
Comparative Transcriptomics of Fat Bodies between Symbiotic and Quasi-Aposymbiotic Adult Females of Blattella germanica with Emphasis on the Metabolic Integration with Its Endosymbiont Blattabacterium and Its Immune System

 , , and
, , and
Abstract
1. Introduction
2. Results
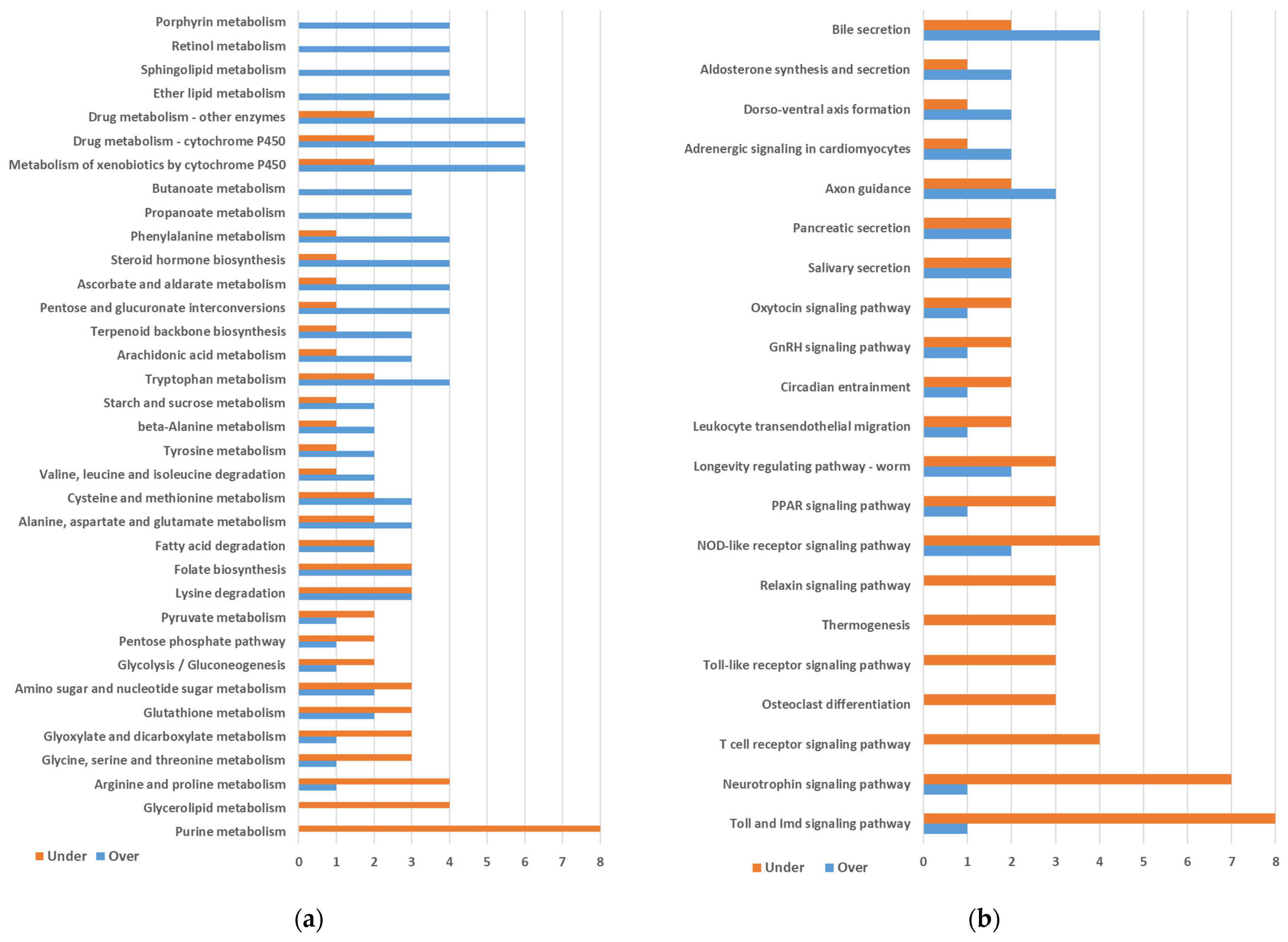
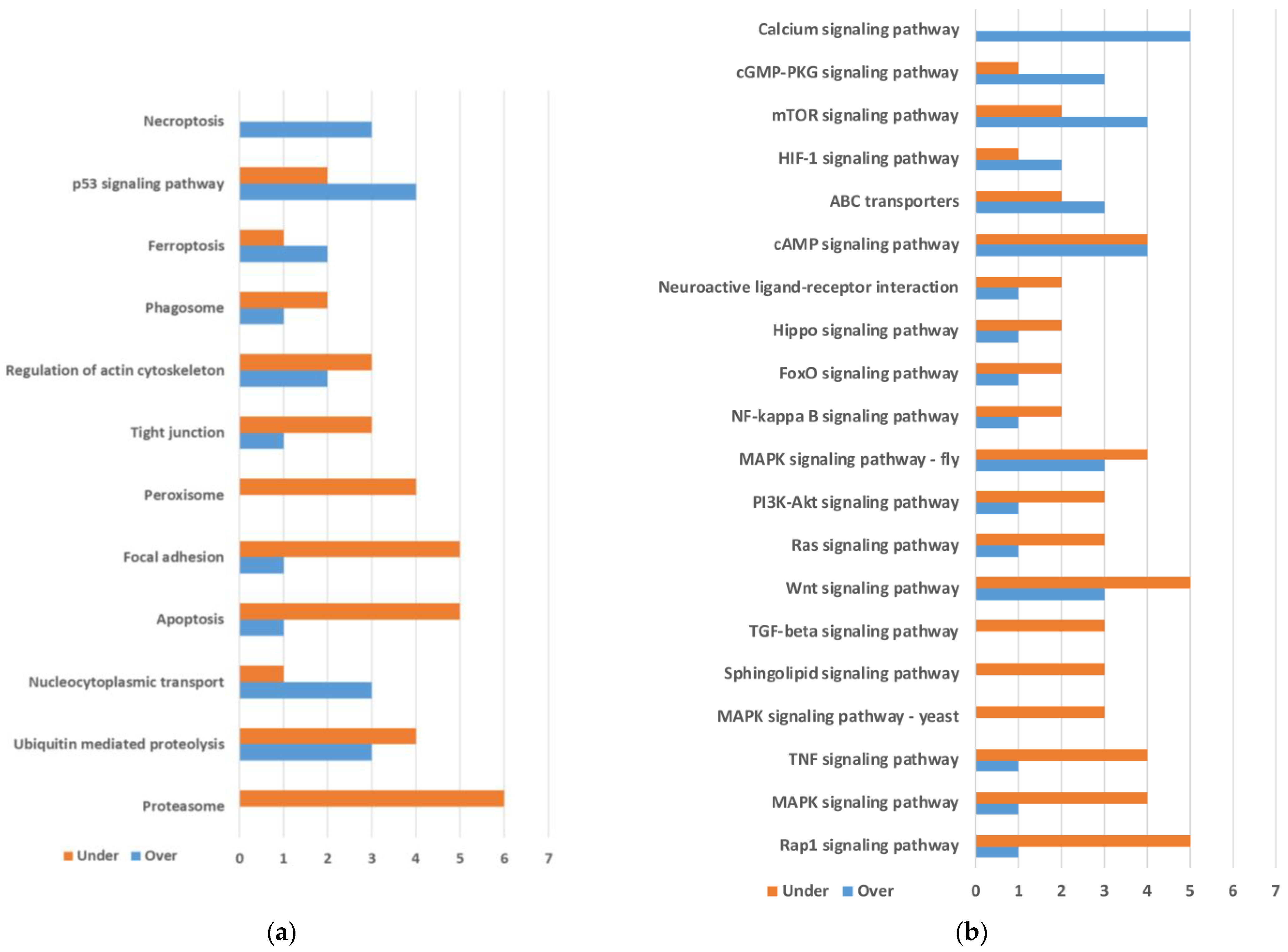
2.1. Differential Gene Expression between Quasi-Aposymbiont and Control Fat Bodies
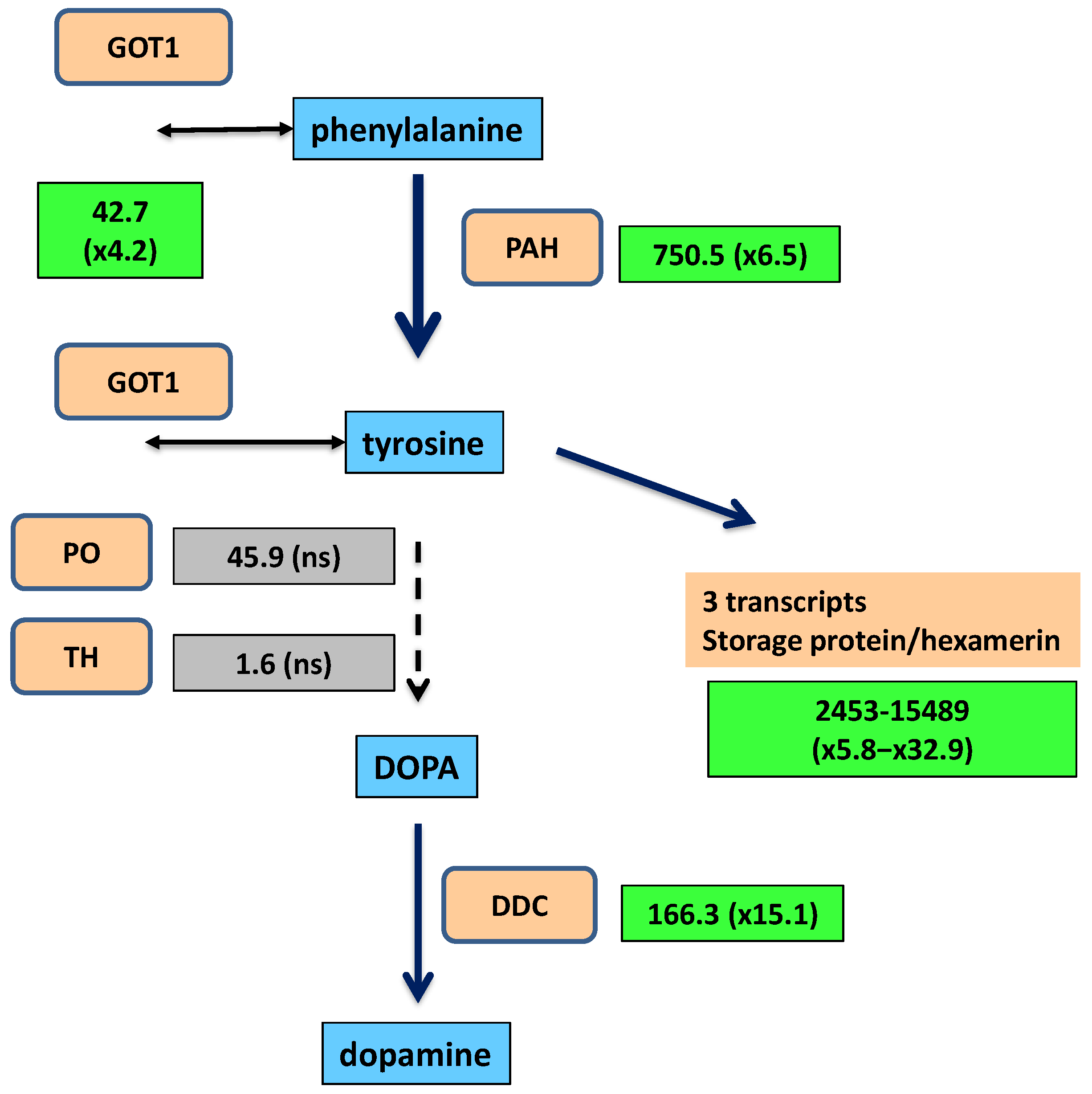
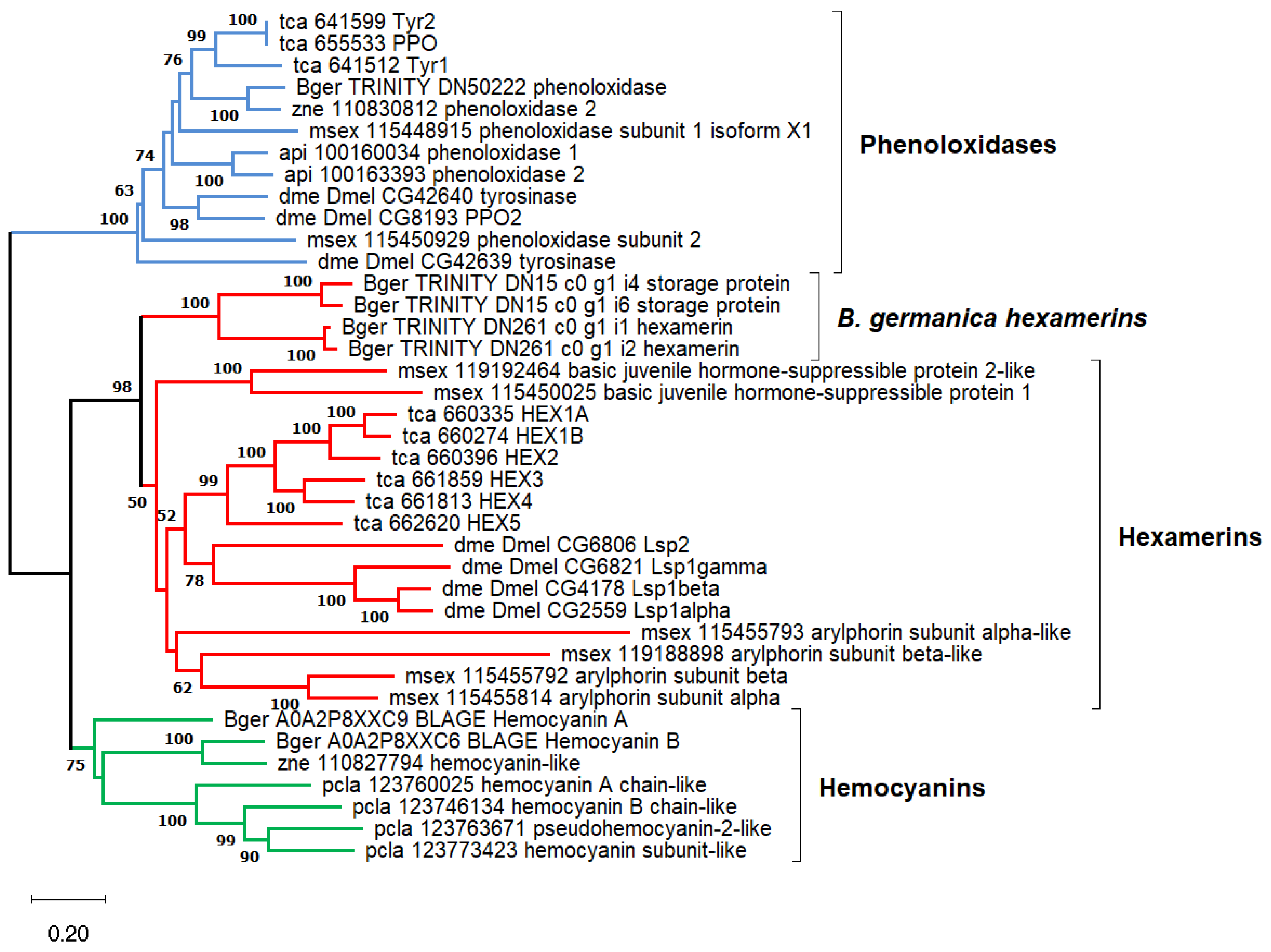
2.2. Synthesis of Tyrosine and Catechol Derivatives for Cuticle Sclerotization
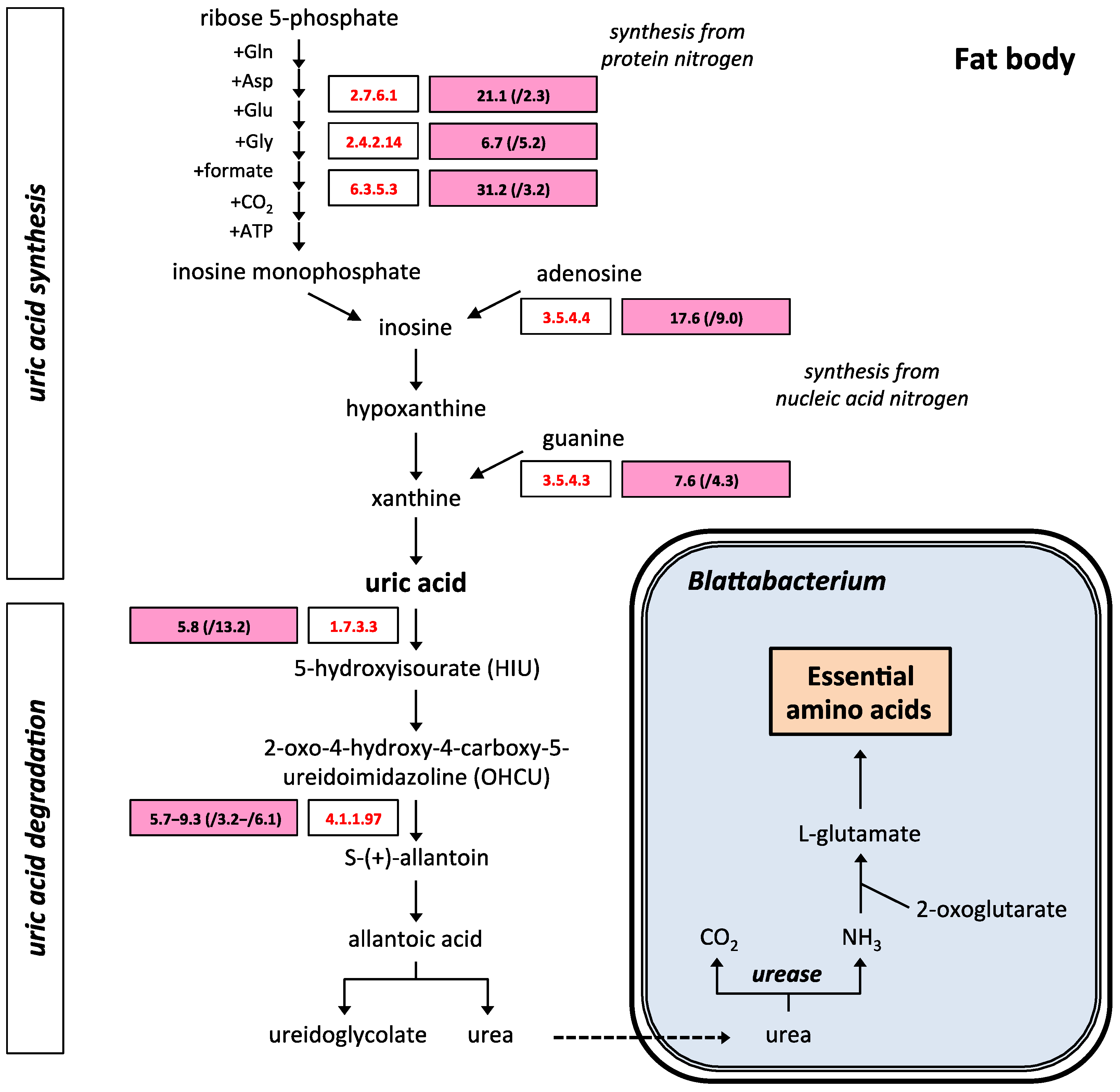
2.3. Uric Acid and Purine Metabolism
2.4. Metabolite Transporters
2.5. Immune System and Antimicrobial Peptides
3. Discussion
4. Materials and Methods
4.1. Insect Rearing
4.2. Insect Dissection and Collection of Samples
4.3. RNA/DNA Extraction and Sequencing
4.4. Read Filtering, Transcriptome Assembly and Expression Analysis
4.5. Functional Annotation
4.6. Signal Prediction and Subcellular Localization
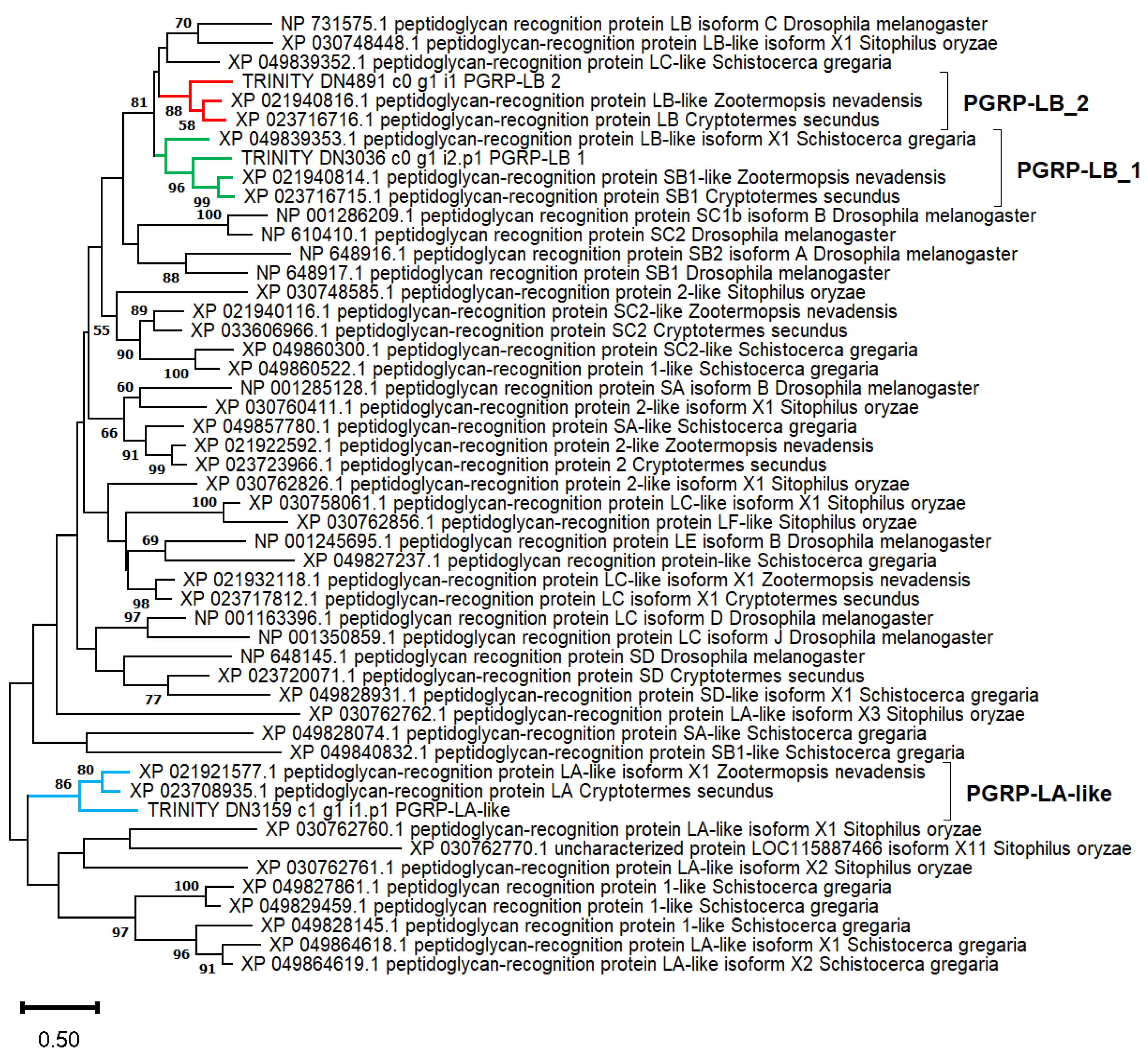
4.7. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moya, A.; Peretó, J.; Gil, R.; Latorre, A. Learning how to live together: Genomic insights into prokaryote-animal symbioses. Nat. Rev. Genet. 