
A Meta-Analysis Approach to Gene Regulatory Network Inference Identifies Key Regulators of Cardiovascular Diseases
 , ,
, ,  and
and
Abstract
1. Introduction
2. Results and Discussion
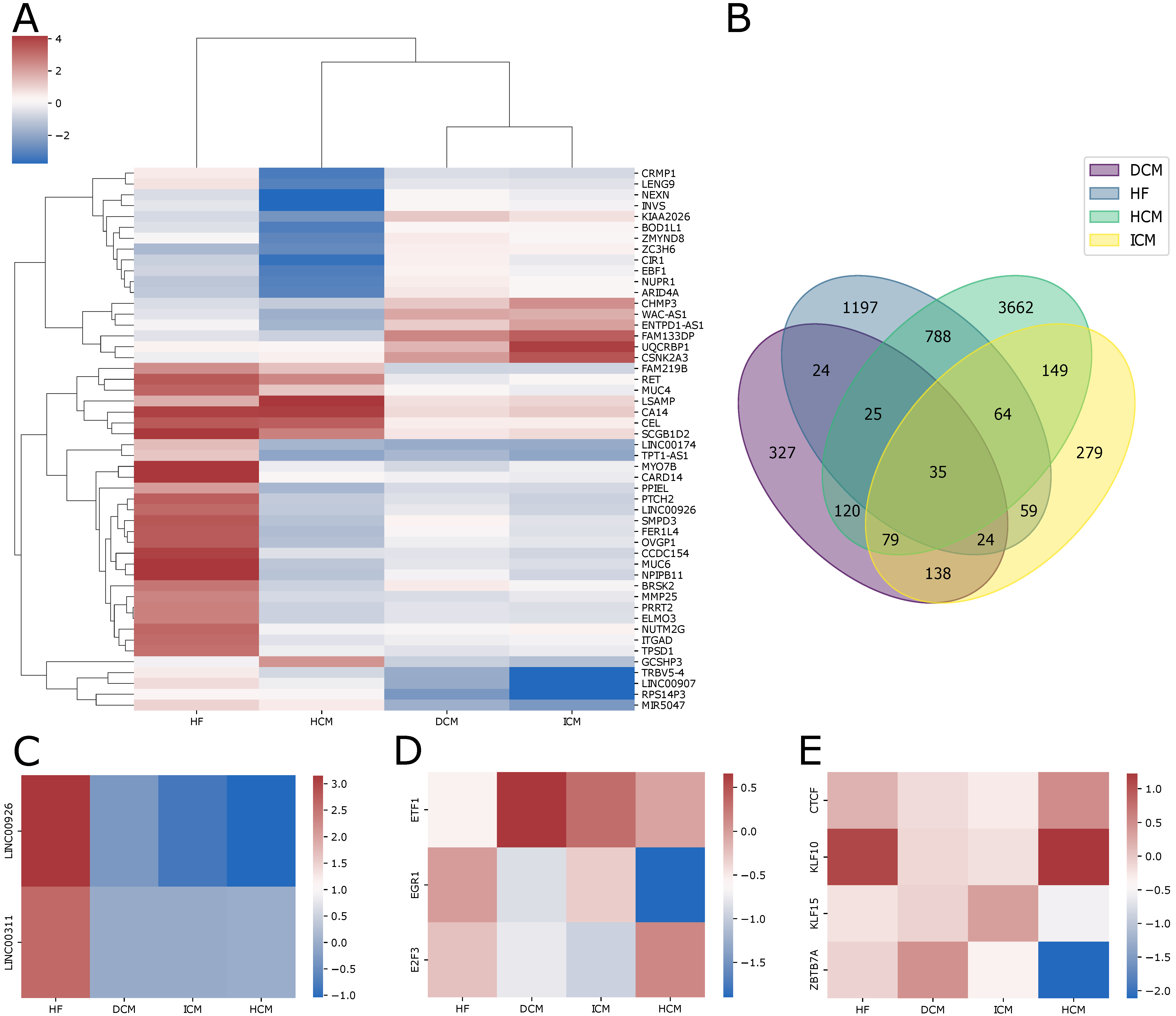
2.1. Transcriptome Changes in Patients with Cardiac Pathologies
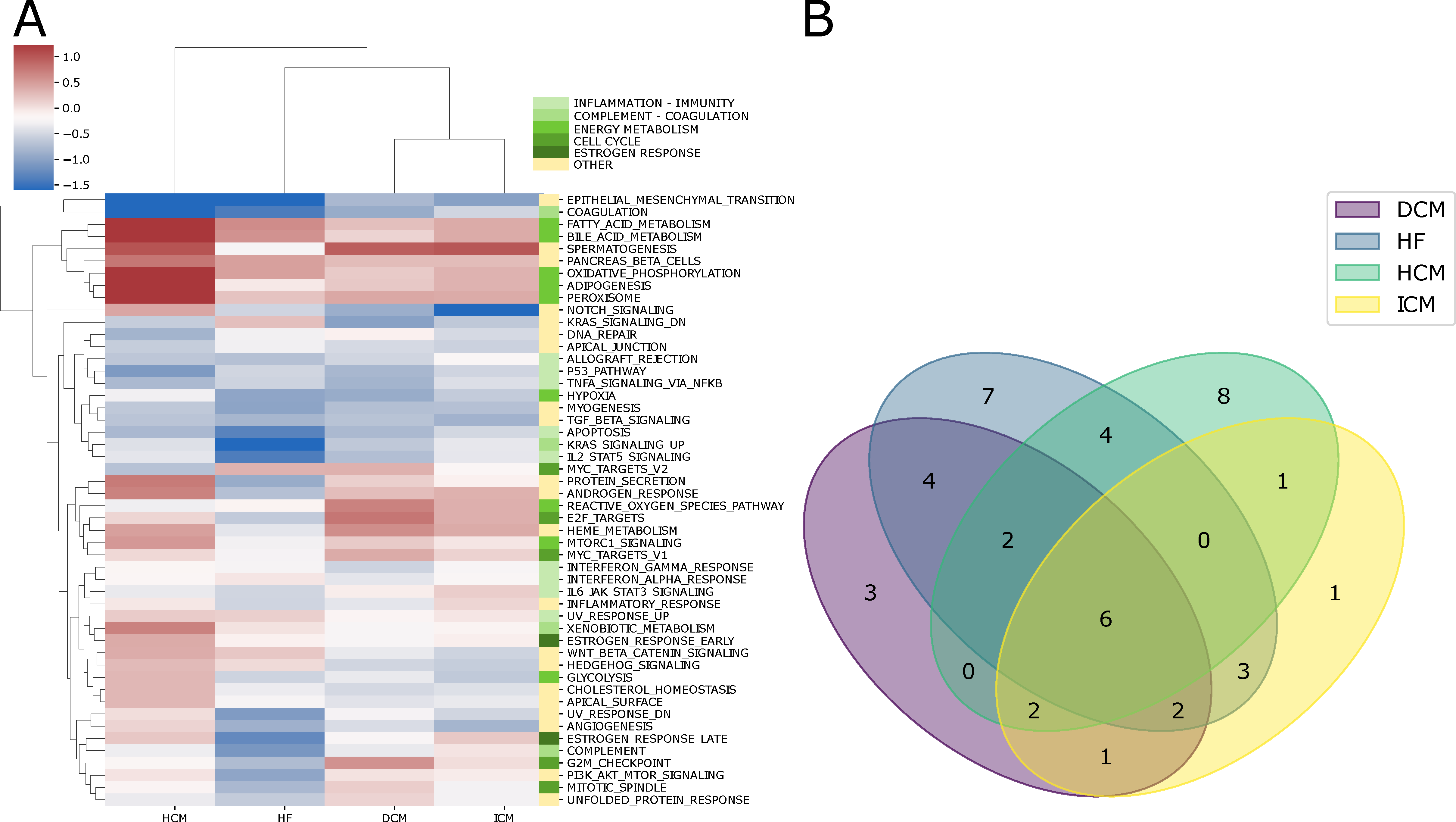
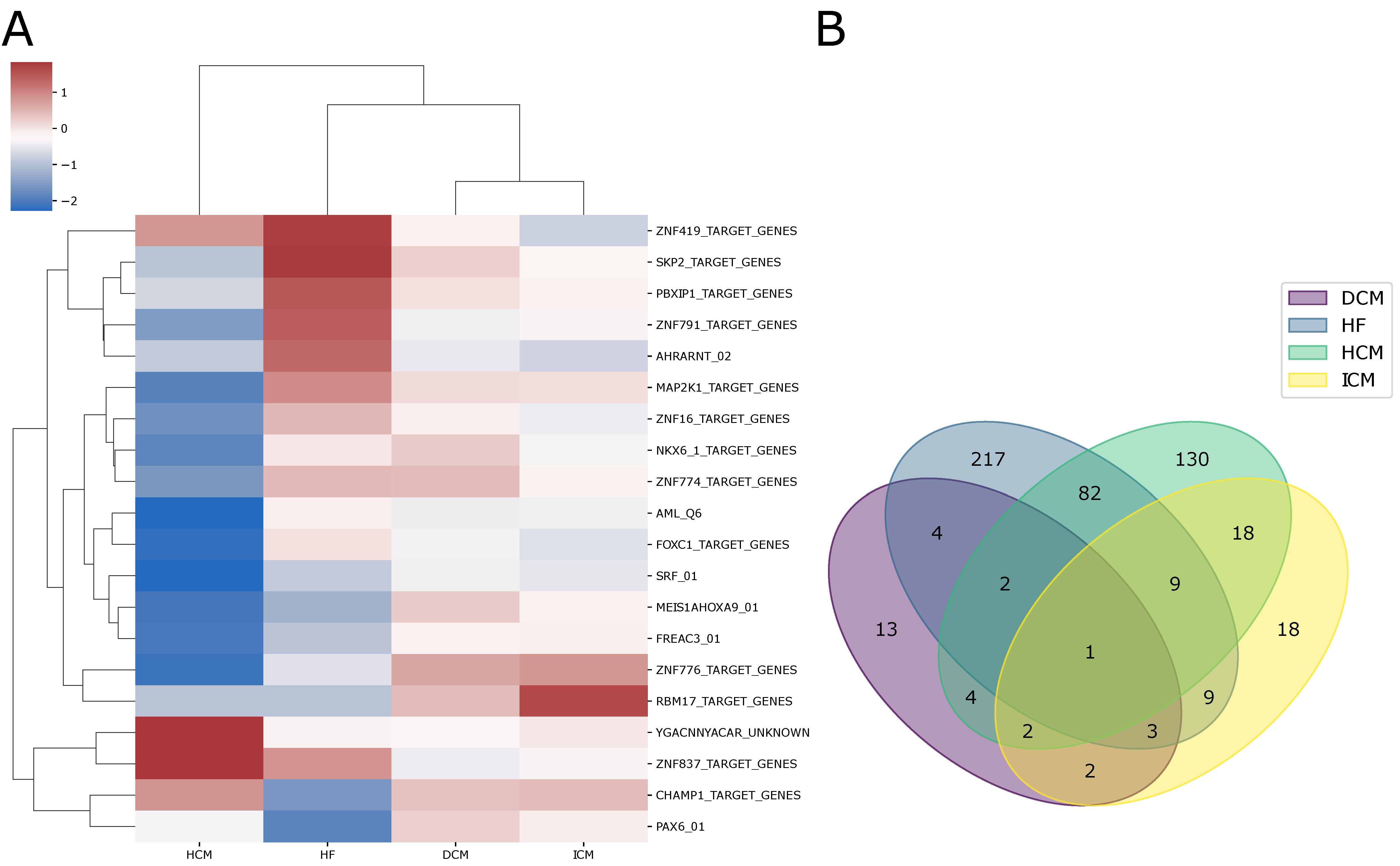
2.2. Alterations in Pathway Activity Profiles
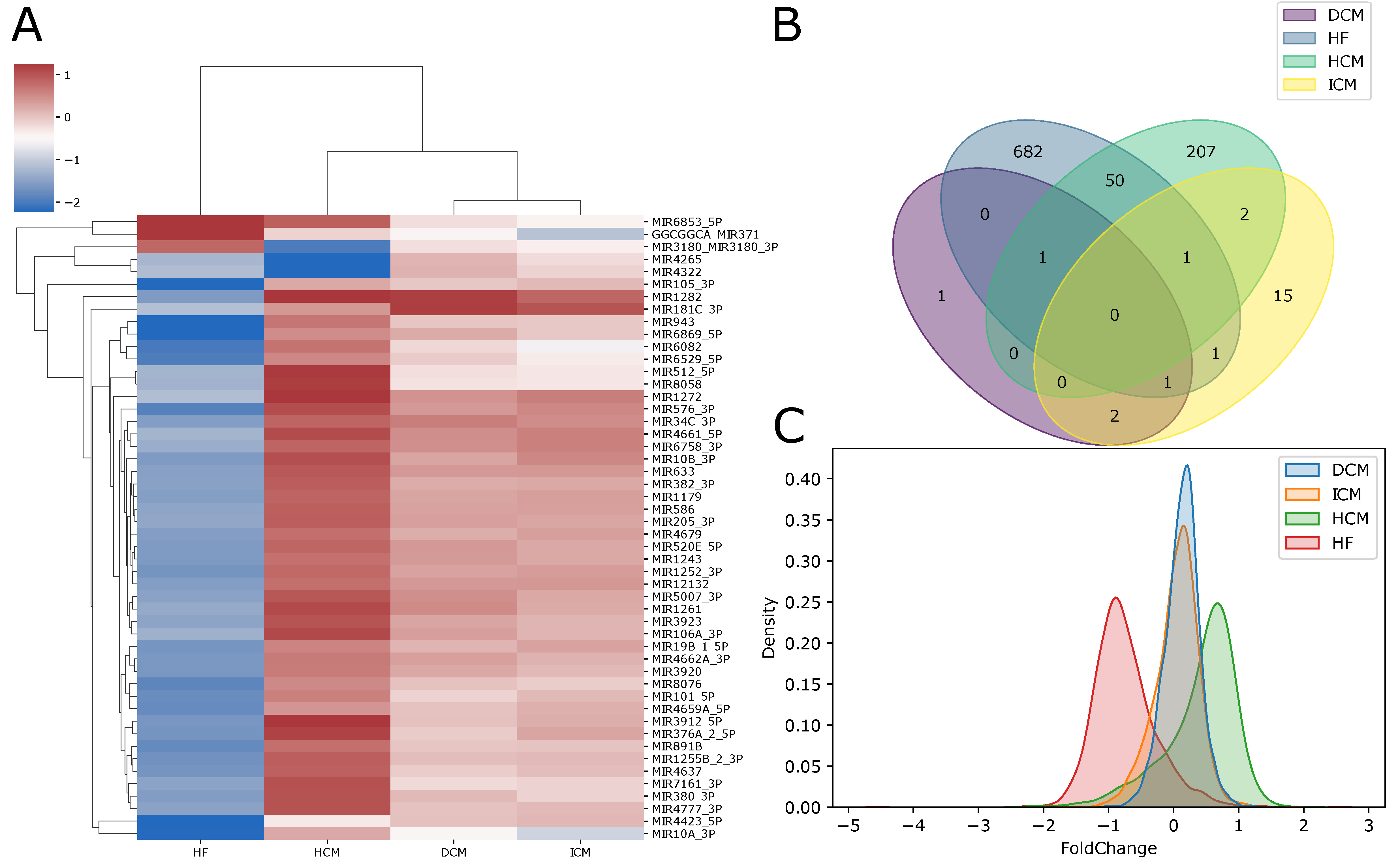
2.3. Differentially Expressed microRNAs
2.4. Variations in Transcription Factor Activity Profiles
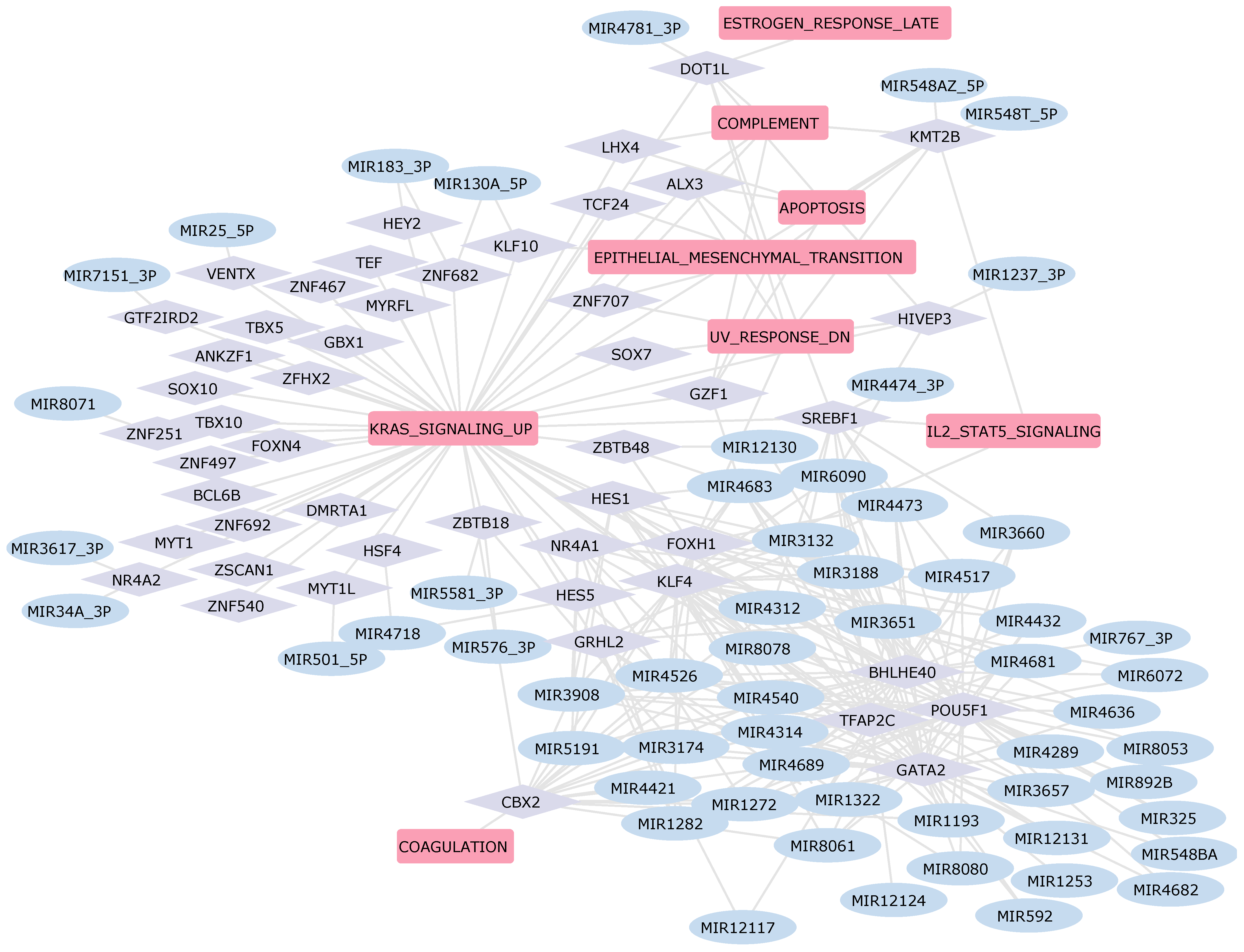
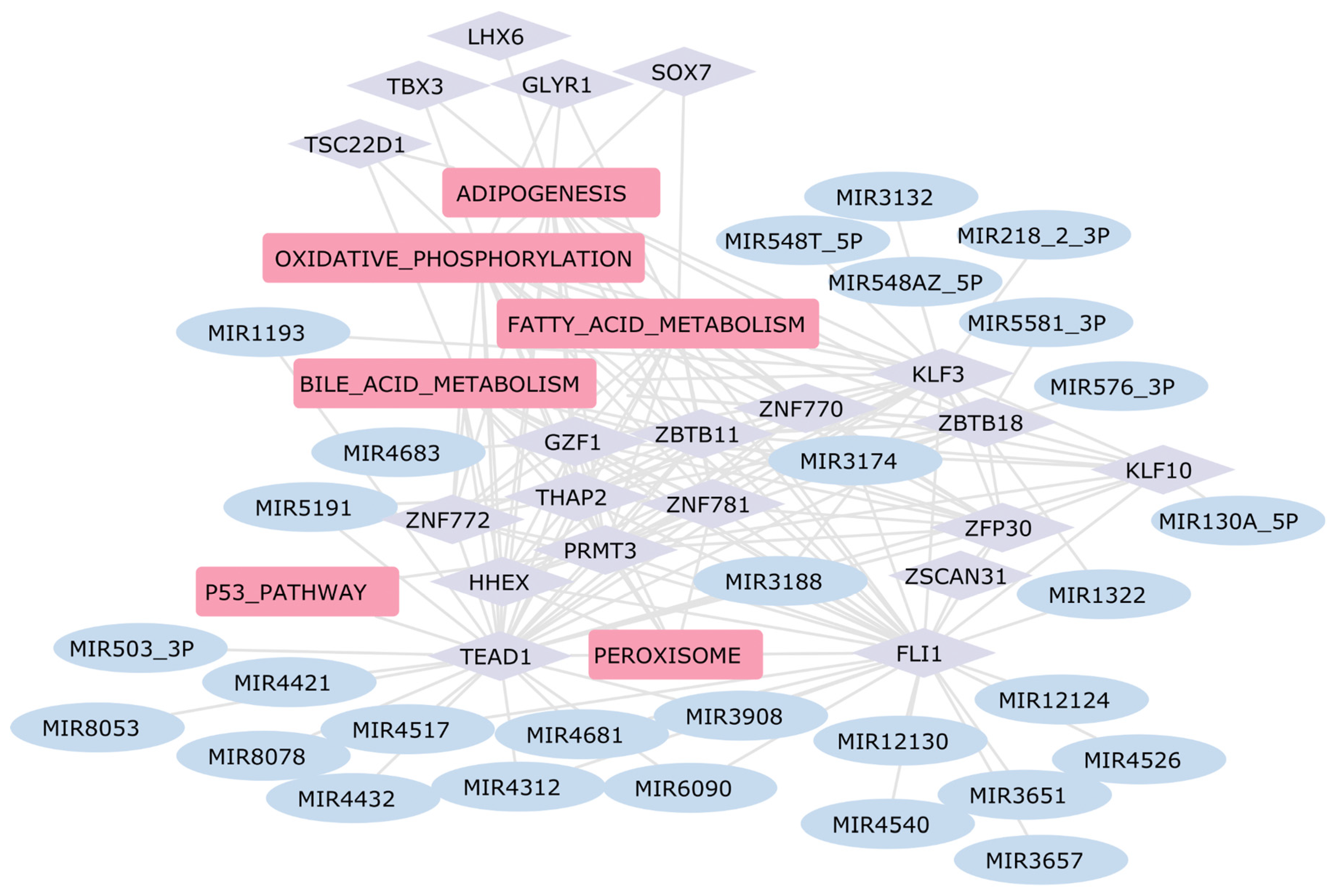
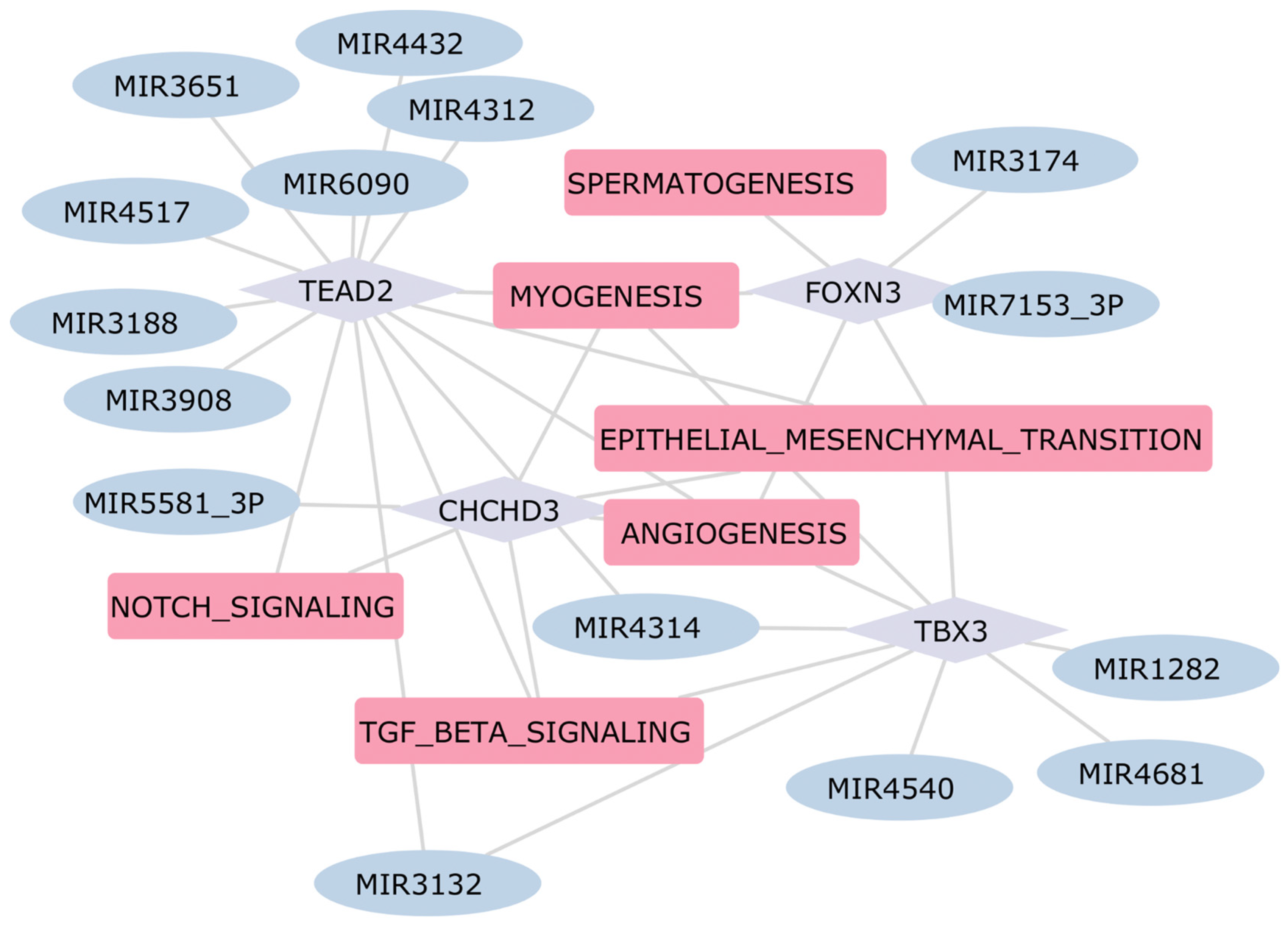
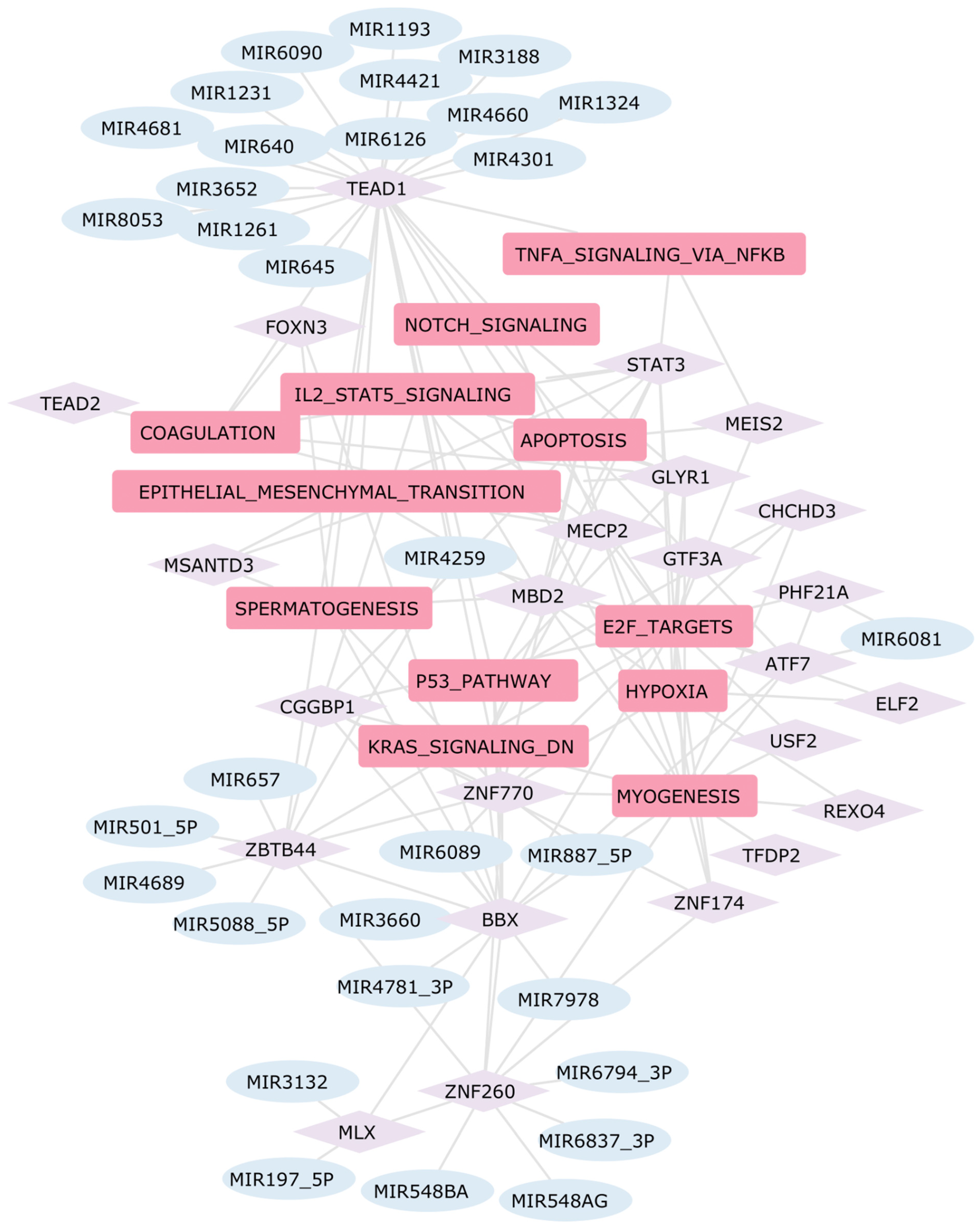
2.5. Construction of the Gene Regulatory Network in Heart Diseases
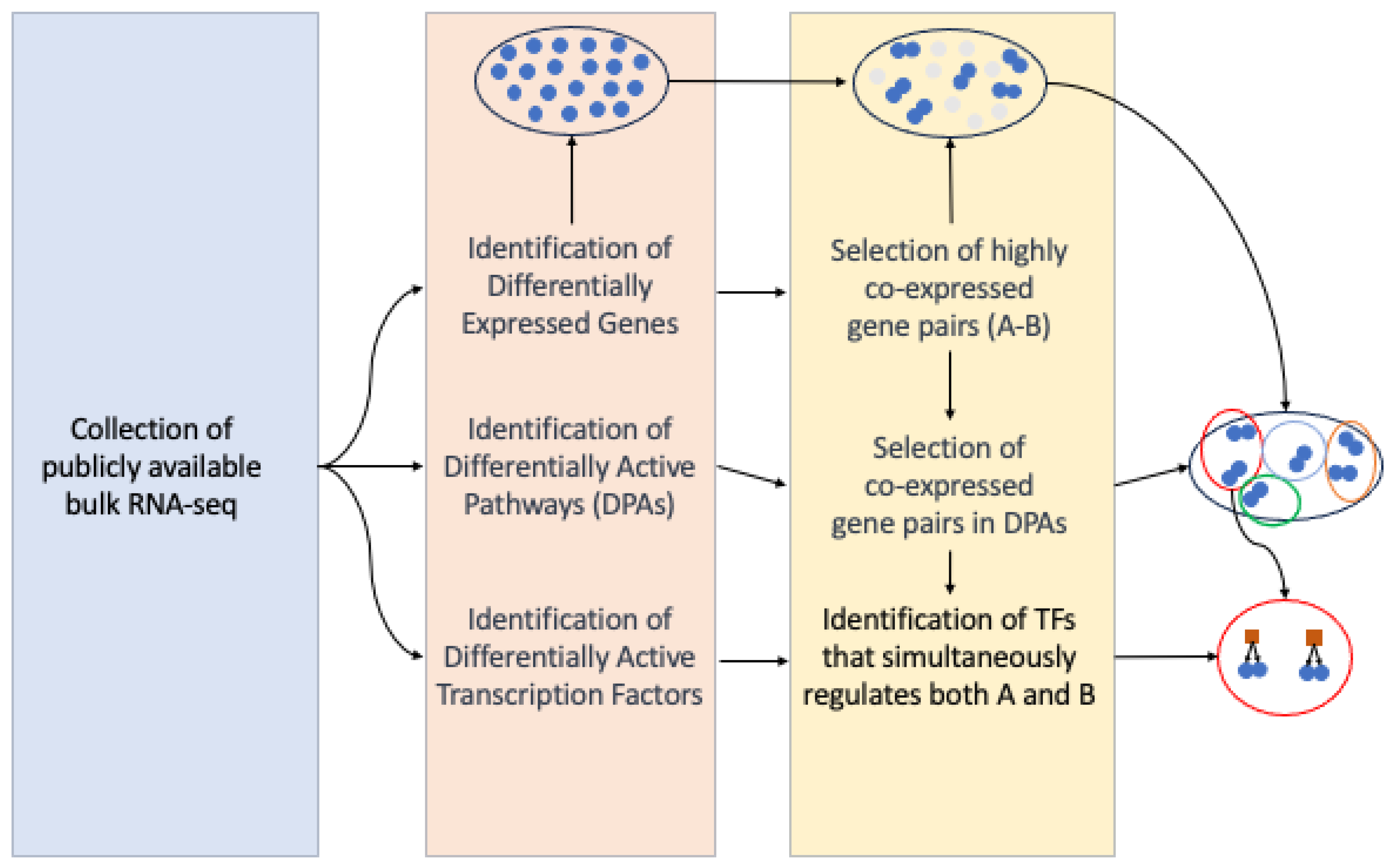
3. Materials and Methods
3.1. Dataset Collection
3.2. Gene Set Variation Analysis (GSVA)
3.3. Meta-Analysis
3.4. Hierarchical Gene Regulatory Network
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Emmons-Bell, S.; Johnson, C.; Roth, G. Prevalence, Incidence and Survival of Heart Failure: A Systematic Review. Heart 2022, 108, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Snipelisky, D.; Chaudhry, S.-P.; Stewart, G.C. The Many Faces of Heart Failure. Card. Electrophysiol. Clin. 2019, 11, 11–20. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure of the European Society of Cardiology (ESC)Developed with the Special Contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [PubMed]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated Cardiomyopathy: The Complexity of a Diverse Genetic Architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef] [PubMed]
- El-Sadr, W.M.; Burman, W.J.; Grant, L.B.; Matts, J.P.; Hafner, R.; Crane, L.; Zeh, D.; Gallagher, B.; Mannheimer, S.B.; Martinez, A.; et al. Discontinuation of Prophylaxis against Mycobacterium Avium Complex Disease in HIV-Infected Patients Who Have a Response to Antiretroviral Therapy. Terry Beirn Community Programs for Clinical Research on AIDS. N. Engl. J. Med. 2000, 342, 1085–1092. [Google Scholar] [CrossRef]
- Del Buono, M.G.; Moroni, F.; Montone, R.A.; Azzalini, L.; Sanna, T.; Abbate, A. Ischemic Cardiomyopathy and Heart Failure After Acute Myocardial Infarction. Curr. Cardiol. Rep. 2022, 24, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A.; Wang, R.-S.; Carnethon, M.R.; Rowin, E.J.; Loscalzo, J.; Maron, B.J.; Maron, M.S. What Causes Hypertrophic Cardiomyopathy? Am. J. Cardiol. 2022, 179, 74–82. [Google Scholar] [CrossRef]
- Liang, W.; Sun, F.; Zhao, Y.; Shan, L.; Lou, H. Identification of Susceptibility Modules and Genes for Cardiovascular Disease in Diabetic Patients Using WGCNA Analysis. J. Diabetes Res. 2020, 2020, 4178639. [Google Scholar] [CrossRef]
- Roth, R.; Kim, S.; Kim, J.; Rhee, S. Single-Cell and Spatial Transcriptomics Approaches of Cardiovascular Development and Disease. BMB Rep. 2020, 53, 393–399. [Google Scholar] [CrossRef]
- Paik, D.T.; Cho, S.; Tian, L.; Chang, H.Y.; Wu, J.C. Single-Cell RNA Sequencing in Cardiovascular Development, Disease and Medicine. Nat. Rev. Cardiol. 2020, 17, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Kuppe, C.; Ramirez Flores, R.O.; Li, Z.; Hayat, S.; Levinson, R.T.; Liao, X.; Hannani, M.T.; Tanevski, J.; Wünnemann, F.; Nagai, J.S.; et al. Spatial Multi-Omic Map of Human Myocardial Infarction. Nature 2022, 608, 766–777. [Google Scholar] [CrossRef] [PubMed]
- Chaffin, M.; Papangeli, I.; Simonson, B.; Akkad, A.-D.; Hill, M.C.; Arduini, A.; Fleming, S.J.; Melanson, M.; Hayat, S.; Kost-Alimova, M.; et al. Single-Nucleus Profiling of Human Dilated and Hypertrophic Cardiomyopathy. Nature 2022, 608, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M. An Overview of Epigenetics in Obesity: The Role of Lifestyle and Therapeutic Interventions. Int. J. Mol. Sci. 2022, 23, 1341. [Google Scholar] [CrossRef] [PubMed]
