

Precise Targeting of Autoantigen-Specific B Cells in Lupus Nephritis with Chimeric Autoantibody Receptor T Cells
 ,
,
Abstract
:1. Introduction
2. Results
2.1. Patient Demographic Characteristics
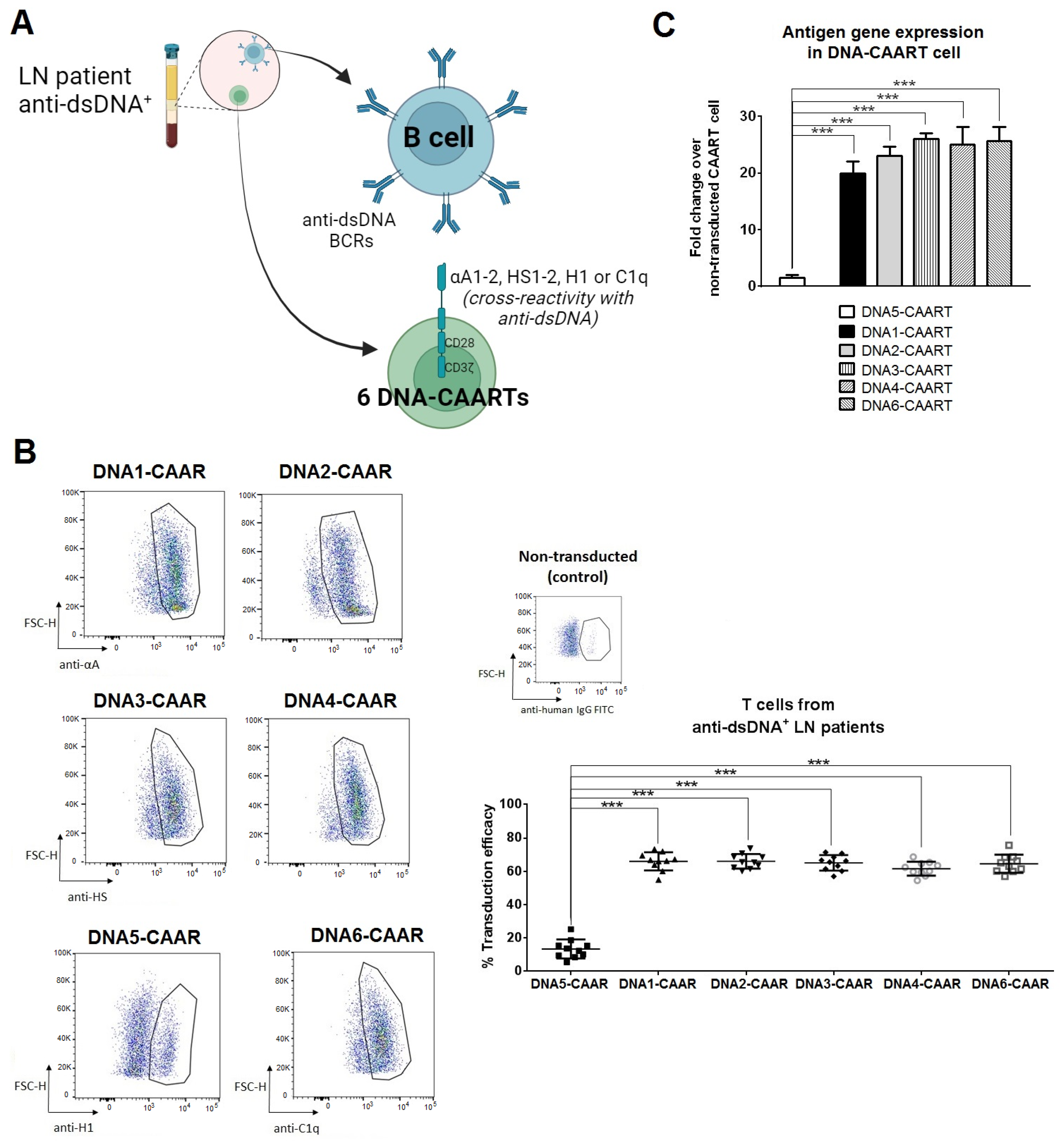
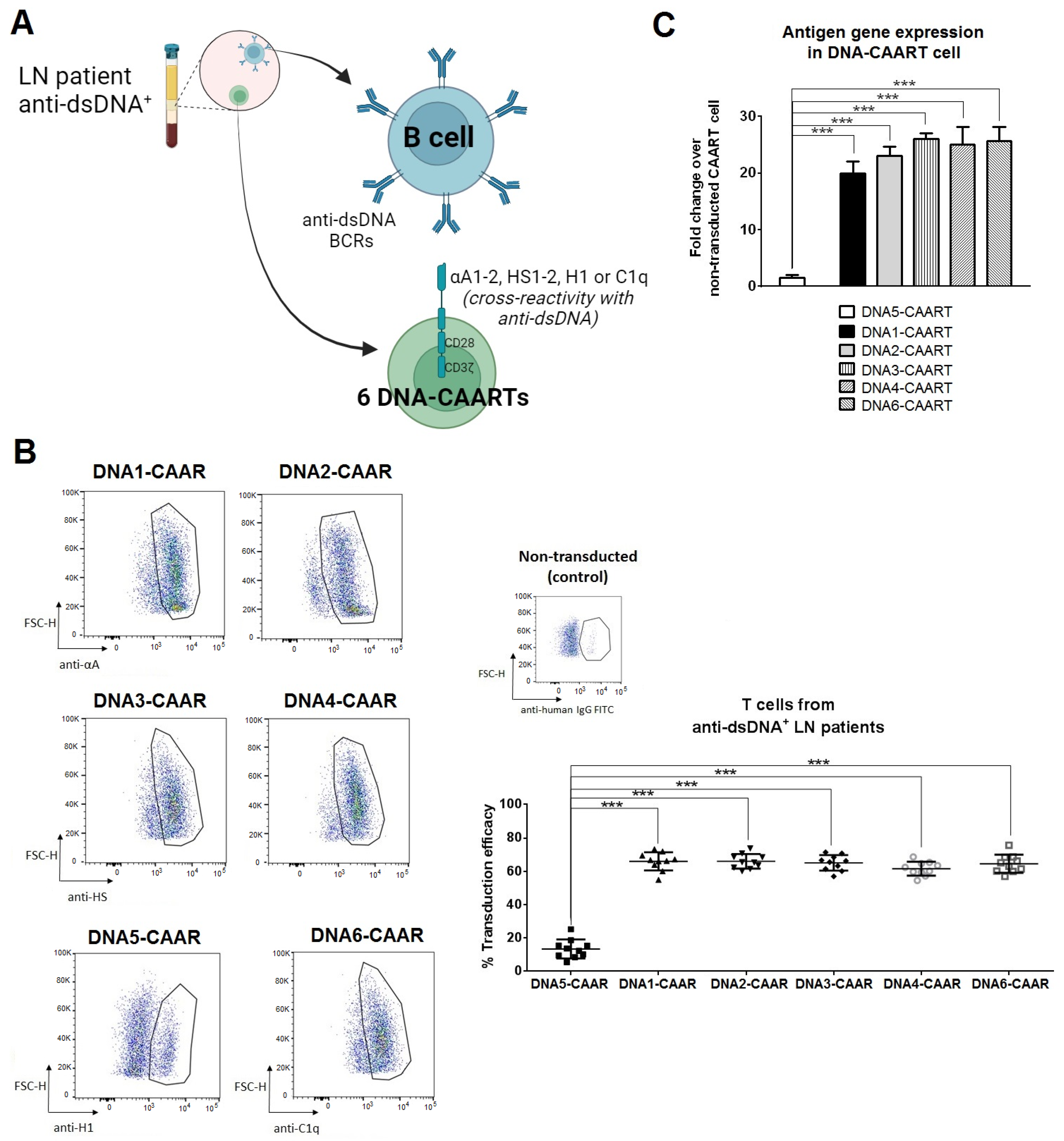
2.2. Design and Construction of Six DNA-CAART
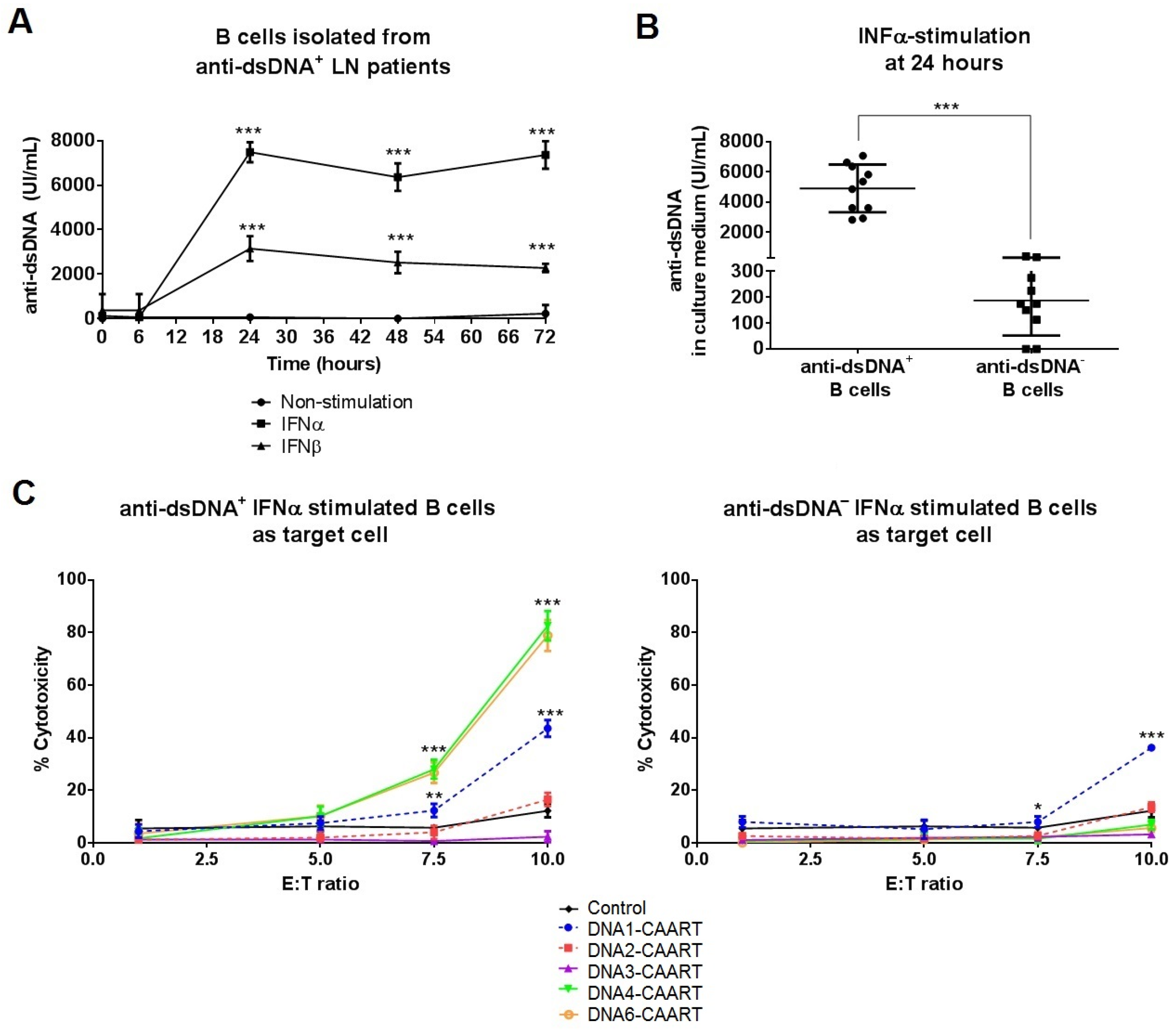
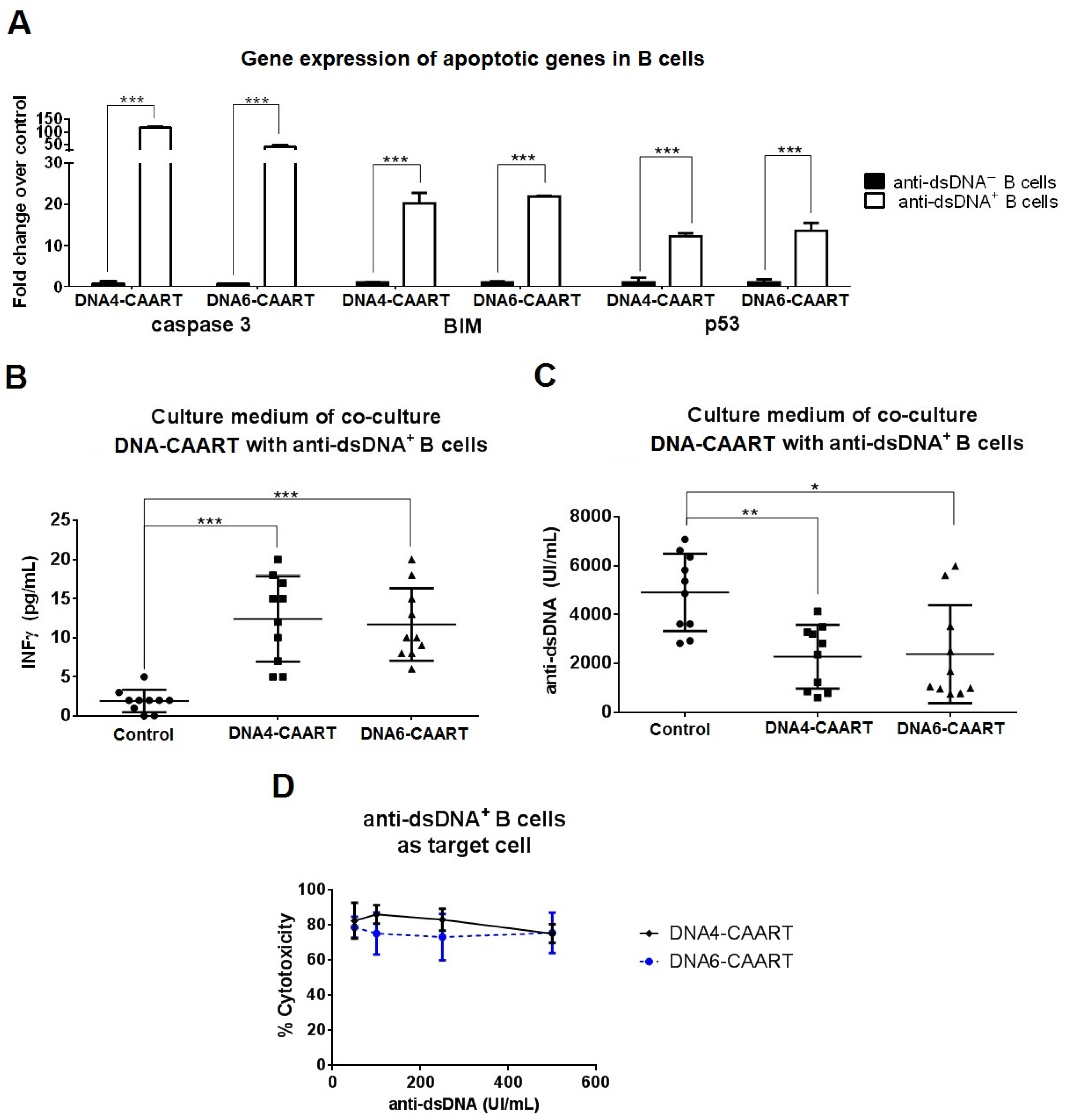
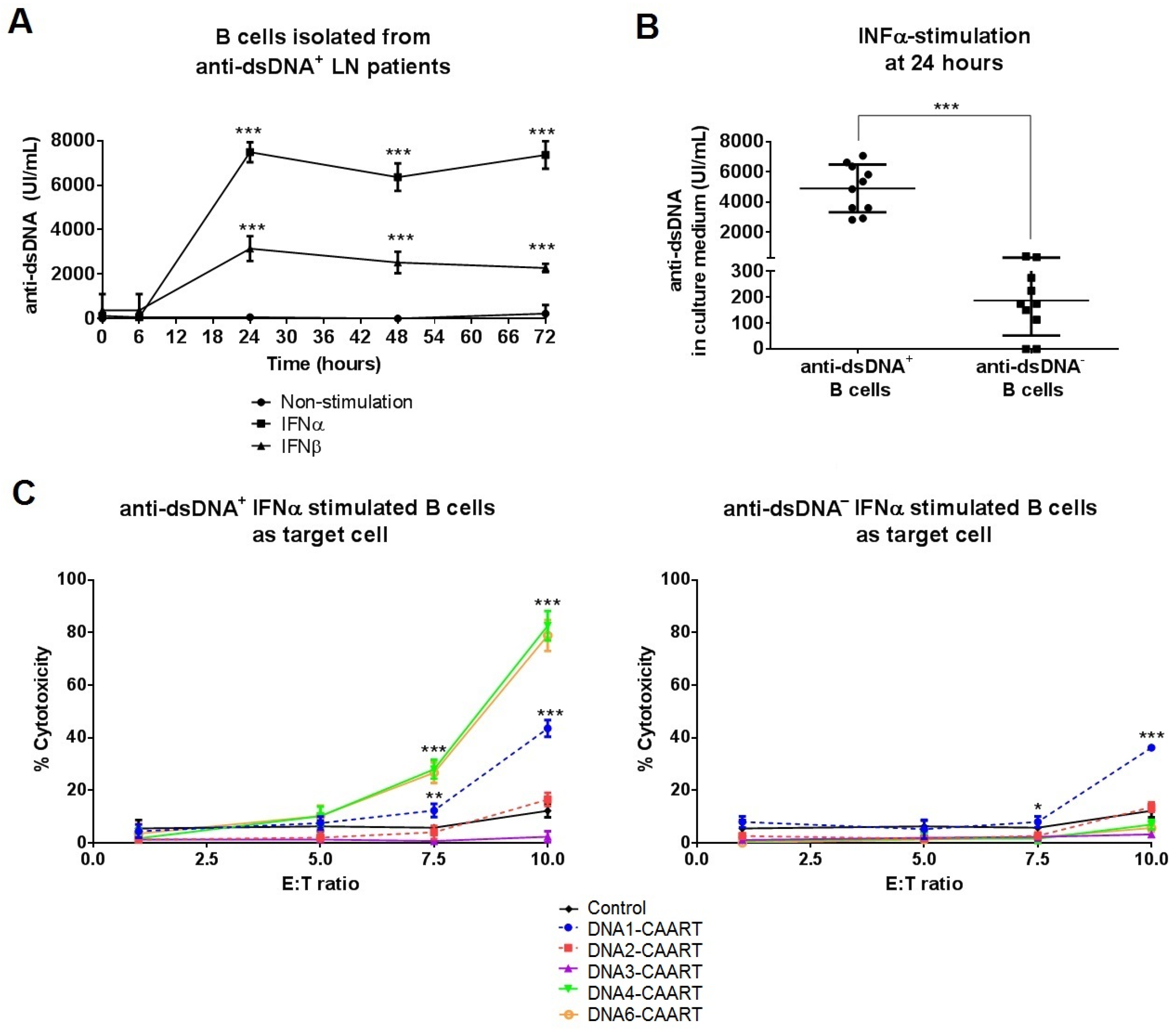
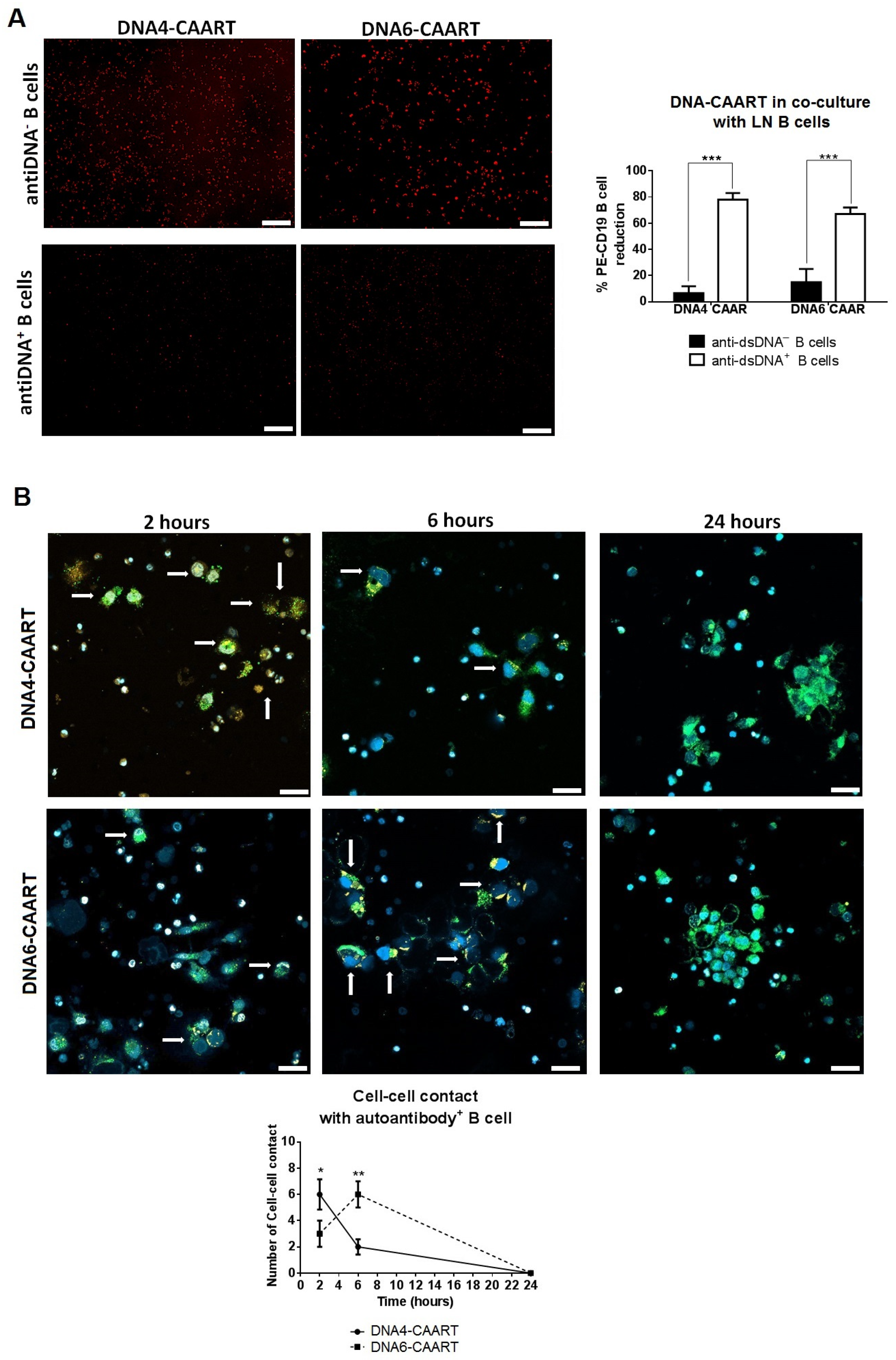
2.3. DNA4 and DNA6-CAART Show Higher Selective Depletion of Anti-dsDNA+ B Cells
2.4. Confocal Live-Cell Imaging Provides Visualization of DNA4 and DNA6-CAART Interaction and Expansion with Anti-dsDNA+ B Cells
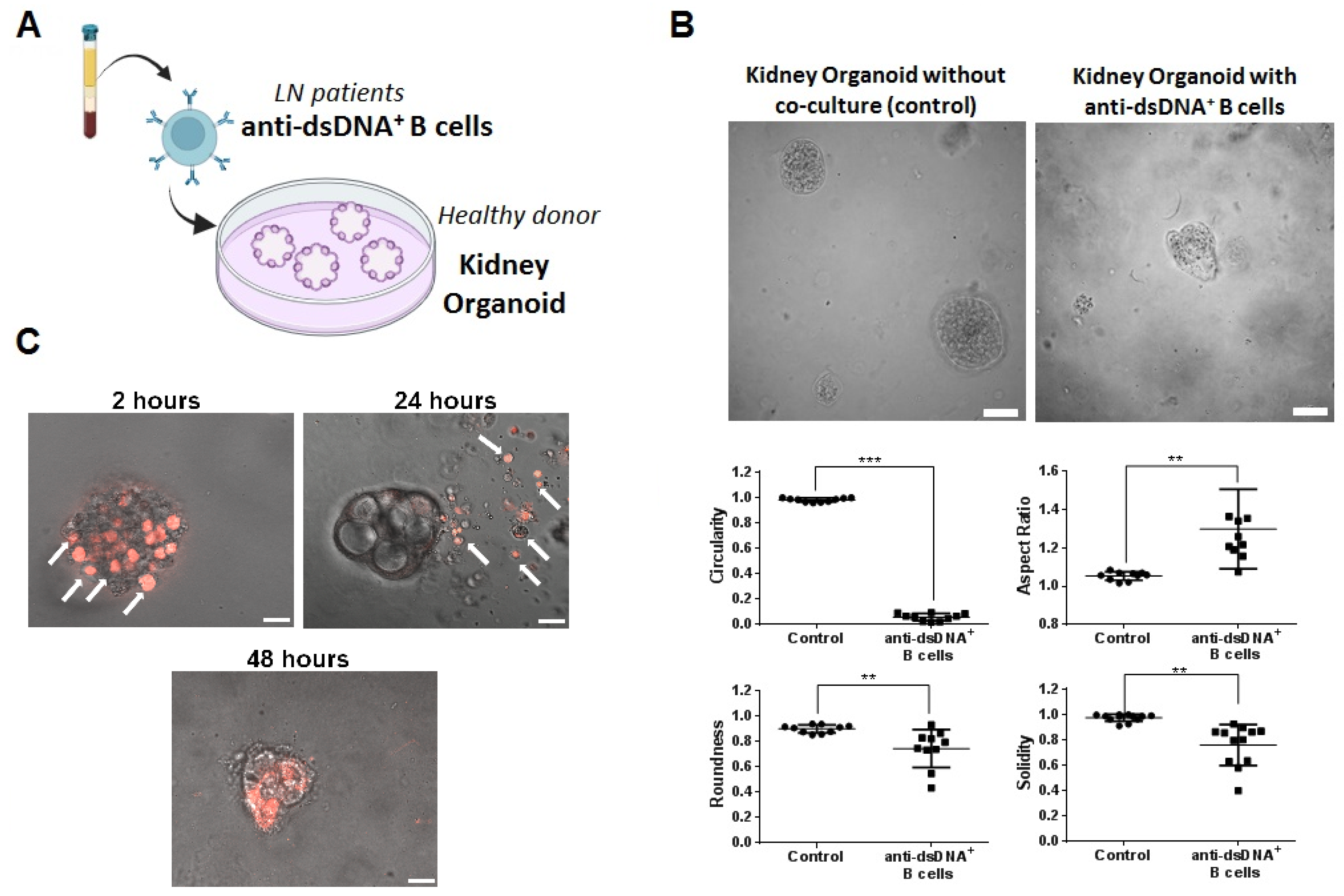
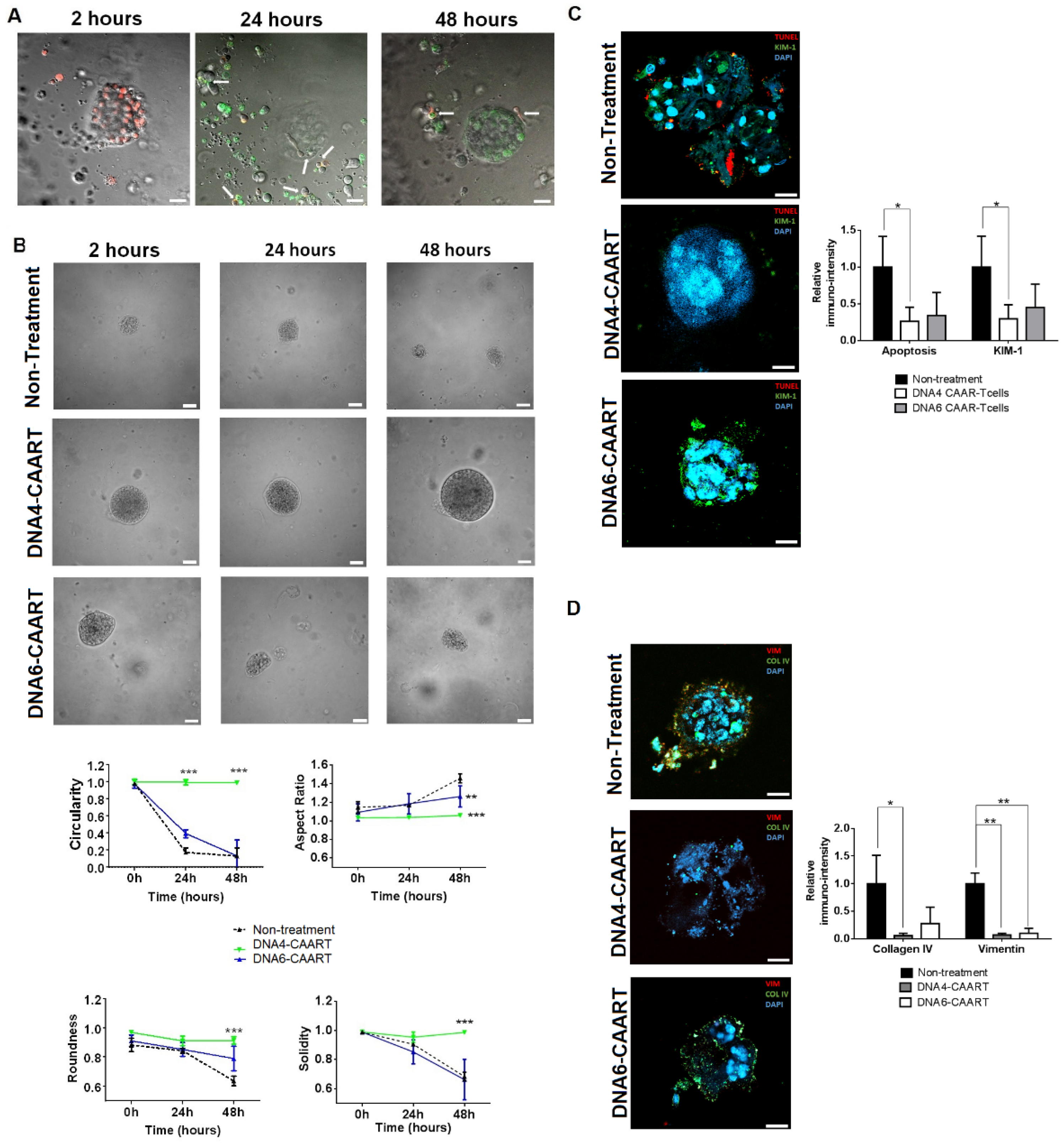
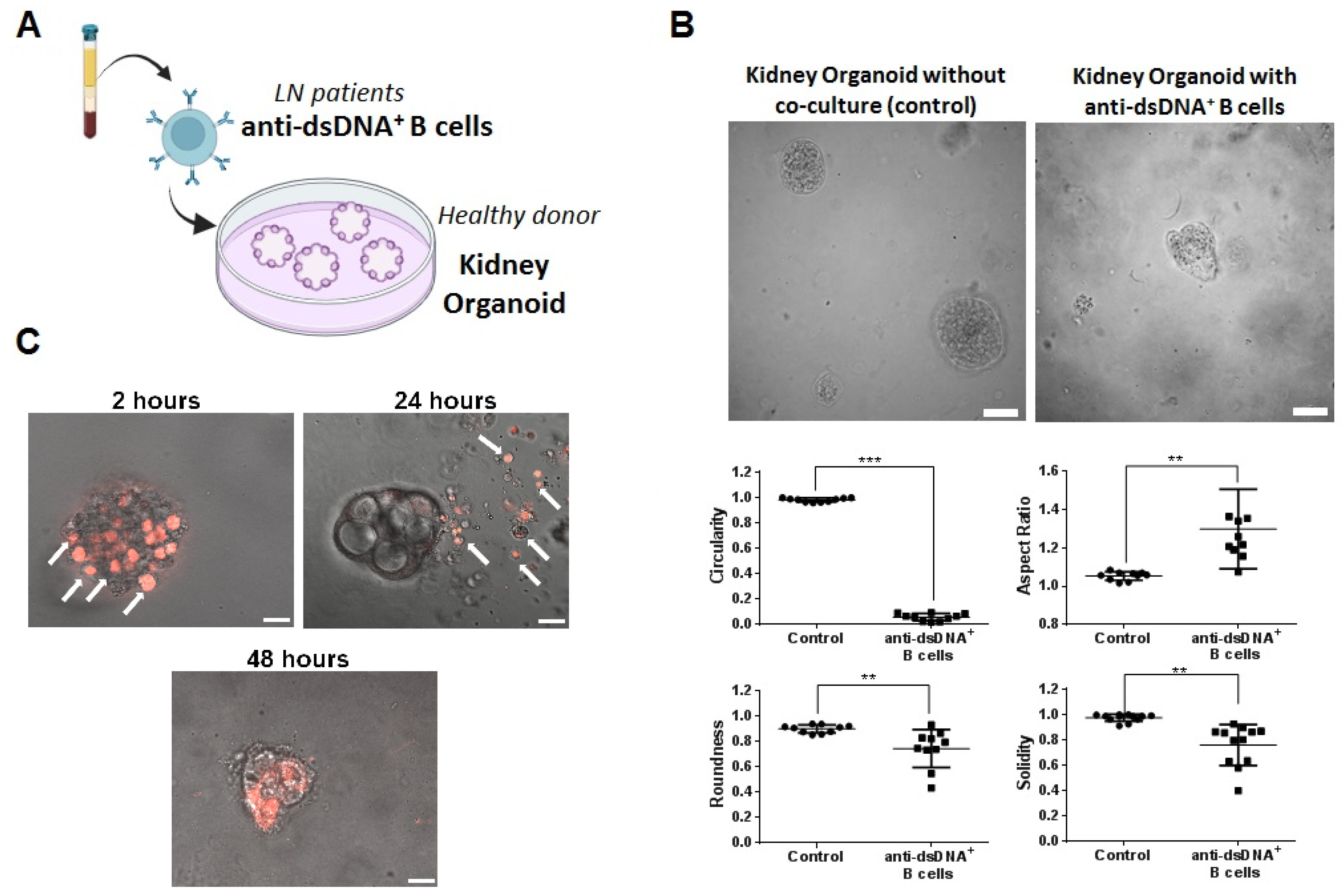
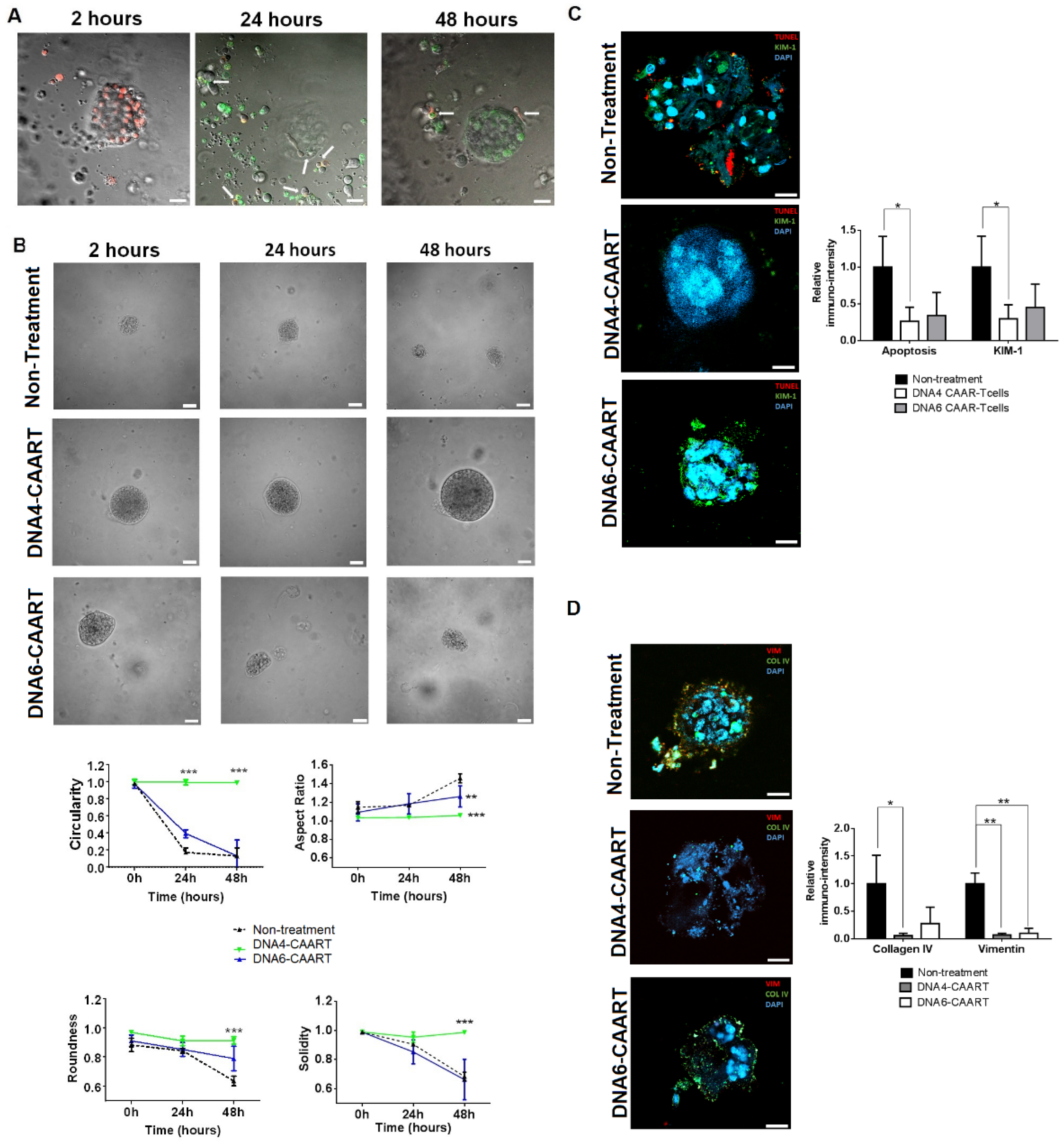
2.5. Therapeutic Evaluation of DNA4 and DNA6-CAART Cells in 3D Immuno-Kidney Model Assay
3. Discussion
4. Materials and Methods
4.1. Design and Construction of CAAR Vectors
4.2. Patient Characteristics
4.3. In Vitro Transduction and Expansion of CAART Cells
4.4. In Vitro Cytotoxicity Assays
4.5. Confocal Live-Cell Microscopy Interaction between B Cells and CAART Cells
4.6. RNA Extraction and RT-qPCR
4.7. Kidney Organoids B Cell Co-Culture to Evaluate Therapeutic CAART Cell Efficacy
4.8. Immunofluorescence Analysis of Kidney Organoids after Exposure to CAART Cells
4.9. Enzyme-Linked Immunosorbent Assay (ELISA)
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tosokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.V.; Almaani, S.; Brodsky, S.; Rovin, B.H. Update on lupus nephritis: Core curriculum 2020. Am. J. Kidney Dis. 2020, 76, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Rosner, S.; Ginzler, E.; Diamond, H. A multicentre study of outcome in systemic lupus erythematosus. II. Causes of death. Arthritis Rheum. 1982, 25, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Adler, M.; Chambers, S.; Edwards, C.; Neild, G.; Isenberg, D. An assessment of renal failure in an SLE cohort with special reference to ethnicity, over a 25-year period. Rheumatology 2006, 45, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Davidson, A. Taming lupus—A new understanding of pathogenesis is leading to clinical advances. Nat. Med. 2012, 18, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.; Anders, H.J. The pathogenesis of lupus nephritis. J. Am. Soc. 2013, 24, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.Y.H.; Chan, T.M. B cell abnormalities in systemic lupus erythematosus and lupus nephritis-role in pathogenesis and effect of immunosuppressive treatments. Int. J. Mol. Sci. 2019, 20, 6231. [Google Scholar] [CrossRef]
