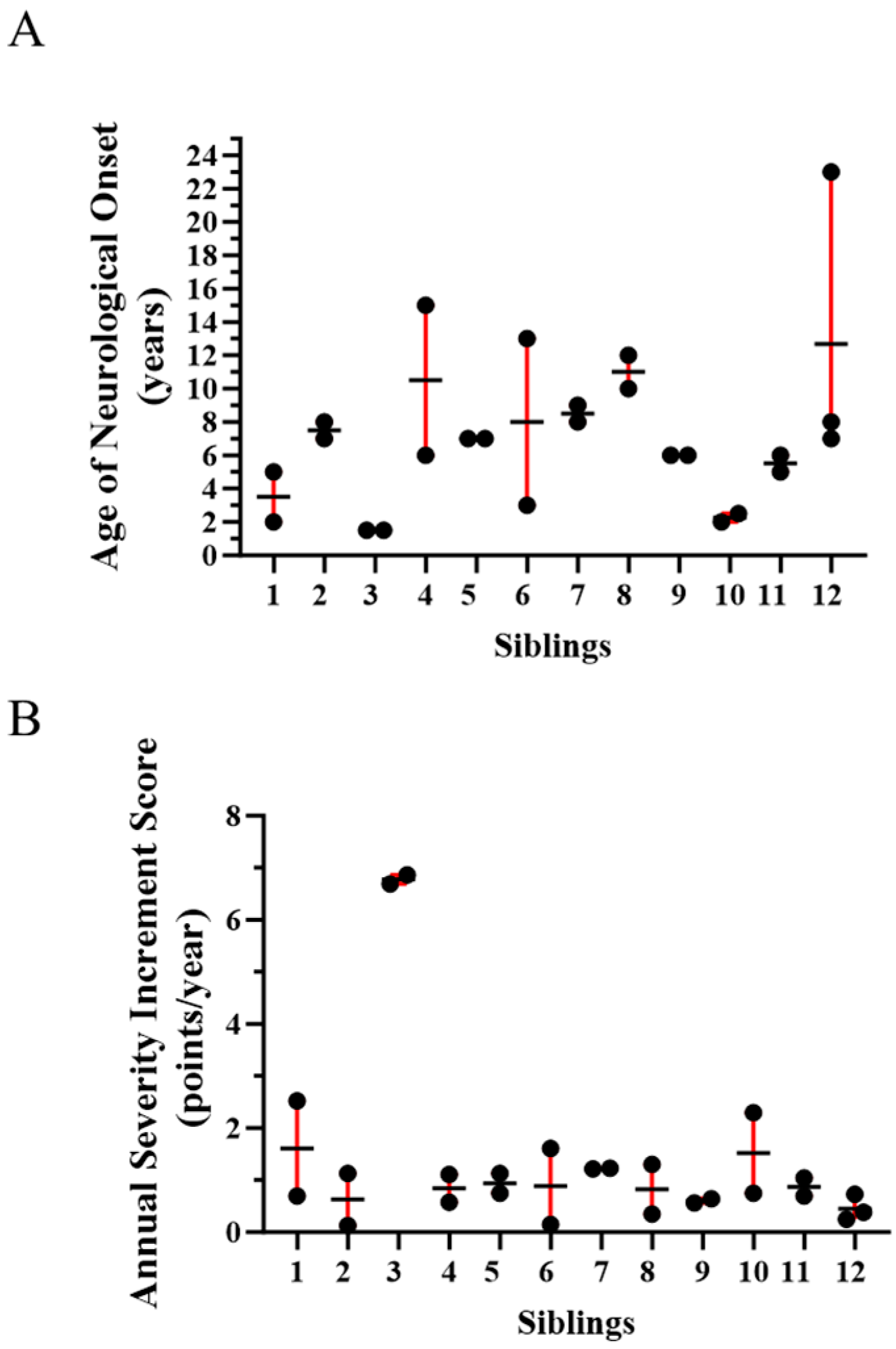
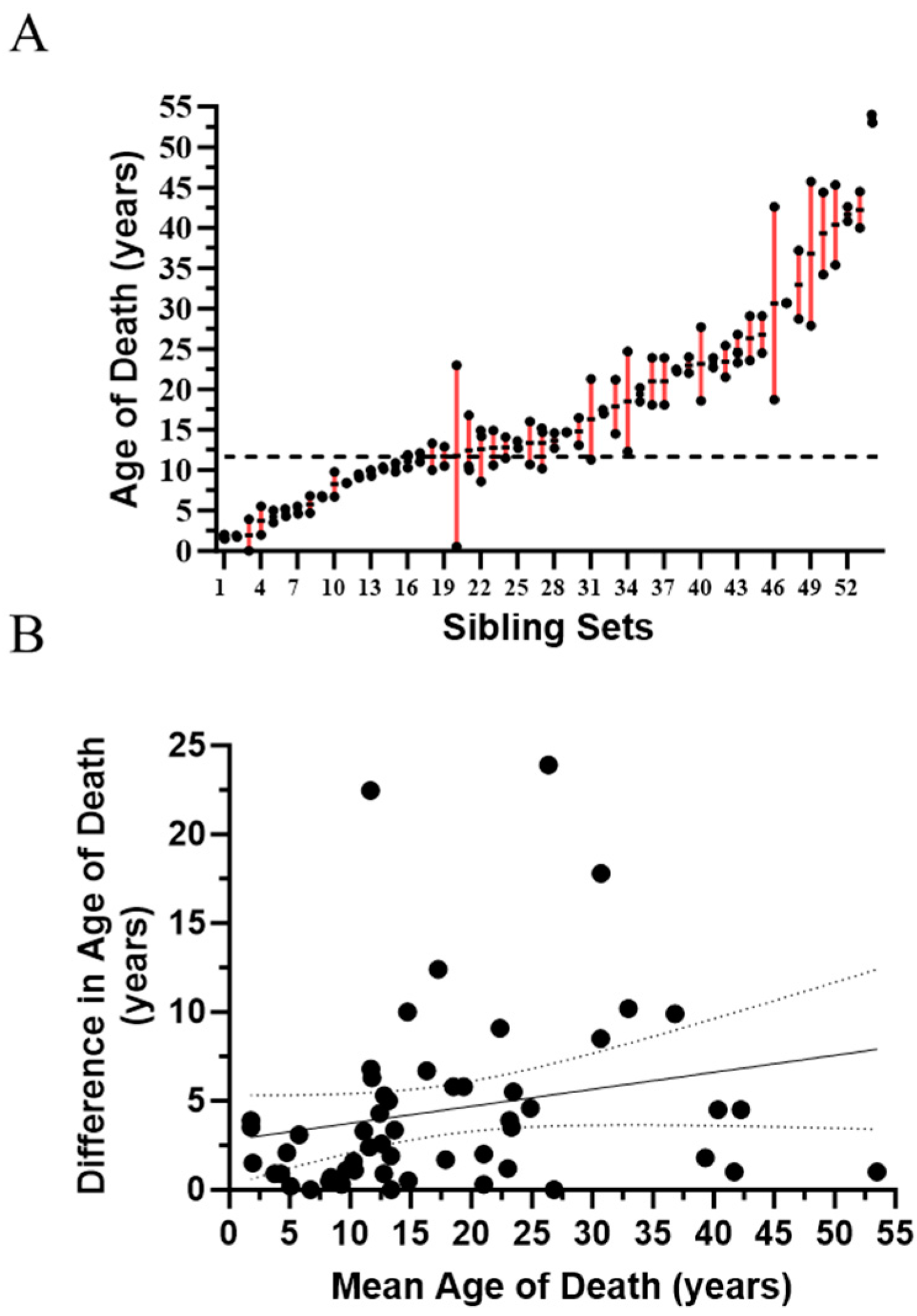
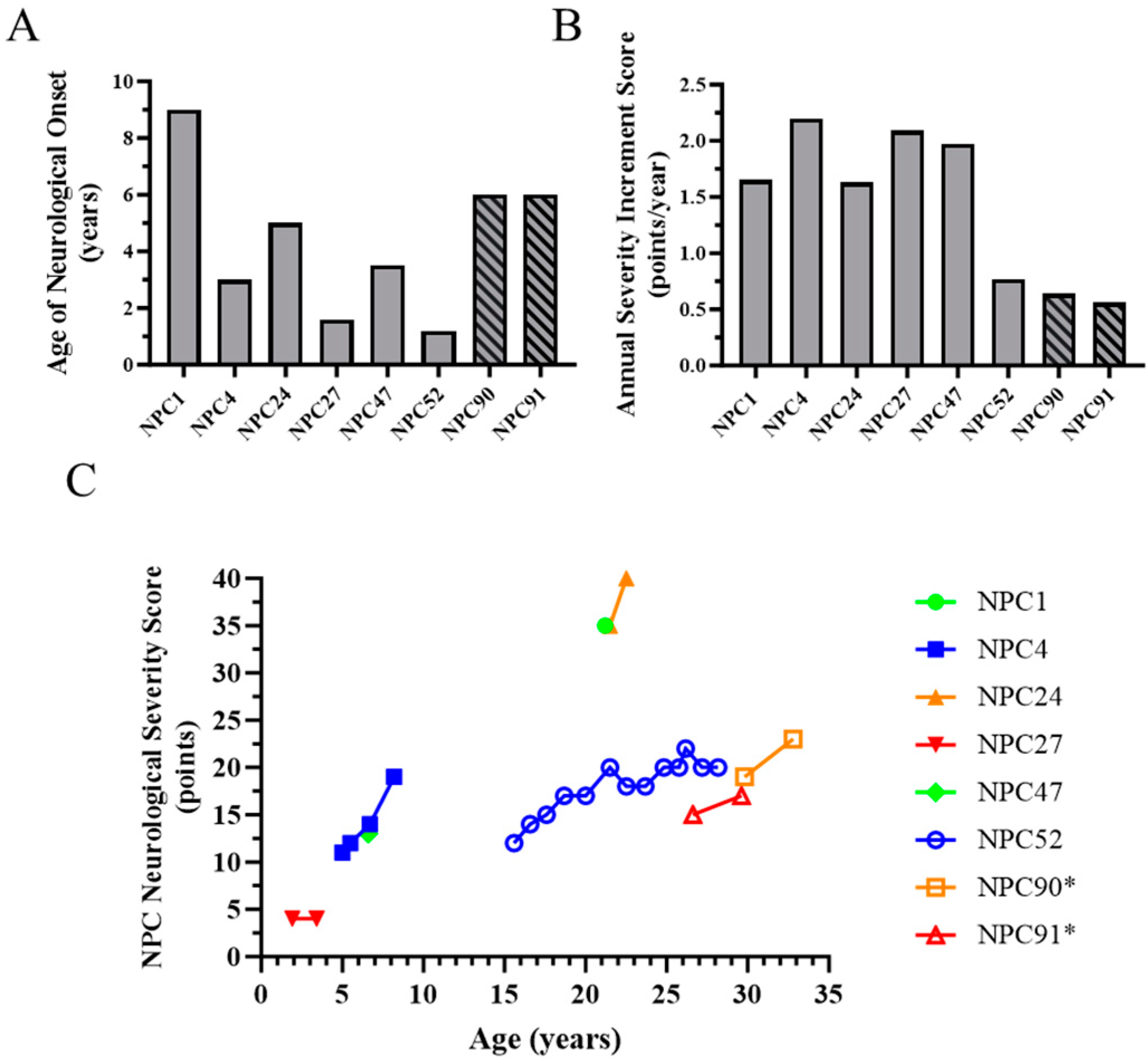
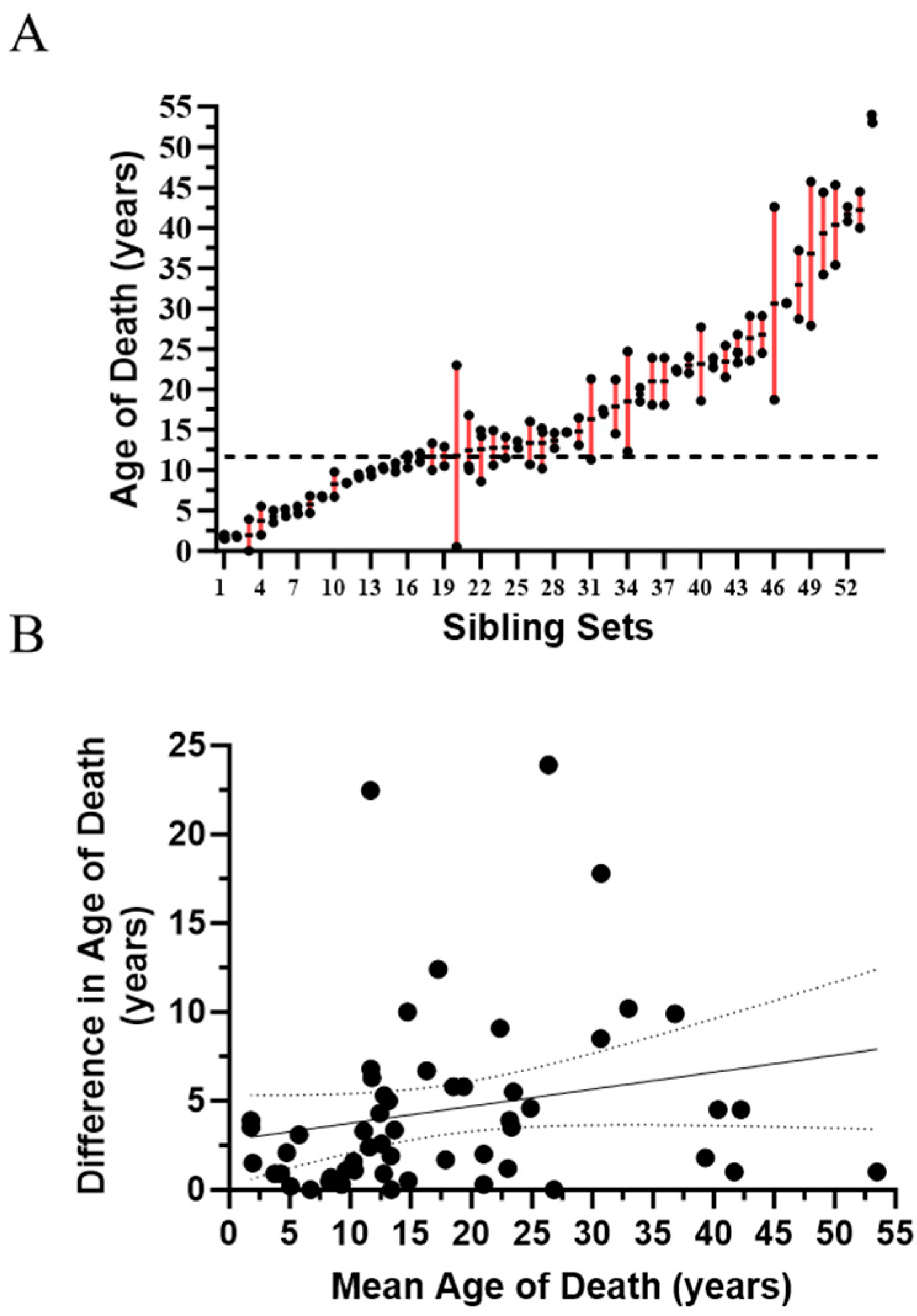
2.1. Evidence for Genetic Modifiers in Individuals with Niemann-Pick Disease, Type C1
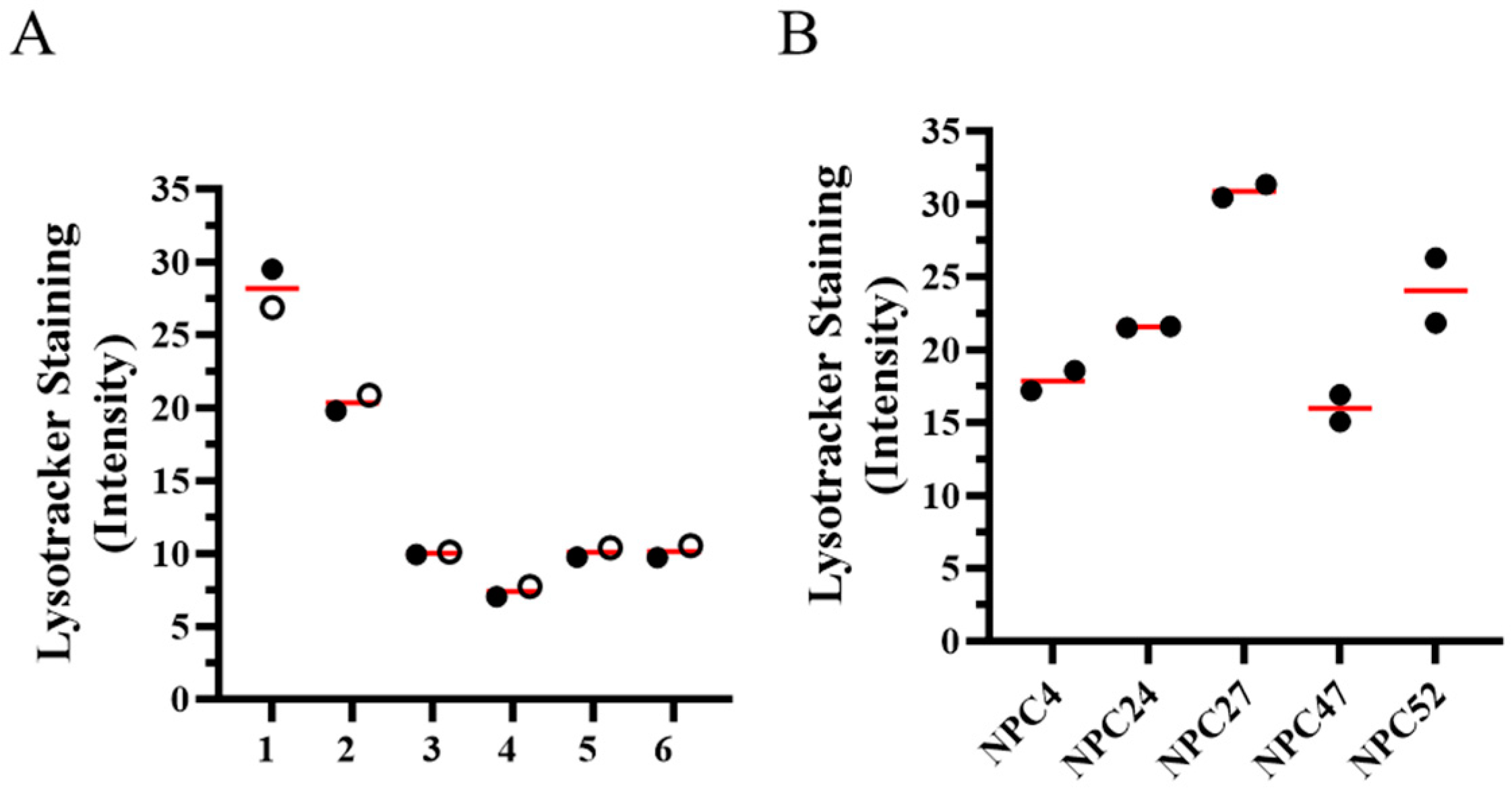
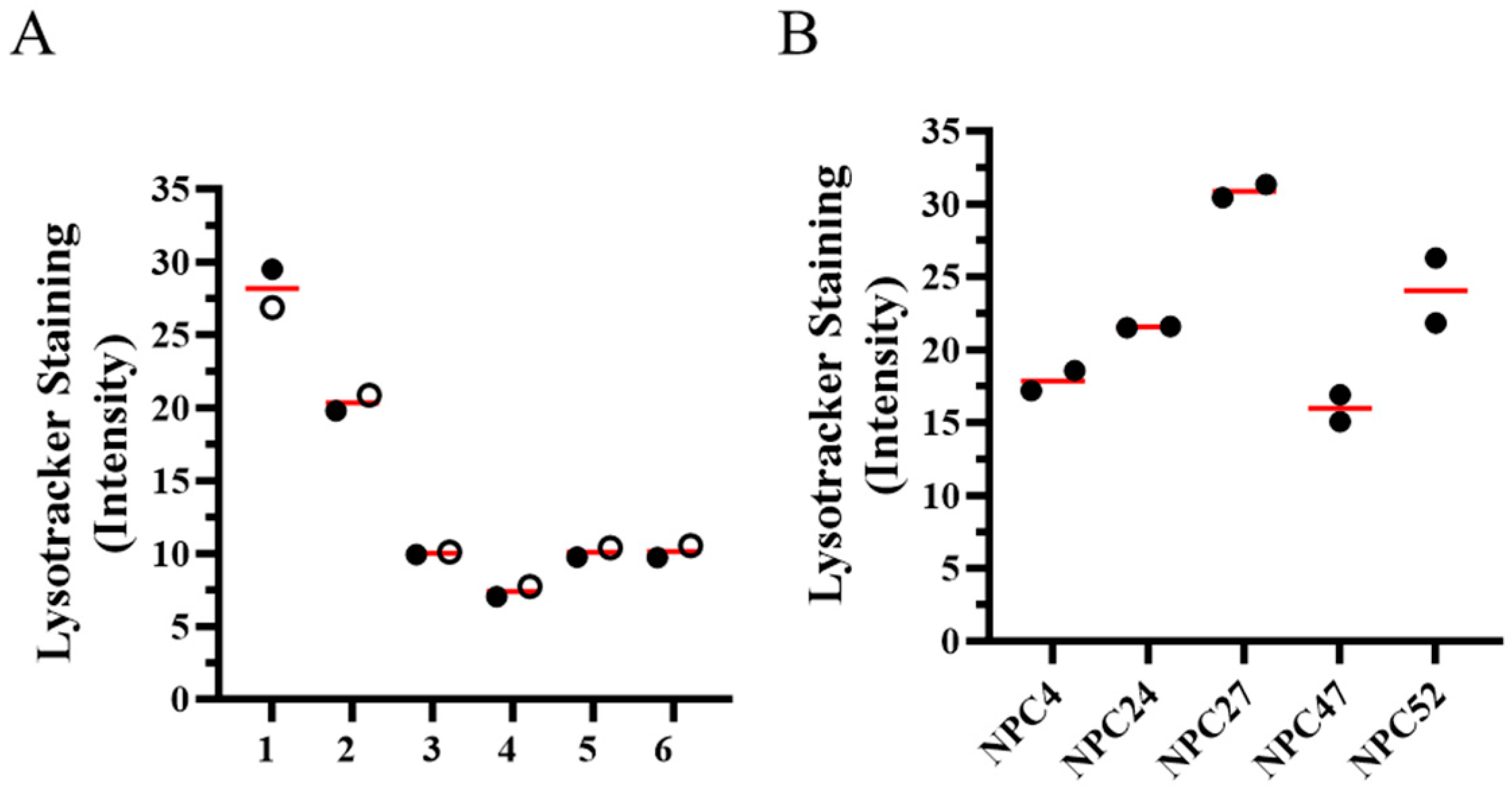
2.2. In Vitro Cellular Phenotype Heterogeneity
2.3. SOAT1 Polymorphism (rs1044925) Genotyping in Individuals with NPC1
2.4. The SOAT1 rs1044925 C-Allele Is Associated with a More Severe NPC1 Phenotype
3. Discussion
4. Materials and Methods
4.1. Study Participants and Ethical Approval
4.2. DNA Samples and Genomic Sequencing
4.3. Statistical Methods and Graphing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haldane, J.B.S. The relative importance of principal and modifying genes in determining some human diseases. J. Genet. 1941, 41, 149–157. [Google Scholar] [CrossRef]
- Chen, R.; Shi, L.; Hakenberg, J.; Naughton, B.; Sklar, P.; Zhang, J.; Zhou, H.; Tian, L.; Prakash, O.; Lemire, M.; et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat. Biotechnol. 2016, 34, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Zhu, J.Y.A.; Matthews, A.; van Karnebeek, C.D.M.; Wasserman, W.W. Assessment of the ExAC data set for the presence of individuals with pathogenic genotypes implicated in severe Mendelian pediatric disorders. Genet. Med. 2017, 19, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.; Feingold, J.; Clerget-Darpoux, F. Identifying modifier genes of monogenic disease: Strategies and difficulties. Hum. Genet. 2008, 124, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Rahit, K.; Tarailo-Graovac, M. Genetic Modifiers and Rare Mendelian Disease. Genes 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Cologna, S.M.; Rosenhouse-Dantsker, A. Insights into the Molecular Mechanisms of Cholesterol Binding to the NPC1 and NPC2 Proteins. Adv. Exp. Med. Biol. 2019, 1135, 139–160. [Google Scholar] [PubMed]