- Światowy, W.J.; Drzewiecka, H.; Kliber, M.; Sąsiadek, M.; Karpiński, P.; Pławski, A.; Jagodziński, P.P. Physical Activity and DNA Methylation in Humans. Int. J. Mol. Sci. 2021, 22, 12989. [Google Scholar] [CrossRef]
- Peterlin, A.; Počivavšek, K.; Petrovič, D.; Peterlin, B. The Role of microRNAs in Heart Failure: A Systematic Review. Front. Cardiovasc. Med. 2020, 7, 161. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, P.; Marzano, F.; Salvatore, M.; Basile, C.; Buonocore, D.; Parlati, A.L.M.; Nardi, E.; Asile, G.; Abbate, V.; Colella, A.; et al. MicroRNAs: Diagnostic, Prognostic and Therapeutic Role in Heart Failure-a Review. ESC Heart Fail. 2023, 10, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-S.; Wu, J.-J.; Xing, X.-X.; Li, Y.-L.; Ma, J.; Duan, Y.-J.; Zhang, J.-P.; Shan, C.-L.; Hua, X.-Y.; Zheng, M.-X.; et al. Focal Ischemic Stroke Modifies Microglia-Derived Exosomal miRNAs: Potential Role of Mir-212-5p in Neuronal Protection and Functional Recovery. Biol. Res. 2023, 56, 52. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Tarazón, E.; Roselló-Lletí, E.; Rivera, M.; Ortega, A.; Molina-Navarro, M.M.; Triviño, J.C.; Lago, F.; González-Juanatey, J.R.; Orosa, P.; Montero, J.A.; et al. RNA Sequencing Analysis and Atrial Natriuretic Peptide Production in Patients with Dilated and Ischemic Cardiomyopathy. PLoS ONE 2014, 9, e90157. [Google Scholar] [CrossRef]
- Sweet, M.E.; Cocciolo, A.; Slavov, D.; Jones, K.L.; Sweet, J.R.; Graw, S.L.; Reece, T.B.; Ambardekar, A.V.; Bristow, M.R.; Mestroni, L.; et al. Transcriptome Analysis of Human Heart Failure Reveals Dysregulated Cell Adhesion in Dilated Cardiomyopathy and Activated Immune Pathways in Ischemic Heart Failure. BMC Genomics 2018, 19, 812. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Yu, P.; Li, D.; Li, Z.; Liao, Y.; Wang, Y.; Zhou, B.; Wang, L. Single-Cell Reconstruction of Progression Trajectory Reveals Intervention Principles in Pathological Cardiac Hypertrophy. Circulation 2020, 141, 1704–1719. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Wang, Y.-Y.; Jia, P.; Xiong, Q.; Hu, Y.; Chang, Y.; Lai, S.; Xu, Y.; Zhao, Z.; Song, J. Multi-Level Transcriptome Sequencing Identifies COL1A1 as a Candidate Marker in Human Heart Failure Progression. BMC Med. 2020, 18, 2. [Google Scholar] [CrossRef]
- Yan, F.-J.; Wang, Y.-J.; Yan, S.-R.; Lu, J.; Zheng, Y.-L. ZNF300 Stimulates Fatty Acid Oxidation and Alleviates Hepatosteatosis through Regulating PPARα. Biochem. J. 2019, 476, 385–404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiao, L.; Sun, L.; Li, Y.; Gao, Y.; Xu, C.; Shao, Y.; Li, M.; Li, C.; Lu, Y.; et al. LncRNA as a SERCA2a Inhibitor to Cause Intracellular Ca Overload and Contractile Dysfunction in a Mouse Model of Myocardial Infarction. Circ. Res. 2018, 122, 1354–1368. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Deng, J.; Ma, W.; Qiao, A.; Xu, S.; Yu, Y.; Boriboun, C.; Kang, X.; Han, D.; Ernst, P.; et al. Ablation of lncRNA Attenuates Pathological Hypertrophy and Heart Failure. Theranostics 2021, 11, 7995–8007. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.; Zhang, Y.-H.; Li, R.-B.; Zhou, L.-Y.; An, T.; Zhang, R.-C.; Zhai, M.; Huang, Y.; Yan, K.-W.; Dong, Y.-H.; et al. LncRNA CAIF Inhibits Autophagy and Attenuates Myocardial Infarction by Blocking p53-Mediated Myocardin Transcription. Nat. Commun. 2018, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Wang, B.; Zhao, H.; Wang, W.; Wang, P.; Deng, Y. LncRNA Promoted Inflammatory Response in Ischemic Heart Failure through Regulation of miR-455-3p/TRAF6 Axis. Inflamm. Res. 2020, 69, 667–681. [Google Scholar] [CrossRef]
- Andenæs, K.; Lunde, I.G.; Mohammadzadeh, N.; Dahl, C.P.; Aronsen, J.M.; Strand, M.E.; Palmero, S.; Sjaastad, I.; Christensen, G.; Engebretsen, K.V.T.; et al. The Extracellular Matrix Proteoglycan Fibromodulin Is Upregulated in Clinical and Experimental Heart Failure and Affects Cardiac Remodeling. PLoS ONE 2018, 13, e0201422. [Google Scholar] [CrossRef]