2008, 9, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Perreau, J.; Moran, N.A. Genetic innovations in animal–microbe symbioses. Nat. Rev. Genet. 2022, 23, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Sazama, E.J.; Ouellette, S.P.; Wesner, J.S. Bacterial endosymbionts are common among, but not necessarily within, insect species. Environ. Entomol. 2019, 48, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P. Biology bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu. Rev. Microbiol. 2005, 59, 155–189. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.W.; Poliakov, A.; Haribal, M.; Jander, G.; van Wijk, K.J.; Douglas, A.E. Matching the supply of bacterial nutrients to the nutritional demand of the animal host. Proc. R. Soc. B 2014, 281, 20141163. [Google Scholar] [CrossRef] [PubMed]
- Latorre, A.; Manzano-Marín, A. Dissecting genome reduction and trait loss in insect endosymbionts. Ann. N. Y. Acad. Sci. 2017, 1389, 52–75. [Google Scholar] [CrossRef] [PubMed]
- Itoh, H.; Tago, K.; Hayatsu, M.; Kikuchi, Y. Detoxifying symbiosis: Microbe-mediated detoxification of phytotoxins and pesticides in insects. Nat. Prod. Rep. 2018, 35, 434–454. [Google Scholar] [CrossRef] [PubMed]
- Brune, A. Symbiotic digestion of lignocellulose in termite guts. Nat. Rev. Microbiol. 2014, 12, 168–180. [Google Scholar] [CrossRef] [PubMed]