- Xie, C.; Turley, S.D.; Pentchev, P.G.; Dietschy, J.M. Cholesterol balance and metabolism in mice with loss of function of Niemann-Pick C protein. Am. J. Physiol. 1999, 276, E336–E344. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ory, D.S. Advancing Diagnosis and Treatment of Niemann-Pick C disease through Biomarker Discovery. Explor. Neuroprotective Ther. 2021, 1, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.C.; Clayton, P.; Gissen, P.; Anheim, M.; Bauer, P.; Bonnot, O.; Dardis, A.; Dionisi-Vici, C.; Klünemann, H.H.; Latour, P.; et al. Recommendations for the detection and diagnosis of Niemann-Pick disease type C: An update. Neurol. Clin. Pract. 2017, 7, 499–511. [Google Scholar] [CrossRef]
- Yanjanin, N.M.; Vélez, J.I.; Gropman, A.; King, K.; Bianconi, S.E.; Conley, S.K.; Brewer, C.C.; Solomon, B.; Pavan, W.J.; Arcos-Burgos, M.; et al. Linear clinical progression, independent of age of onset, in Niemann-Pick disease, type C. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153B, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Las Heras, M.; Szenfeld, B.; Ballout, R.A.; Buratti, E.; Zanlungo, S.; Dardis, A.; Klein, A.D. Understanding the phenotypic variability in Niemann-Pick disease type C (NPC): A need for precision medicine. NPJ Genom. Med. 2023, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Suzuki, K.; Nanba, E.; Yamamoto, T.; Ohno, K.; Murayama, S. Niemann-Pick type C disease: Accelerated neurofibrillary tangle formation and amyloid beta deposition associated with apolipoprotein E epsilon 4 homozygosity. Ann. Neurol. 2002, 52, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Yanjanin, N.M.; Elrick, M.J.; Ware, C.; Lieberman, A.P.; Porter, F.D. Apolipoprotein E genotype and neurological disease onset in Niemann-Pick disease, type C1. Am. J. Med. Genet. A 2012, 158A, 2775–2780. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.I.; Smith, A.C.; Sinaii, N.; Farhat, N.; King, M.C.; Machielse, L.; Porter, F.D. Association of Miglustat With Swallowing Outcomes in Niemann-Pick Disease, Type C1. JAMA Neurol. 2020, 77, 1564–1568. [Google Scholar] [CrossRef]