- Elsik, M. Myocardial Fibrosis and Extracellular Matrix Remodelling in Chronic Heart Failure: Novel Treatment and Monitoring Strategies. Ph.D. Thesis, Monash University, Clayton, VIC, Australia, 2010. [Google Scholar]
- Belviso, I.; Angelini, F.; Di Meglio, F.; Picchio, V.; Sacco, A.M.; Nocella, C.; Romano, V.; Nurzynska, D.; Frati, G.; Maiello, C.; et al. The Microenvironment of Decellularized Extracellular Matrix from Heart Failure Myocardium Alters the Balance between Angiogenic and Fibrotic Signals from Stromal Primitive Cells. Int. J. Mol. Sci. 2020, 21, 7903. [Google Scholar] [CrossRef]
- Guertl, B.; Noehammer, C.; Hoefler, G. Metabolic Cardiomyopathies. Int. J. Exp. Pathol. 2000, 81, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Perestrelo, A.R.; Silva, A.C.; La Cruz, J.O.-D.; Martino, F.; Horváth, V.; Caluori, G.; Polanský, O.; Vinarský, V.; Azzato, G.; de Marco, G.; et al. Multiscale Analysis of Extracellular Matrix Remodeling in the Failing Heart. Circ. Res. 2021, 128, 24–38. [Google Scholar] [CrossRef]
- Kapelko, V.I. Extracellular Matrix Alterations in Cardiomyopathy: The Possible Crucial Role in the Dilative Form. Exp. Clin. Cardiol. 2001, 6, 41–49. [Google Scholar] [PubMed]
- Chen, K.; Shi, Y.; Zhu, H. Analysis of the Role of Glucose Metabolism-Related Genes in Dilated Cardiomyopathy Based on Bioinformatics. J. Thorac. Dis. 2023, 15, 3870–3884. [Google Scholar] [CrossRef]
- Calderon-Dominguez, M.; Belmonte, T.; Quezada-Feijoo, M.; Ramos, M.; Calderon-Dominguez, J.; Campuzano, O.; Mangas, A.; Toro, R. Plasma Microrna Expression Profile for Reduced Ejection Fraction in Dilated Cardiomyopathy. Sci. Rep. 2021, 11, 7517. [Google Scholar] [CrossRef]
- Burke, M.A.; Chang, S.; Wakimoto, H.; Gorham, J.M.; Conner, D.A.; Christodoulou, D.C.; Parfenov, M.G.; DePalma, S.R.; Eminaga, S.; Konno, T.; et al. Molecular Profiling of Dilated Cardiomyopathy That Progresses to Heart Failure. JCI Insight 2016, 1, e86898. [Google Scholar] [CrossRef]
- Marín-García, J.; Goldenthal, M.J. Fatty Acid Metabolism in Cardiac Failure: Biochemical, Genetic and Cellular Analysis. Cardiovasc. Res. 2002, 54, 516–527. [Google Scholar] [CrossRef]
- Unezaki, S.; Horai, R.; Sudo, K.; Iwakura, Y.; Ito, S. Ovol2/Movo, a Homologue of Drosophila Ovo, Is Required for Angiogenesis, Heart Formation and Placental Development in Mice. Genes Cells 2007, 12, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Sunny, S.S.; Lachova, J.; Kasparek, P.; Palkova, M.; Spoutil, F.; Prochazka, J.; Sedlacek, R.; Liskova, P.; Kozmik, Z. Ovol2 Promoter Mutations in Mice and Human Illuminate Species-Specific Phenotypic Divergence. Hum. Mol. Genet. 2024, 33, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Geng, G.-J.; Guo, J.-G.; Mi, Y.-J.; Zhu, X.-L.; Li, N.; Liu, H.-M.; Lin, J.-F.; Wang, J.-W.; Zhao, G.; et al. An NF-κB/OVOL2 Circuit Regulates Glucose Import and Cell Survival in Non-Small Cell Lung Cancer. Cell Commun. Signal. 2022, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Gugnoni, M.; Manzotti, G.; Vitale, E.; Sauta, E.; Torricelli, F.; Reggiani, F.; Pistoni, M.; Piana, S.; Ciarrocchi, A. OVOL2 Impairs RHO GTPase Signaling to Restrain Mitosis and Aggressiveness of Anaplastic Thyroid Cancer. J. Exp. Clin. Cancer Res. 2022, 41, 108. [Google Scholar] [CrossRef] [PubMed]
- Ozcelik, C.; Erdmann, B.; Pilz, B.; Wettschureck, N.; Britsch, S.; Hübner, N.; Chien, K.R.; Birchmeier, C.; Garratt, A.N. Conditional Mutation of the ErbB2 (HER2) Receptor in Cardiomyocytes Leads to Dilated Cardiomyopathy. Proc. Natl. Acad. Sci. USA 2002, 99, 8880–8885. [Google Scholar] [CrossRef] [PubMed]