- Ma, K.; Du, W.; Wang, X.; Yuan, S.; Cai, X.; Liu, D.; Li, J.; Lu, L. Multiple functions of B cells in the pathogenesis of systemic lupus erythematosus. Int. J. Mol. Sci. 2019, 20, 6021. [Google Scholar] [CrossRef]
- Chang, A.; Clark, M.R.; Ko, K. Cellular aspects of the pathogenesis of lupus nephritis. Curr. Opin. Rheumatol. 2021, 33, 197–204. [Google Scholar] [CrossRef]
- Nashi, E.; Wang, Y.; Diamond, B. The role of B cells in lupus pathogenesis. Int. J. Biochem. Cell Biol. 2010, 42, 543–550. [Google Scholar] [CrossRef]
- Yu, F.; Haas, M.; Glassock, R.; Zhao, M.H. Redefining lupus nephritis: Clinical implications of pathophysiologic subtypes. Nat. Rev. Nephrol. 2017, 13, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Almaani, S.; Rovin, B.H. B-cell therapy in lupus nephritis: An overview. Nephrol. Dial. Transplant. 2019, 34, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Navarra, S.V.; Guzmán, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.-M.; Thomas, M.; Kim, H.-Y.; León, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef] [PubMed]
- Doria, A.; Stohl, W.; Schwarting, A.; Okada, M.; Scheinberg, M.; van Vollenhoven, R.; Hammer, A.E.; Groark, J.; Bass, D.; Fox, N.L.; et al. Efficacy and safety of subcutaneous belimumab in anti-double-stranded DNA-positive, hypocomplementemic patients with systemic lupus erythematosus. Arthritis Rheumatol. 2018, 70, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Kamburova, E.G.; Koenen, H.J.P.M.; Borgman, K.J.E.; Berge, I.J.; Joosten, I.; Hilbrands, L.B. A single dose of rituximab does not deplete B cells in secondary lymphoid organs but alters phenotype and function. Am. J. Transplant. 2013, 13, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Thurlings, R.M.; Vos, K.; Wijbrandts, C.A.; Zwinderman, A.H.; Gerlag, D.M.; Tak, P.P. Synovial tissue response to rituximab: Mechanism of action and identification of biomarkers of reponse. Ann. Rheum. Dis. 2008, 67, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barret, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, J.; Liu, H.; Yin, G.; Xie, Q. Immunotherapy deriving from CAR-T cell treatment in autoimmune disease. J. Immunol. Res. 2019, 2019, 5727516. [Google Scholar] [CrossRef]