- Patterson, M.C.; Mengel, E.; Vanier, M.T.; Moneuse, P.; Rosenberg, D.; Pineda, M. Treatment outcomes following continuous miglustat therapy in patients with Niemann-Pick disease Type C: A final report of the NPC Registry. Orphanet J. Rare Dis. 2020, 15, 104. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick disease type C patients: A review. Orphanet J. Rare Dis. 2018, 13, 140. [Google Scholar] [CrossRef]
- Patterson, M.C.; Garver, W.S.; Giugliani, R.; Imrie, J.; Jahnova, H.; Meaney, F.J.; Nadjar, Y.; Vanier, M.T.; Moneuse, P.; Morand, O.; et al. Long-term survival outcomes of patients with Niemann-Pick disease type C receiving miglustat treatment: A large retrospective observational study. J. Inherit. Metab. Dis. 2020, 43, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Munkacsi, A.B.; Chen, F.W.; Brinkman, M.A.; Higaki, K.; Gutiérrez, G.D.; Chaudhari, J.; Layer, J.V.; Tong, A.; Bard, M.; Boone, C.; et al. An “exacerbate-reverse” strategy in yeast identifies histone deacetylase inhibition as a correction for cholesterol and sphingolipid transport defects in human Niemann-Pick type C disease. J. Biol. Chem. 2011, 286, 23842–23851. [Google Scholar] [CrossRef] [PubMed]
- Pipalia, N.H.; Cosner, C.C.; Huang, A.; Chatterjee, A.; Bourbon, P.; Farley, N.; Helquist, P.; Wiest, O.; Maxfield, F.R. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. USA 2011, 108, 5620–5625. [Google Scholar] [CrossRef] [PubMed]
- Pugach, E.K.; Feltes, M.; Kaufman, R.J.; Ory, D.S.; Bang, A.G. High-content screen for modifiers of Niemann-Pick type C disease in patient cells. Hum. Mol. Genet. 2018, 27, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Bartz, F.; Kern, L.; Erz, D.; Zhu, M.; Gilbert, D.; Meinhof, T.; Wirkner, U.; Erfle, H.; Muckenthaler, M.; Pepperkok, R.; et al. Identification of cholesterol-regulating genes by targeted RNAi screening. Cell Metab. 2009, 10, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Lu, A. Sorting (Nexin-13) out Novel Insights into Endolysosomal Cholesterol Export. Contact 2022, 5, 25152564221114513. [Google Scholar] [CrossRef] [PubMed]