- Szmit, S.; Jank, M.; Maciejewski, H.; Balsam, P.; Łój, M.; Filipiak, K.J.; Motyl, T.; Opolski, G. Relationship between Clinical Data and Gene Expression in the HER2/ErbB2-Dependent Signaling Pathway in Patients with Acute Heart Failure. J. Appl. Genet. 2013, 54, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Crone, S.A.; Zhao, Y.-Y.; Fan, L.; Gu, Y.; Minamisawa, S.; Liu, Y.; Peterson, K.L.; Chen, J.; Kahn, R.; Condorelli, G.; et al. ErbB2 Is Essential in the Prevention of Dilated Cardiomyopathy. Nat. Med. 2002, 8, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Negro, A.; Brar, B.K.; Lee, K.-F. Essential Roles of Her2/erbB2 in Cardiac Development and Function. Recent Prog. Horm. Res. 2004, 59, 1–12. [Google Scholar] [CrossRef]
- Gacita, A.M.; Dellefave-Castillo, L.; Page, P.G.T.; Barefield, D.Y.; Wasserstrom, J.A.; Puckelwartz, M.J.; Nobrega, M.A.; McNally, E.M. Altered Enhancer and Promoter Usage Leads to Differential Gene Expression in the Normal and Failed Human Heart. Circ. Heart Fail. 2020, 13, e006926. [Google Scholar] [CrossRef] [PubMed]
- Cen, M.; Hu, P.; Cai, Z.; Fang, T.; Zhang, J.; Lu, M. TIEG1 Deficiency Confers Enhanced Myocardial Protection in the Infarcted Heart by Mediating the Pten/Akt Signalling Pathway. Int. J. Mol. Med. 2017, 39, 569–578. [Google Scholar] [CrossRef][Green Version]
- Li, L.; Li, H.; Tien, C.-L.; Jain, M.K.; Zhang, L. Kruppel-Like Factor 15 Regulates the Circadian Susceptibility to Ischemia Reperfusion Injury in the Heart. Circulation 2020, 141, 1427–1429. [Google Scholar] [CrossRef] [PubMed]
- Simkhovich, B.Z.; Marjoram, P.; Poizat, C.; Kedes, L.; Kloner, R.A. Brief Episode of Ischemia Activates Protective Genetic Program in Rat Heart: A Gene Chip Study. Cardiovasc. Res. 2003, 59, 450–459. [Google Scholar] [CrossRef][Green Version]
- Chen, K.; Yu, G.; Gumireddy, K.; Li, A.; Yao, W.; Gao, L.; Chen, S.; Hao, J.; Wang, J.; Huang, Q.; et al. ZBRK1, a Novel Tumor Suppressor, Activates VHL Gene Transcription through Formation of a Complex with VHL and p300 in Renal Cancer. Oncotarget 2015, 6, 6959–6976. [Google Scholar] [CrossRef][Green Version]
- Wu, J.; Eni, A.; Roussuri, E.; Ma, B. Correlation between ZBRK1/ZNF350 Gene Polymorphism and Breast Cancer. BMC Med. Genomics 2021, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Rutter, J.L.; Smith, A.M.; Dávila, M.R.; Sigurdson, A.J.; Giusti, R.M.; Pineda, M.A.; Doody, M.M.; Tucker, M.A.; Greene, M.H.; Zhang, J.; et al. Mutational Analysis of the BRCA1-Interacting Genes ZNF350/ZBRK1 and BRIP1/BACH1 among BRCA1 and BRCA2-Negative Probands from Breast-Ovarian Cancer Families and among Early-Onset Breast Cancer Cases and Reference Individuals. Hum. Mutat. 2003, 22, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kuwano, Y.; Nishikawa, T.; Rokutan, K.; Nishida, K. Promoter Methylation Accelerates Colon Cancer Cell Migration. Oncotarget 2018, 9, 36750–36769. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cao, Y.-Q.; Yuan, L.; Zhao, Q.; Yuan, J.-L.; Miao, C.; Chang, Y.-F.; Wen, X.-T.; Wu, R.; Huang, X.-B.; Wen, Y.-P.; et al. Hsp40 Protein DNAJB6 Interacts with Viral NS3 and Inhibits the Replication of the Japanese Encephalitis Virus. Int. J. Mol. Sci. 2019, 20, 5719. [Google Scholar] [CrossRef]
- Fazio, G.; Vernuccio, F.; Grassedonio, E.; Grutta, G.; Lo Re, G.; Midiri, M. Ischemic and Non-Ischemic Dilated Cardiomyopathy. Open Med. 2014, 9, 15–20. [Google Scholar] [CrossRef]
- Freeman, K.; Colon-Rivera, C.; Charlotte Olsson, M.; Moore, R.L.; Weinberger, H.D.; Grupp, I.L.; Vikstrom, K.L.; Iaccarino, G.; Koch, W.J.; Leinwand, L.A. Progression from Hypertrophic to Dilated Cardiomyopathy in Mice That Express a Mutant Myosin Transgene. Am. J. Physiol.-Heart Circ. Physiol. 2001, 280, H151–H159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lui, K.O.; Zhou, B. Endocardial Cell Plasticity in Cardiac Development, Diseases and Regeneration. Circ. Res. 2018, 122, 774–789. [Google Scholar] [CrossRef] [PubMed]
- von Gise, A.; Pu, W.T. Endocardial and Epicardial Epithelial to Mesenchymal Transitions in Heart Development and Disease. Circ. Res. 2012, 110, 1628–1645. [Google Scholar] [CrossRef]