- Kansal, R.; Richardson, N.; Neeli, I.; Khawaja, S.; Chamberlain, D.; Ghani, M.; Ghani, Q.-U.-A.; Balazs, L.; Beranova-Giorgianni, S.; Giorgianni, F.; et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci. Transl. Med. 2019, 11, eaav1648. [Google Scholar] [CrossRef]
- Jin, X.; Xu, Q.; Pu, C.; Zhu, K.; Lu, C.; Jiang, Y.; Xiao, L.; Han, Y.; Lu, L. Therapeutic efficacy of anti-CD19 CAR-T cells in a mouse model of systemic lupus erythematosus. Cell. Mol. Immunol. 2021, 18, 1896–1903. [Google Scholar] [CrossRef]
- Mougiakakos, D.; Krönke, G.; Völkl, S.; Kretschmann, S.; Aigner, M.; Kharboutli, S.; Böltz, S.; Manger, B.; Mackenser, A.; Schett, G. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N. Engl. J. Med. 2021, 385, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Mackensen, A.; Müller, F.; Mougiakakos, D.; Böltz, S.; Wilhelm, A.; Aigner, M.; Völkl, S.; Simon, D.; Kleyer, A.; Munoz, L.; et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med. 2022, 28, 2124–2132. [Google Scholar] [CrossRef]
- Nezhad, M.S.; Seifalian, A.; Bagheri, N.; Yaghoubi, S.; Karimi, M.H.; Adbollahpour-Alitappeh, M. Chimeric antigen receptor based therapy as a potential approach in autoimmune diseases: How close are we to the treatment? Front. Immunol. 2020, 11, 603237. [Google Scholar] [CrossRef]
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.; Cho, M.J.; Zenzo, G.D.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016, 353, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Mao, X.; Manfredo-Vieria, S.; Lee, J.; Patel, D.; Choi, E.J.; Alvarado, A.; Cottman-Thomas, E.; Maseda, D.; Tsao, P.Y.; et al. Precision targeting of autoantigen-specific B cells in muscle-specific tyrosine kinase myasthenia gravis with chimeric autoantibody receptor T cells. Nat. Biotechnol. 2023, 41, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Reincke, S.M.; Wardernburg, N.; Homeyer, M.A.; Kornau, H.-C.; Spagni, G.; Li, L.Y.; Kreye, J.; Sánchez-Sendín, E.; Blumenau, S.; Stappert, D.; et al. Chimeric autoantibody receptor T cells deplete NMDA receptor-specific B cells. Cell 2023, 186, 5084–5097. [Google Scholar] [CrossRef]
- Sahlolbei, M.; Azangou-Khyavy, M.; Khanali, J.; Khorsand, B.; Shiralipour, A.; Ahmadbeigi, N.; Madjd, Z.; Ghanbarian, H.; Ardjmand, A.; Hashemi, S.M.; et al. Engineering chimeric autoantibody receptor T cells for targeted B cell depletion in multiple sclerosis model: An in-vitro study. Heliyon 2023, 9, e19763. [Google Scholar] [CrossRef]
- Hahn, B.H. Antibodies to DNA. N. Engl. J. Med. 1998, 338, 1359–1368. [Google Scholar] [CrossRef]