- López-Sánchez, M.J.; Neef, A.; Peretó, J.; Patiño-Navarrete, R.; Pignatelli, M.; Latorre, A.; Moya, A. Evolutionary convergence and nitrogen metabolism in Blattabacterium strain Bge, primary endosymbiont of the cockroach Blattella germanica. PLoS Genet. 2009, 5, e1000721. [Google Scholar] [CrossRef]
- Anbutsu, H.; Moriyama, M.; Nikoh, N.; Hosokawa, T.; Futahashi, R.; Tanahashi, M.; Meng, X.Y.; Kuriwada, T.; Mori, N.; Oshima, K.; et al. Small genome symbiont underlies cuticle hardness in beetles. Proc. Natl. Acad. Sci. USA 2017, 114, E8382–E8391. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, J.S.T.; Bauer, E.; Okude, G.; Fukatsu, T.; Kaltenpoth, M.; Engl, T. Cuticle supplementation and nitrogen recycling by a dual bacterial symbiosis in a family of xylophagous beetles. ISME J. 2023, 17, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Engel, P. Mechanisms underlying gut microbiota-host interactions in insects. J. Exp. Biol. 2021, 224, jeb207696. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.E. Multiorganismal insects: Diversity and function of resident microorganisms. Annu. Rev. Entomol. 2015, 60, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Bandi, C.; Damiani, G.; Magrassi, L.; Grigolo, A.; Fani, R.; Sacchi, L. Flavobacteria as intracellular symbionts in cockroaches. Proc. R. Soc. Lond. B 1994, 257, 43–48. [Google Scholar] [CrossRef]