- Luxán, G.; D’Amato, G.; MacGrogan, D.; de la Pompa, J.L. Endocardial Notch Signaling in Cardiac Development and Disease. Circ. Res. 2016, 118, e1–e18. [Google Scholar] [CrossRef]
- Zhang, H.; von Gise, A.; Liu, Q.; Hu, T.; Tian, X.; He, L.; Pu, W.; Huang, X.; He, L.; Cai, C.-L.; et al. Yap1 Is Required for Endothelial to Mesenchymal Transition of the Atrioventricular Cushion. J. Biol. Chem. 2014, 289, 18681–18692. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, J.; Lange, A.W.; Yutzey, K.E. Hearts and Bones: Shared Regulatory Mechanisms in Heart Valve, Cartilage, Tendon, and Bone Development. Dev. Biol. 2006, 294, 292–302. [Google Scholar] [CrossRef]
- Previs, M.J.; O’Leary, T.S.; Morley, M.P.; Palmer, B.M.; LeWinter, M.; Yob, J.M.; Pagani, F.D.; Petucci, C.; Kim, M.-S.; Margulies, K.B.; et al. Defects in the Proteome and Metabolome in Human Hypertrophic Cardiomyopathy. Circ. Heart Fail. 2022, 15, e009521. [Google Scholar] [CrossRef] [PubMed]
- Ranjbarvaziri, S.; Kooiker, K.B.; Ellenberger, M.; Fajardo, G.; Zhao, M.; Vander Roest, A.S.; Woldeyes, R.A.; Koyano, T.T.; Fong, R.; Ma, N.; et al. Altered Cardiac Energetics and Mitochondrial Dysfunction in Hypertrophic Cardiomyopathy. Circulation 2021, 144, 1714–1731. [Google Scholar] [CrossRef]
- Ramos-Kuri, M.; Meka, S.H.; Salamanca-Buentello, F.; Hajjar, R.J.; Lipskaia, L.; Chemaly, E.R. Molecules Linked to Ras Signaling as Therapeutic Targets in Cardiac Pathologies. Biol. Res. 2021, 54, 23. [Google Scholar] [CrossRef] [PubMed]
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The miRNA-212/132 Family Regulates Both Cardiac Hypertrophy and Cardiomyocyte Autophagy. Nat. Commun. 2012, 3, 1078. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-D.; Song, X.-W.; Li, Q.; Wang, G.-K.; Jing, Q.; Qin, Y.-W. Attenuation of microRNA-22 Derepressed PTEN to Effectively Protect Rat Cardiomyocytes from Hypertrophy. J. Cell. Physiol. 2012, 227, 1391–1398. [Google Scholar] [CrossRef]
- Kavazis, A.N.; Smuder, A.J.; Powers, S.K. Effects of Short-Term Endurance Exercise Training on Acute Doxorubicin-Induced FoxO Transcription in Cardiac and Skeletal Muscle. J. Appl. Physiol. 2014, 117, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Ching, J.K.; Elizabeth, S.V.; Ju, J.-S.; Lusk, C.; Pittman, S.K.; Weihl, C.C. mTOR Dysfunction Contributes to Vacuolar Pathology and Weakness in Valosin-Containing Protein Associated Inclusion Body Myopathy. Hum. Mol. Genet. 2013, 22, 1167–1179. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, H.; Zhang, L. Identification of the Tumor-suppressive Function of Circular RNA FOXO3 in Non-small Cell Lung Cancer through Sponging miR-155. Mol. Med. Rep. 2018, 17, 7692–7700. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; He, P.; Wang, Y. MicroRNA-223-3p Regulates Cell Chemo-Sensitivity by Targeting FOXO3 in Prostatic Cancer. Gene 2018, 658, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, J.; Lu, C. Circular RNA Foxo3 Enhances Progression of Ovarian Carcinoma Cells. Aging 2021, 13, 22432–22443. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Hartmann, D.; Braren, R.; Gupta, A.; Wang, B.; Wang, Y.; Mogler, C.; Cheng, Z.; Wirth, T.; Friess, H.; et al. Oncogenic Akt-FOXO3 Loop Favors Tumor-Promoting Modes and Enhances Oxidative Damage-Associated Hepatocellular Carcinogenesis. BMC Cancer 2019, 19, 887. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Cai, G.; Jiang, L.; Gao, L.; Yang, Z.; Zhang, W. Identification of Key Pathways and RNAs Associated with Skeletal Muscle Atrophy after Spinal Cord Injury. J. Musculoskelet. Neuronal Interact. 2021, 21, 550–559. [Google Scholar] [PubMed]
- Li, Z.; Song, Y.; Liu, L.; Hou, N.; An, X.; Zhan, D.; Li, Y.; Zhou, L.; Li, P.; Yu, L.; et al. miR-199a Impairs Autophagy and Induces Cardiac Hypertrophy through mTOR Activation. Cell Death Differ. 2017, 24, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Liu, J.; Zhang, Y.; Ding, L.; Ye, Q. MiR-3180 Inhibits Hepatocellular Carcinoma Growth and Metastasis by Targeting Lipid Synthesis and Uptake. Cancer Cell Int. 2023, 23, 66. [Google Scholar] [CrossRef]