- Van der Vlag, J.; Berden, J.H.M. Lupus nephritis: Role of antinucleosome autoantibodies. Semin. Nephrol. 2011, 31, 376–389. [Google Scholar] [CrossRef]
- Kianmehr, N.; Khoshmirsafa, M.; Shekarabi, M.; Fala, R.; Haghighi, A.; Masoodian, M.; Seif, F.; Omidi, F.; Shirani, F.; Dadfar, N. High frequency of concurrent anti-C1q and anti-dsDNA but not anti-C3b antibodies in patients with lupus nephritis. J. Immunoass. Immunochem. 2021, 42, 406–423. [Google Scholar] [CrossRef]
- Bock, M.; Heijnen, I.; Trendelenburg, M. Anti-C1q antibodies as a follow-up marker in SLE patients. PLoS ONE 2015, 10, e0123572. [Google Scholar] [CrossRef]
- Conti, F.; Ceccarelli, F.; Perricone, C.; Massaro, L.; Marocchi, E.; Miranda, F.; Spinelli, F.R.; Truglia, S.; Alessandri, C.; Valesini, G. Systemic lupus erythematosus with and without anti-dsDNA antibodies: Analysis from a large monocentric cohort. Mediat. Inflamm. 2015, 2015, 328078. [Google Scholar]
- Yung, S.; Chan, T.M. Mechanisms of kidney injury in lupus nephritis—The role of anti-dsDNA antibodies. Front. Immunol. 2015, 6, 475. [Google Scholar] [CrossRef]
- Linnik, M.D.; Hu, J.Z.; Heilbrunn, K.R.; Strand, V.; Hurley, F.L.; Joh, T.; LJP 394 Investigator Consortium. Relationship between anti-double-stranded DNA antibodies and exacerbation of renal disease in patients with systemic lupus erythematosus. Arthritis Rheum. 2005, 52, 1129–1137. [Google Scholar] [CrossRef]
- Chi, S.; Yu, Y.; Shi, J.; Zhang, Y.; Yan, J.; Yang, L.; Liu, X. Marker for identification of systemic lupus erythematosus patients with active lupus nephritis. Dis. Markers 2015, 2015, 450351. [Google Scholar] [CrossRef]
- Mostoslavsky, G.; Fisherl, R.; Yachimovich, N.; Yarkoni, Y.; Rosenmann, E.; Monestier, M.; Baniyash, M.; Eilat, D. Lupus anti-DNA autoantibodies cross-react with a glomerular structural protein: A case for tissue injury by molecular mimicry. Eur. J. Immunol. 2001, 31, 1221–1227. [Google Scholar] [CrossRef]
- Mjelle, J.E.; Rekvig, O.P.; Van der Vlag, J.; Fenton, K.A. Nephrogenic antibodies bind in glomeruli through interaction with exposed chromatin fragments and not with renal cross-reactive antigens. Autoimmunity 2011, 44, 373–383. [Google Scholar] [CrossRef]
- Krishnan, M.R.; Wang, C.; Marion, T.N. Anti-DNA autoantibodies initiate experimental lupus nephritis by binding directly to the glomerular basement membrane in mice. Kidney Int. 2012, 82, 184–192. [Google Scholar] [CrossRef]