- Latorre, A.; Domínguez-Santos, R.; García-Ferris, C.; Gil, R. Of cockroaches and symbionts: Recent advances in the characterization of the relationship between Blattella germanica and its dual symbiotic system. Life 2022, 12, 290. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, L.; Grigolo, A.; Mazzini, M.; Bigliardi, E.; Baccetti, B.; Laudani, U. Symbionts in the oocytes of Blattella germanica (L.) (Dictyoptera: Blattellidae): Their mode of transmission. Int. J. Insect Morphol. Embryol. 1988, 17, 437–446. [Google Scholar] [CrossRef]
- Sacchi, L.; Grigolo, A.; Laudani, U.; Ricevuti, G.; Dealessi, F. Behavior of symbionts during oogenesis and early stages of development in the German cockroach, Blattella germanica (Blattodea). J. Invertebr. Pathol. 1985, 46, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, P.; Pérez-Cobas, A.E.; van de Pol, C.; Baixeras, J.; Moya, A.; Latorre, A. Succession of the gut microbiota in the cockroach Blattella germanica. Int. Microbiol. 2014, 17, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, D.A.; Wipfler, B.; Béthoux, O.; Donath, A.; Fujita, M.; Kohli, M.K.; Legendre, F.; Liu, S.; Machida, R.; Misof, B.; et al. An integrative phylogenomic approach illuminates the evolutionary history of cockroaches and termites (Blattodea). Proc. R. Soc. B 2019, 286, 20182076. [Google Scholar] [CrossRef]
- Bandi, C.; Sironi, M.; Damiani, G.; Magrassi, L.; Nalepa, C.A.; Laudani, U.; Sacchi, L. The establishment of intracellular symbiosis in an ancestor of cockroaches and termites. Proc. R. Soc. Lond. B 1995, 259, 293–299. [Google Scholar] [CrossRef]
- Arab, D.A.; Bourguignon, T.; Wang, Z.; Ho, S.Y.W.; Lo, N. Evolutionary rates are correlated between cockroach symbionts and mitochondrial genomes. Biol. Lett. 2020, 16, 20190702. [Google Scholar] [CrossRef] [PubMed]
- Lo, N.; Beninati, T.; Stone, F.; Walker, J.; Sacchi, L. Cockroaches that lack Blattabacterium endosymbionts: The phylogenetically divergent genus Nocticola. Biol. Lett. 2007, 3, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, G.; Elbourne, L.D.H.H.; Kinjo, Y.; Saitoh, S.; Sabree, Z.; Hojo, M.; Yamada, A.; Hayashi, Y.; Shigenobu, S.; Bandi, C.; et al. Maintenance of essential amino acid synthesis pathways in the Blattabacterium cuenoti symbiont of a wood-feeding cockroach. Biol. Lett. 2013, 9, 20121153. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, C.A. Origin of mutualism between termites and flagellated gut protists: Transition from horizontal to vertical transmission. Front. Ecol. Evol. 2020, 8, 14. [Google Scholar] [CrossRef]