- Lin, F.; Yang, Y.; Guo, Q.; Xie, M.; Sun, S.; Wang, X.; Li, D.; Zhang, G.; Li, M.; Wang, J.; et al. Analysis of the Molecular Mechanism of Acute Coronary Syndrome Based on circRNA-miRNA Network Regulation. Evid. Based Complement. Alternat. Med. 2020, 2020, 1584052. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, M.; Holzem, A.; Kron, A.; Nogova, L.; Ihle, M.A.; von Levetzow, C.; Fassunke, J.; Wömpner, C.; Bitter, E.; Koleczko, S.; et al. Co-Occurrence of Targetable Mutations in Non-Small Cell Lung Cancer (NSCLC) Patients Harboring MAP2K1 Mutations. Lung Cancer 2020, 144, 40–48. [Google Scholar] [CrossRef]
- Ye, T.; Zhang, J.-Y.; Liu, X.-Y.; Zhou, Y.-H.; Yuan, S.-Y.; Yang, M.-M.; Xie, W.-Z.; Gao, C.; Chen, Y.-X.; Huang, M.-L.; et al. The Predictive Value of Mutations on Efficiency of Immunotherapy in Melanoma. Front. Immunol. 2021, 12, 785526. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Dai, X.; Bianchi, A.; De Castro Silva, I.; Mehra, S.; Garrido, V.T.; Lamichhane, P.; Singh, S.P.; Zhou, Z.; Dosch, A.R.; et al. Combined MEK and STAT3 Inhibition Uncovers Stromal Plasticity by Enriching for Cancer-Associated Fibroblasts With Mesenchymal Stem Cell-Like Features to Overcome Immunotherapy Resistance in Pancreatic Cancer. Gastroenterology 2022, 163, 1593–1612. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.; Fan, L.; Sahgal, N.; Qi, J.; Filipp, F.V. The Histone Demethylase KDM3A Regulates the Transcriptional Program of the Androgen Receptor in Prostate Cancer Cells. Oncotarget 2017, 8, 30328–30343. [Google Scholar] [CrossRef]
- Feng, B.; Li, X.; Zhang, Q.; Wang, Y.; Gu, S.; Yao, R.-E.; Li, Z.; Gao, S.; Chang, G.; Li, Q.; et al. Molecular and Phenotypic Spectrum of Cardio-Facio-Cutaneous Syndrome in Chinese Patients. Orphanet J. Rare Dis. 2023, 18, 284. [Google Scholar] [CrossRef] [PubMed]
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular Basis of Physiological Heart Growth: Fundamental Concepts and New Players. Nat. Rev. Mol. Cell Biol. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Heineke, J.; Molkentin, J.D. Regulation of Cardiac Hypertrophy by Intracellular Signalling Pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, C.J.; McDowell, S.A.; Clerk, A. Kinases as Therapeutic Targets for Heart Failure. Nat. Rev. Drug Discov. 2003, 2, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Fuller, S.J.; Osborne, S.A.; Leonard, S.J.; Hardyman, M.A.; Vaniotis, G.; Allen, B.G.; Sugden, P.H.; Clerk, A. Cardiac Protein Kinases: The Cardiomyocyte Kinome and Differential Kinase Expression in Human Failing Hearts. Cardiovasc. Res. 2015, 108, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Xu, C.-E. Integrated Bioinformatics and Machine Learning Algorithms Reveal the Critical Cellular Senescence-Associated Genes and Immune Infiltration in Heart Failure due to Ischemic Cardiomyopathy. Front. Immunol. 2023, 14, 1150304. [Google Scholar] [CrossRef]
- Xu, J.; Keeton, A.B.; Franklin, J.L.; Li, X.; Venable, D.Y.; Frank, S.J.; Messina, J.L. Insulin Enhances Growth Hormone Induction of the MEK/ERK Signaling Pathway. J. Biol. Chem. 2006, 281, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Frangogiannis, N.G. Diabetes-Associated Cardiac Fibrosis: Cellular Effectors, Molecular Mechanisms and Therapeutic Opportunities. J. Mol. Cell. Cardiol. 2016, 90, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Gao, C.; Qian, D.; Cao, D.; Han, L.; Yang, L. Regulatory Mechanism of Fibrosis-Related Genes in Patients with Heart Failure. Front. Genet. 2022, 13, 1032572. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-P.; Yang, R.-H.; Shang, J.; Gao, T.; Wang, R.; Peng, X.-D.; Miao, X.; Pan, L.; Yuan, W.-J.; Lin, L.; et al. FOXC1 up-Regulates the Expression of Toll-like Receptors in Myocardial Ischaemia. J. Cell. Mol. Med. 2019, 23, 7566–7580. [Google Scholar] [CrossRef]