- Mageed, R.A.; Zack, D.J. Cross-reactivity and pathogenicity of anti-DNA autoantibodies in systemic lupus erythematosus. Lupus 2002, 11, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.D.; Ibañez, D.; Urowitz, M.B. Systemic lupus erythematosus disease activity index 2000. J. Rheumatol. 2002, 29, 288–291. [Google Scholar] [PubMed]
- Schutgens, F.; Rookmaaker, M.B.; Margaritis, T.; Rios, A.; Ammerlaan, C.; Jansen, J.; Gijzen, L.; Vormann, M.; Vonk, A.; Viveen, M.; et al. Tubuloids derived from human adult kidney and urine for personalized disease modeling. Nat. Biotechnol. 2019, 37, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Fenton, K.; Fismen, S.; Hedberg, A.; Seredkina, N.; Fenton, C.; Mortensen, E.S.; Rekvig, O.P. Anti-dsDNA antibodies promote initiation, and acquired loss of renal dnase 1 promotes progression of lupus nephritis in autoinmune (NZBxNZW)F1 mice. PLoS ONE 2009, 4, e8474. [Google Scholar] [CrossRef] [PubMed]
- Waldman, M.; Madaio, M.P. Pathogenic autoantibodies in lupus nephritis. Lupus 2005, 14, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xia, Y. Anti-double stranded DNA antibodies: Origin, pathogenicity, and targeted therapies. Front. Immunol. 2019, 10, 1667. [Google Scholar] [CrossRef] [PubMed]
- Touloupi, E.; Routsias, J.G.; Tzioufas, A.G. Cross-recognition between histones and La/SSB may account for anti-DNA reactivity in SLE patients. Clin. Exp. Immunol. 2005, 142, 172–179. [Google Scholar] [CrossRef]
- Franchin, G.; Son, M.; Kim, S.J.; Ben-Zvi, I.; Zhang, J.; Diamond, B. Anti-DNA antibodies cross-react with C1q. J. Autoimmun. 2013, 44, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Weinstein, E.; Tuzova, M.; Davidson, A.; Mundel, P.; Marambio, P.; Putterman, C. Cross-reactivity of human lupus anti-DNA antibodies with alpha-actinin and neprhitogenic potential. Arhtritis Rheum. 2005, 52, 522–530. [Google Scholar] [CrossRef]
- Termaat, R.M.; Assman, K.J.; Dijkman, H.B.; Gompel, F.; Smeenk, R.J.; Berden, J.H. Anti-DNA antibodies can bidn to the glomerulus via two distinct mechanisms. Kidney Int. 1992, 42, 1363–1371. [Google Scholar] [CrossRef]
- Yung, S.; Chan, T.M. Anti-dsDNA antibodies and resident renal cells—Their putative roles in pathogenesis of renal lesions in lupus nephritis. Clin. Immunol. 2017, 185, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Faaber, P.; Rijke, T.P.; Van de Putte, L.B.; Capel, P.J.; Berden, J.H. Cross-reactivity of human and murine anti-DNA antibodies with heparan sulfate. The major glycosaminoglycan in glomerular basement membranes. J. Clin. Investig. 1986, 77, 1824–1830. [Google Scholar] [CrossRef] [PubMed]