- Praggastis, M.; Tortelli, B.; Zhang, J.; Fujiwara, H.; Sidhu, R.; Chacko, A.; Chen, Z.; Chung, C.; Lieberman, A.P.; Sikora, J.; et al. A murine Niemann-Pick C1 I1061T knock-in model recapitulates the pathological features of the most prevalent human disease allele. J. Neurosci. 2015, 35, 8091–8106. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gil, J.L.; Watkins-Chow, D.E.; Baxter, L.L.; Elliot, G.; Harper, U.L.; Wincovitch, S.M.; Wedel, J.C.; Incao, A.A.; Huebecker, M.; Boehm, F.J.; et al. Genetic background modifies phenotypic severity and longevity in a mouse model of Niemann-Pick disease type C1. Dis. Model. Mech. 2020, 13, dmm042614. [Google Scholar] [CrossRef] [PubMed]
- Parra, J.; Klein, A.D.; Castro, J.; Morales, M.G.; Mosqueira, M.; Valencia, I.; Cortés, V.; Rigotti, A.; Zanlungo, S. Npc1 deficiency in the C57BL/6J genetic background enhances Niemann-Pick disease type C spleen pathology. Biochem. Biophys. Res. Commun. 2011, 413, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Erickson, R.P. A modifier of Niemann Pick C 1 maps to mouse chromosome 19. Mamm. Genome 2000, 11, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Gondre-Lewis, M.C.; McGlynn, R.; Walkley, S.U. Cholesterol accumulation in NPC1-deficient neurons is ganglioside dependent. Curr. Biol. 2003, 13, 1324–1329. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, J.K.; Bae, Y.C.; Yang, S.H.; Okino, N.; Schuchman, E.H.; Yamashita, T.; Bae, J.S.; Jin, H.K. Inhibition of GM3 synthase attenuates neuropathology of Niemann-Pick disease Type C. by affecting sphingolipid metabolism. Mol. Cells. 2014, 37, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Sugimoto, Y.; Ohsaki, Y.; Ueno, M.; Kato, S.; Kitamura, Y.; Hosokawa, H.; Davies, J.P.; Ioannou, Y.A.; Vanier, M.T.; et al. Endosomal accumulation of Toll-like receptor 4 causes constitutive secretion of cytokines and activation of signal transducers and activators of transcription in Niemann-Pick disease type C (NPC) fibroblasts: A potential basis for glial cell activation in the NPC brain. J. Neurosci. 2007, 27, 1879–1891. [Google Scholar]