- Katare, P.B.; Nizami, H.L.; Paramesha, B.; Dinda, A.K.; Banerjee, S.K. Activation of Toll like Receptor 4 (TLR4) Promotes Cardiomyocyte Apoptosis through SIRT2 Dependent p53 Deacetylation. Sci. Rep. 2020, 10, 19232. [Google Scholar] [CrossRef]
- Meng, X.; Nie, Y.; Wang, K.; Fan, C.; Zhao, J.; Yuan, Y. Identification of Atrial Fibrillation-Associated Genes and Using Genome-Wide Association and Transcriptome Expression Profile Data on Left-Right Atrial Appendages. Front. Genet. 2021, 12, 696591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, G.; Zhong, N.; Shan, J.; Li, X.; Wu, Y.; Xu, Y.; Yuan, Y. Possible Key microRNAs and Corresponding Molecular Mechanisms for Atrial Fibrillation. Anatol. J. Cardiol. 2020, 23, 324–333. [Google Scholar] [CrossRef]
- Li, F.; Yang, Y.; Xue, C.; Tan, M.; Xu, L.; Gao, J.; Xu, L.; Zong, J.; Qian, W. Zinc Finger Protein ZBTB20 Protects against Cardiac Remodelling Post-Myocardial Infarction via ROS-TNFα/ASK1/JNK Pathway Regulation. J. Cell. Mol. Med. 2020, 24, 13383–13396. [Google Scholar] [CrossRef]
- Wei, L.; He, F.; Zhang, W.; Chen, W.; Yu, B. Analysis of Master Transcription Factors Related to Parkinson’s Disease through the Gene Transcription Regulatory Network. Arch. Med. Sci. 2021, 17, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Odumpatta, R.; Arumugam, M. Integrative Analysis of Gene Expression and Regulatory Network Interaction Data Reveals the Protein Kinase C Family of Serine/Threonine Receptors as a Significant Druggable Target for Parkinson’s Disease. J. Mol. Neurosci. 2021, 71, 466–480. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.; Shi, G. The circRNA-miRNA-mRNA Regulatory Network in Systemic Lupus Erythematosus. Clin. Rheumatol. 2021, 40, 331–339. [Google Scholar] [CrossRef]
- Christopoulos, P.F.; Gjølberg, T.T.; Krüger, S.; Haraldsen, G.; Andersen, J.T.; Sundlisæter, E. Targeting the Notch Signaling Pathway in Chronic Inflammatory Diseases. Front. Immunol. 2021, 12, 668207. [Google Scholar] [CrossRef]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9, a022236. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Ferreira, A.; Reis, C.A.; Sousa, M.J.; Oliveira, M.J.; Preto, A. KRAS as a Modulator of the Inflammatory Tumor Microenvironment: Therapeutic Implications. Cells 2022, 11, 398. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Zhao, H. Reconstructing Transcriptional Regulatory Networks through Genomics Data. Stat. Methods Med. Res. 2009, 18, 595–617. [Google Scholar]
- Subramaniam, M.; Hawse, J.R.; Rajamannan, N.M.; Ingle, J.N.; Spelsberg, T.C. Functional Role of KLF10 in Multiple Disease Processes. Biofactors 2010, 36, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Rajamannan, N.M.; Subramaniam, M.; Abraham, T.P.; Vasile, V.C.; Ackerman, M.J.; Monroe, D.G.; Chew, T.-L.; Spelsberg, T.C. TGFbeta Inducible Early Gene-1 (TIEG1) and Cardiac Hypertrophy: Discovery and Characterization of a Novel Signaling Pathway. J. Cell. Biochem. 2007, 100, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.; Krakowiak, P.A.; Watkins, W.S.; Root, S.; Carey, J.C.; Jorde, L.B. A Gene for Ulnar-Mammary Syndrome Maps to 12q23-q24.1. Hum. Mol. Genet. 1995, 4, 1973–1977. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.; Root, S.; Carey, J.C. Clinical Analysis of a Large Kindred with the Pallister Ulnar-Mammary Syndrome. Am. J. Med. Genet. 1996, 65, 325–331. [Google Scholar] [CrossRef]
- Wolf, C.M.; Berul, C.I. Inherited Conduction System Abnormalities--One Group of Diseases, Many Genes. J. Cardiovasc. Electrophysiol. 2006, 17, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, M.E.; Basson, C.T.; Woodrow Benson, D., Jr.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L. Genetic Basis for Congenital Heart Defects: Current Knowledge. Circulation 2007, 115, 3015–3038. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, P.; Sdek, P.; MacLellan, W.R. Cardiac Myocyte Cell Cycle Control in Development, Disease, and Regeneration. Physiol. Rev. 2007, 87, 521–544. [Google Scholar] [CrossRef]