- Termaat, R.M.; Brinkman, K.; Van Gompel, F.; Van den Heuvel, L.P.; Veerkamp, J.H.; Smeenk, R.J.; Berden, J.H.M. Croos-reactivity of monoclonal anti-DNA antibodies with heparan sulfate is mediated via bound DNA/histone complexes. J. Autoimmun. 1990, 3, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Meneghetti, M.C.Z.; Hughes, A.J.; Rudd, T.R.; Nader, H.B.; Power, A.K.; Yates, E.A.; Lima, M.A. Heparan sulfate and heparin interactions with proteins. J. R. Soc. Interface 2015, 12, 20150589. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Luo, C.; Li, C.; Huang, J.; Liu, F. Organoids to study immune functions, immunological disease and immunotherapy. Cancer Lett. 2020, 477, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Mu, P.; Zhou, S.; Lv, T.; Xia, F.; Shen, L.; Wan, J.; Wang, Y.; Zhang, H.; Cai, S.; Peng, J.; et al. Newly developed 3D in vitro models to study tumor-immune interaction. J. Exp. Clin. Cancer Res. 2023, 42, 81. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Li, Z.; Mei, H.; Li, W.; Chen, D.; Liu, L.; Zhang, Z.; Sun, Y.; Song, F.; Chen, W.; et al. Patient-derived organoids of bladder cancer recapitulate antigen expression profiles and serve as a personal evaluation model for CAR-T cells in vitro. Clin. Transl. Immunol. 2021, 10, e1248. [Google Scholar] [CrossRef]
- Nozaki, Y.; Kinoshita, K.; Yano, T.; Shiga, T.; Hino, S.; Niki, K.; Kishimoto, K.; Funauchi, M.; Matsumura, I. Estimation of kidney injury molecule-1 (Kim-1) in patients with lupus nephritis. Lupus 2014, 23, 769–777. [Google Scholar] [CrossRef]
- Yan, Q.; Sui, W.; Xie, S.; Chen, H.; Xie, S.; Zou, G.; Guo, J.; Zou, H. Expression and role of integrin-linked kinase and collagen IV in human renal allografts with interstitial fibrosis and tubular atrophy. Transpl. Immunol. 2010, 23, 1–5. [Google Scholar] [CrossRef]
- Guarino, M.; Tosoni, A.; Nebuloni, M. Direct contribution of epithelium to organ fibrosis: Epithelial-mesenchymal transition. Hum. Pathol. 2009, 40, 1365–1376. [Google Scholar] [CrossRef]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory process in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Almqvist, N.; Winkler, T.H.; Martensson, I. Autoantibodies: Focus on anti-DNA antibodies. Self Nonself 2011, 2, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Vanhecke, D.; Roumenina, L.T.; Wan, H.; Osthoff, M.; Schaller, M.; Trendelenburg, M. Identification of a major linear C1q epitope allows detection of systemic lupus erythematosus anti-C1q antibodies by a specific peptide-based enzyme-linked immunosorbent assay. Arthritis Rheum. 