- Lopez, M.E.; Klein, A.D.; Scott, M.P. Complement is dispensable for neurodegeneration in Niemann-Pick disease type C. J. Neuroinflammation. 2012, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.E.; Klein, A.D.; Hong, J.; Dimbil, U.J.; Scott, M.P. Neuronal and epithelial cell rescue resolves chronic systemic inflammation in the lipid storage disorder Niemann-Pick C. Hum. Mol. Genet. 2012, 21, 2946–2960. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.T.; Tu, X.; Yang, K.; Wu, J.; Repa, J.J.; Yan, N. Tonic prime-boost of STING signalling mediates Niemann-Pick disease type C. Nature 2021, 596, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Chen, C.; Mei, C.; Liu, S.; Xiang, C.; Fang, W.; Zhang, F.; Xu, Y.; Chen, S.; Zhang, Q.; et al. Innate immune sensing of lysosomal dysfunction drives multiple lysosomal storage disorders. Nat. Cell Biol. 2024, 26, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.; Pressey, S.N.; Cooper, J.D.; Soriano, S. Loss of amyloid precursor protein in a mouse model of Niemann-Pick type C disease exacerbates its phenotype and disrupts tau homeostasis. Neurobiol. Dis. 2011, 42, 349–359. [Google Scholar] [CrossRef]
- Pacheco, C.D.; Elrick, M.J.; Lieberman, A.P. Tau deletion exacerbates the phenotype of Niemann-Pick type C mice and implicates autophagy in pathogenesis. Hum. Mol. Genet. 2009, 18, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.; Aten, J.; Ottenhoff, R.; van Roomen, C.P.; Herrera Moro, D.; Claessen, N.; Vinueza Veloz, M.F.; Zhou, K.; Lin, Z.; Mirzaian, M.; et al. Reducing GBA2 Activity Ameliorates Neuropathology in Niemann-Pick Type C Mice. PLoS ONE 2015, 10, e0135889. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.; Chang, C.C.; Maue, R.A.; Melton, E.M.; Peden, A.A.; Garver, W.S.; Lee, J.; Schroen, P.; Huang, M.; Chang, T.Y. Acat1/Soat1 knockout extends the mutant Npc1 mouse lifespan and ameliorates functional deficiencies in multiple organelles of mutant cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2201646119. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.M.; Jones, R.D.; Repa, J.J.; Turley, S.D. Niemann-Pick C1-deficient mice lacking sterol O-acyltransferase 2 have less hepatic cholesterol entrapment and improved liver function. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G454–G463. [Google Scholar] [CrossRef]
- Wu, D.F.; Yin, R.X.; Aung, L.H.H.; Hu, X.J.; Cao, X.L.; Miao, L.; Li, Q.; Yan, T.T.; Wu, J.Z.; Pan, S.L. Polymorphism of rs1044925 in the acyl-CoA:cholesterol acyltransferase-1 gene and serum lipid levels in the Guangxi Bai Ku Yao and Han populations. Lipids Health Dis. 2010, 9, 139. [Google Scholar] [CrossRef]
- Wu, D.F.; Yin, R.X.; Aung, L.H.H.; Li, Q.; Yan, T.T.; Zeng, X.N.; Huang, K.K.; Huang, P.; Wu, J.Z.; Pan, S.L. Sex-specific association of ACAT-1 rs1044925 SNP and serum lipid levels in the hypercholesterolemic subjects. Lipids Health Dis. 2012, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Wang, Y.H.; Ma, Y.T.; Fu, Z.Y.; Yang, Y.N.; Ma, X.; Li, X.M.; Adi, D.; Liu, F.; Chen, B.D. ACAT-1 gene polymorphism is associated with increased susceptibility to coronary artery disease in Chinese Han population: A case-control study. Oncotarget 2017, 8, 89055–89063. [Google Scholar] [CrossRef] [PubMed]
- Alavez-Rubio, J.S.; Martinez-Rodriguez, N.; Escobedo-de-la-Pena, J.; Garrido-Acosta, O.; Juarez-Cedillo, T. Relationship Between Genetic Variants of ACAT1 and APOE with the Susceptibility to Dementia (SADEM Study). Mol. Neurobiol. 2021, 58, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Lämsä, R.; Helisalmi, S.; Herukka, S.K.; Tapiola, T.; Pirttilä, T.; Vepsäläinen, S.; Hiltunen, M.; Soininen, H. Study on the association between SOAT1 polymorphisms, Alzheimer’s disease risk and the level of CSF biomarkers. Dement. Geriatr. Cogn. Disord. 