- Liu, R.; Lee, J.; Kim, B.S.; Wang, Q.; Buxton, S.K.; Balasubramanyam, N.; Kim, J.J.; Dong, J.; Zhang, A.; Li, S.; et al. Tead1 Is Required for Maintaining Adult Cardiomyocyte Function, and Its Loss Results in Lethal Dilated Cardiomyopathy. JCI Insight 2017, 2, 93343. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinformatics 2013, 14, 7. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Haynes, W.A.; Vallania, F.; Liu, C.; Bongen, E.; Tomczak, A.; Andres-Terrè, M.; Lofgren, S.; Tam, A.; Deisseroth, C.A.; Li, M.D.; et al. Empowering Multi-Cohort Gene Expression Analysis to Increase Reproducibility. Pac. Symp. Biocomput. 2017, 22, 144–153. [Google Scholar]
- Kim, S. Ppcor: An R Package for a Fast Calculation to Semi-Partial Correlation Coefficients. Commun. Stat. Appl. Methods 2015, 22, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Bonett, D.G.; Wright, T.A. Sample Size Requirements for Estimating Pearson, Kendall and Spearman Correlations. Psychometrika 2000, 65, 23–28. [Google Scholar] [CrossRef]
- Willmer, T.; Cooper, A.; Peres, J.; Omar, R.; Prince, S. The T-Box Transcription Factor 3 in Development and Cancer. Biosci. Trends 2017, 11, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Giraud, J.; Molina-Castro, S.; Seeneevassen, L.; Sifré, E.; Izotte, J.; Tiffon, C.; Staedel, C.; Boeuf, H.; Fernandez, S.; Barthelemy, P.; et al. Verteporfin Targeting YAP1/TAZ-TEAD Transcriptional Activity Inhibits the Tumorigenic Properties of Gastric Cancer Stem Cells. Int. J. Cancer 2020, 146, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Young, N.; Punkosdy, G.; Cavanaugh, J.; Bantle, C.; Constan, A.; Li, B.; Conley, J.; Sanchez-Martin, M.; Xu, L.; McGovern, K.; et al. Abstract 1646: IK-930, a Paralog-Selective TEAD Inhibitor for Treating YAP/TAZ-TEAD Dependent Cancers. Cancer Res. 2023, 83, 1646. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO | Disease | # of Diseased Samples | # of Healthy Samples |
|---|---|---|---|
| GSE55296 [20] | Dilated Cardiomyopathy | 13 | 10 |
| GSE116250 [21] | Dilated Cardiomyopathy | 37 | 14 |
| GSE133054 [22] | Hypertrophic Cardiomyopathy | 8 | 8 |
| GSE135055 [23] | Heart Failure | 21 | 9 |
| GSE133054 [22] | Heart Failure | 7 | 8 |
| GSE48166 | Ischemic Cardiomyopathy | 15 | 15 |
| GSE55296 [20] | Ischemic Cardiomyopathy | 13 | 10 |
| GSE116250 [21] | Ischemic Cardiomyopathy | 13 | 14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pepe, G.; Appierdo, R.; Ausiello, G.; Helmer-Citterich, M.; Gherardini, P.F. A Meta-Analysis Approach to Gene Regulatory Network Inference Identifies Key Regulators of Cardiovascular Diseases. Int. J. Mol. Sci. 2024, 25, 4224. https://doi.org/10.3390/ijms25084224
Pepe G, Appierdo R, Ausiello G, Helmer-Citterich M, Gherardini PF. A Meta-Analysis Approach to Gene Regulatory Network Inference Identifies Key Regulators of Cardiovascular Diseases. International Journal of Molecular Sciences. 2024; 25(8):4224. https://doi.org/10.3390/ijms25084224
Chicago/Turabian StylePepe, Gerardo, Romina Appierdo, Gabriele Ausiello, Manuela Helmer-Citterich, and Pier Federico Gherardini. 2024. "A Meta-Analysis Approach to Gene Regulatory Network Inference Identifies Key Regulators of Cardiovascular Diseases" International Journal of Molecular Sciences 25, no. 8: 4224. https://doi.org/10.3390/ijms25084224
APA StylePepe, G., Appierdo, R., Ausiello, G., Helmer-Citterich, M., & Gherardini, P. F. (2024). A Meta-Analysis Approach to Gene Regulatory Network Inference Identifies Key Regulators of Cardiovascular Diseases. International Journal of Molecular Sciences, 25(8), 4224. https://doi.org/10.3390/ijms25084224