2012, 64, 3706–3714. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Metharom, P.; Jullie, H.; Ellem, K.A.O.; Cleghorn, G.; West, M.J.; Wei, M.Q. The significance of controlled conditions in lentiviral vector titration and in the use of multiplicity of infection (MOI) for predicting gene transfer events. Genet. Vaccines Ther. 2004, 2, 6. [Google Scholar] [CrossRef]
- Cortés-Hernández, J.; Ordi-Ros, J.; Labrador, M.; Buján, S.; Balada, E.; Segarra, A.; Vilardell-Tarrés, M. Antihistone and anti-double-stranded deoxyribonucleic acid antibodies are associated with renal disease in systemic lupus erythematosus. Am. J. Med. 2004, 116, 165–173. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lupus Nephritis Anti-DNA+ (n = 10) | Lupus Nephritis Anti-DNA− (n = 10) | p Value a | |
|---|---|---|---|
| Demographic | |||
| Age (years) | 41.2 ± 8.3 | 42.8 ± 12.8 | 0.744 |
| Gender (Female/male) | 10/10 | 8/10 | 0.136 |
| Race/ethnicity, n (%) | |||
| White | 4 (40) | 6 (60) | 0.371 |
| Hispanic | 6 (60) | 4 (40) | |
| Other | 0 (0) | 0 (0) | |
| Laboratory parameters | |||
| Serum creatinine, mg/dL | 1.1 ± 0.9 | 0.8 ± 0.2 | 0.317 |
| eGFR (mL/min) | 87 ± 6.6 | 88 ± 3.4 | 0.675 |
| Serum C3, mg/dL | 75 ± 32 | 96 ± 16 | 0.08 |
| Serum C4, mg/dL | 13.9 ± 8.8 | 16.4 ± 11.9 | 0.600 |
| Hypocomplementemia | 6 (60) | 3 (30) | 0.178 |
| Proteinuria, g/24 h | 1.9 ± 1.5 | 1.3 ± 1.5 | 0.383 |
| Autoantibody status at inclusion | |||
| ANA titre ≥ 1/80, n (%) | 10 (100) | 8 (80) | 0.474 |
| Anti-dsDNA antibodies ≥ 15 IU/mL, n (%) | 10 (100) | 0 (0) | <0.0001 |
| Anti-dsDNA antibodies, IU/mL | 268.5 ± 237.6 | 2.8 ± 8.8 | 0.0024 |
| Anti-SSA/Ro antibodies, n (%) | 6 (60) | 3 (30) | 0.177 |
| Anti-SSA/La antibodies, n (%) | 2 (20) | 0 (0) | 0.136 |
| Anti-RNP antibodies, n (%) | 4 (40) | 4 (40) | 1.000 |
| Anti-Sm antibodies, n (%) | 6 (60) | 4 (40) | 0.371 |
| Anti-C1q, n (%) | 6 (60) | 0 (0) | 0.011 |
| Anti-Histone1, n (%) | 6 (60) | 1 (10) | 0.057 |
| Anti-α-actinin, n (%) | 6 (60) | 1 (10) | 0.057 |
| Anti-heparan sulphate, n (%) | 9 (90) | 1 (10) | 0.0011 |
| Disease index (SLEDAI-2K) | |||
| Total score | 5.8 ± 1.8 | 4.4 ± 2.0 | 0.117 |
| Treatment at inclusion | |||
| Antimalarial agents, n (%) | 7 (70) | 4 (40) | 0.177 |