2007, 24, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.G.; Wang, Y.H.; Yang, J.F.; Ma, Q.L.; Tang, Z.; Dong, X.M.; Chan, P. Association between acyl-coenzyme A: Cholesterol acyltransferase gene and risk for Alzheimer’s disease in Chinese. Neurosci. Lett. 2005, 388, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Brunner, F.S.M. Effects of a SOAT1 Haplotype on SOAT1 mRNA Expression and on the Risk for Sporadic Alzheimer’s Disease; Swill Federal Institute of Technology Zurich: Zürich, Switzerland, 2006. [Google Scholar]
- Pfrieger, F.W. The Niemann-Pick type diseases—A synopsis of inborn errors in sphingolipid and cholesterol metabolism. Prog. Lipid Res. 2023, 90, 101225. [Google Scholar] [CrossRef] [PubMed]
- Cortina-Borja, M.; Te Vruchte, D.; Mengel, E.; Amraoui, Y.; Imrie, J.; Jones, S.A.; i Dali, C.; Fineran, P.; Kirkegaard, T.; Runz, H.; et al. Annual severity increment score as a tool for stratifying patients with Niemann-Pick disease type C and for recruitment to clinical trials. Orphanet J. Rare Dis. 2018, 13, 143. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.; Patterson, M.; Platt, F.; Guldberg, C.; Mathieson, T.; Pacey, J. International consensus on clinical severity scale use in evaluating Niemann-Pick disease Type C in paediatric and adult patients: Results from a Delphi Study. Orphanet J. Rare Dis. 2021, 16, 482. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.C.; Lloyd-Price, L.; Guldberg, C.; Doll, H.; Burbridge, C.; Chladek, M.; íDali, C.; Mengel, E.; Symonds, T. Validation of the 5-domain Niemann-Pick type C Clinical Severity Scale. Orphanet J. Rare Dis. 2021, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Marçais, C.; Rafi, M.A.; Yamamoto, T.; Morris, J.A.; Pentchev, P.G.; Ohno, K.; Wenger, D.A.; Vanier, M.T. Niemann-Pick C1 disease: The I1061T substitution is a frequent mutant allele in patients of Western European descent and correlates with a classic juvenile phenotype. Am. J. Hum. Genet. 