- Patiño-Navarrete, R.; Moya, A.; Latorre, A.; Peretó, J. Comparative genomics of Blattabacterium cuenoti: The frozen legacy of an ancient endosymbiont genome. Genome Biol. Evol. 2013, 5, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Kinjo, Y.; Bourguignon, T.; Hongoh, Y.; Lo, N.; Tokuda, G.; Ohkuma, M. Coevolution of metabolic pathways in Blattodea and their Blattabacterium endosymbionts, and comparisons with other insect-bacteria symbioses. Microbiol. Spectr. 2022, 10, e0277922. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, L.; Nalepa, C.; Bigliardi, E.; Corona, S.; Grigolo, A.; Laudani, U.; Bandi, C. Ultrastructural studies of the fat body and bacterial endosymbionts of Cryptocercus punctulatus Scudder (Blattaria: Cryptocercidae). Symbiosis 1998, 25, 251–269. [Google Scholar]
- Park, M.S.; Park, P.; Takeda, M. Roles of fat body trophocytes, mycetocytes and urocytes in the American cockroach, Periplaneta americana under starvation conditions: An ultrastructural study. Arthropod Struct. Dev. 2013, 42, 287–295. [Google Scholar] [CrossRef]
- Patiño-Navarrete, R.; Piulachs, M.-D.; Belles, X.; Moya, A.; Latorre, A.; Peretó, J. The cockroach Blattella germanica obtains nitrogen from uric acid through a metabolic pathway shared with its bacterial endosymbiont. Biol. Lett. 2014, 10, 20140407. [Google Scholar] [CrossRef]
- Sabree, Z.L.; Kambhampati, S.; Moran, N.A. Nitrogen recycling and nutritional provisioning by Blattabacterium, the cockroach endosymbiont. Proc. Natl. Acad. Sci. USA 2009, 106, 19521–19526. [Google Scholar] [CrossRef]
- Arrese, E.L.; Soulages, J.L. Insect fat body: Energy, metabolism, and regulation. Annu. Rev. Entomol. 2010, 55, 207–225. [Google Scholar] [CrossRef] [PubMed]
- Roma, G.C.; Bueno, O.C.; Camargo-Mathias, M.I. Morpho-physiological analysis of the insect fat body: A review. Micron 2010, 41, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Skowronek, P.; Wójcik, Ł.; Strachecka, A. Fat body-multifunctional insect tissue. Insects 2021, 12, 547. [Google Scholar] [CrossRef] [PubMed]
- Arakane, Y.; Noh, M.Y.; Asano, T.; Kramer, K.J. Tyrosine metabolism for insect cuticle pigmentation and sclerotization. In Extracellular Composite Matrices in Arthropods; Cohen, E., Moussian, B., Eds.; Springer: Cham, Switzerland, 2016; pp. 165–220. [Google Scholar] [CrossRef]
- Burmester, T. Evolution and function of the insect hexamerins. Eur. J. Entomol. 1999, 96, 213–225. [Google Scholar]