| Immunosuppressive therapy, n (%) | 10 (100) | 10 (100) | 1.000 |
| MMF/EC-MPS | 10 (100) | 10 (100) | 1.000 |
| Anticalcineurinic agents | 2 (20) | 2 (20) | 1.000 |
| Corticosteroid use, n (%) | 8 (80) | 9 (90) | 0.531 |
| Daily prednisone dose, mg/day | 2.5 ± 0.8 | 2.0 ± 0.5 | 0.111 |
| Type of GMN at flare, n (%) | |||
| Class III | 3 (30) | 2 (20) | 1.000 |
| Class IV | 5 (50) | 6 (60) | 1.000 |
| Class III/IV-V | 2 (20) | 2 (20) | 1.000 |
| Activity Index, mean | 6.3 ± 5.2 | 3.3 ± 1.9 | 0.104 |
| Chronicity Index, mean | 3.7 ± 1.7 | 2.8 ± 1.0 | 0.166 |
| Autoantibody status at renal biopsy | |||
| ANA titre ≥ 1/80, n (%) | 10 (100) | 10 (100) | 1.000 |
| Anti-dsDNA antibodies ≥ 15 IU/mL, n (%) | 10 (100) | 7 (70) | 0.210 |
| Anti-dsDNA antibodies, IU/mL | 384.6 ± 165.9 | 179.14 ± 297.35 | 0.073 |
| Anti-SSA/Ro antibodies, n (%) | 6 (60) | 3 (30) | 0.177 |
| Anti-SSA/La antibodies, n (%) | 2 (20) | 0 (0) | 0.136 |
| Anti-RNP antibodies, n (%) | 4 (40) | 4 (40) | 1.000 |
| Anti-Sm antibodies, n (%) | 6 (60) | 4 (40) | 0.371 |
| Anti-C1q, n (%) | 6 (60) | 2 (20) | 0.169 |
| Anti-Histone1, n (%) | 5 (50) | 2 (20) | 0.349 |
| Anti-α-actinin, n (%) | 7 (70) | 5 (50) | 0.649 |
| Anti-heparan sulphate, n (%) | 10 (100) | 7 (70) | 0.210 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solé, C.; Royo, M.; Sandoval, S.; Moliné, T.; Gabaldón, A.; Cortés-Hernández, J. Precise Targeting of Autoantigen-Specific B Cells in Lupus Nephritis with Chimeric Autoantibody Receptor T Cells. Int. J. Mol. Sci. 2024, 25, 4226. https://doi.org/10.3390/ijms25084226
Solé C, Royo M, Sandoval S, Moliné T, Gabaldón A, Cortés-Hernández J. Precise Targeting of Autoantigen-Specific B Cells in Lupus Nephritis with Chimeric Autoantibody Receptor T Cells. International Journal of Molecular Sciences. 2024; 25(8):4226. https://doi.org/10.3390/ijms25084226
Chicago/Turabian StyleSolé, Cristina, Maria Royo, Sebastian Sandoval, Teresa Moliné, Alejandra Gabaldón, and Josefina Cortés-Hernández. 2024. "Precise Targeting of Autoantigen-Specific B Cells in Lupus Nephritis with Chimeric Autoantibody Receptor T Cells" International Journal of Molecular Sciences 25, no. 8: 4226. https://doi.org/10.3390/ijms25084226
APA StyleSolé, C., Royo, M., Sandoval, S., Moliné, T., Gabaldón, A., & Cortés-Hernández, J. (2024). Precise Targeting of Autoantigen-Specific B Cells in Lupus Nephritis with Chimeric Autoantibody Receptor T Cells. International Journal of Molecular Sciences, 25(8), 4226. https://doi.org/10.3390/ijms25084226