1999, 65, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Te Vruchte, D.; Speak, A.O.; Wallom, K.L.; Al Eisa, N.; Smith, D.A.; Hendriksz, C.J.; Simmons, L.; Lachmann, R.H.; Cousins, A.; Hartung, R.; et al. Relative acidic compartment volume as a lysosomal storage disorder-associated biomarker. J. Clin. Investig. 2014, 124, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Baxter, L.L.; Watkins-Chow, D.E.; Johnson, N.L.; Farhat, N.Y.; Platt, F.M.; Dale, R.K.; Porter, F.D.; Pavan, W.J.; Rodriguez-Gil, J.L. Correlation of age of onset and clinical severity in Niemann-Pick disease type C1 with lysosomal abnormalities and gene expression. Sci. Rep. 2022, 12, 2162. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farhat, N.Y.; Alexander, D.; McKee, K.; Iben, J.; Rodriguez-Gil, J.L.; Wassif, C.A.; Cawley, N.X.; Balch, W.E.; Porter, F.D. Sterol O-Acyltransferase 1 (SOAT1): A Genetic Modifier of Niemann-Pick Disease, Type C1. Int. J. Mol. Sci. 2024, 25, 4217. https://doi.org/10.3390/ijms25084217
Farhat NY, Alexander D, McKee K, Iben J, Rodriguez-Gil JL, Wassif CA, Cawley NX, Balch WE, Porter FD. Sterol O-Acyltransferase 1 (SOAT1): A Genetic Modifier of Niemann-Pick Disease, Type C1. International Journal of Molecular Sciences. 2024; 25(8):4217. https://doi.org/10.3390/ijms25084217
Chicago/Turabian StyleFarhat, Nicole Y., Derek Alexander, Kyli McKee, James Iben, Jorge L. Rodriguez-Gil, Christopher A. Wassif, Niamh X. Cawley, William E. Balch, and Forbes D. Porter. 2024. "Sterol O-Acyltransferase 1 (SOAT1): A Genetic Modifier of Niemann-Pick Disease, Type C1" International Journal of Molecular Sciences 25, no. 8: 4217. https://doi.org/10.3390/ijms25084217
APA StyleFarhat, N. Y., Alexander, D., McKee, K., Iben, J., Rodriguez-Gil, J. L., Wassif, C. A., Cawley, N. X., Balch, W. E., & Porter, F. D. (2024). Sterol O-Acyltransferase 1 (SOAT1): A Genetic Modifier of Niemann-Pick Disease, Type C1. International Journal of Molecular Sciences, 25(8), 4217. https://doi.org/10.3390/ijms25084217