- Burmester, T. Origin and evolution of arthropod hemocyanins and related proteins. J. Comp. Physiol. B 2002, 172, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.E. Molecular dissection of nutrient exchange at the insect-microbial interface. Curr. Opin. Insect Sci. 2014, 4, 23–28. [Google Scholar] [CrossRef] [PubMed]
- González-Domenech, C.; Belda, E.; Patiño-Navarrete, R.; Moya, A.; Peretó, J.; Latorre, A. Metabolic stasis in an ancient symbiosis: Genome-scale metabolic networks from two Blattabacterium cuenoti strains, primary endosymbionts of cockroaches. BMC Microbiol. 2012, 12, S5. [Google Scholar] [CrossRef] [PubMed]
- Ponce-de-León, M.; Montero, F.; Peretó, J. Solving gap metabolites and blocked reactions in genome-scale models: Application to the metabolic network of Blattabacterium cuenoti. BMC Syst. Biol. 2013, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Alleman, A.; Hertweck, K.L.; Kambhampati, S. Random genetic drift and selective pressures shaping the Blattabacterium genome. Sci. Rep. 2018, 8, 13427. [Google Scholar] [CrossRef]
- Kinjo, Y.; Lo, N.; Martín, P.V.; Tokuda, G.; Pigolotti, S.; Bourguignon, T. Enhanced mutation rate, relaxed selection, and the “Domino Effect” are associated with gene loss in Blattabacterium, a cockroach endosymbiont. Mol. Biol. Evol. 2021, 38, 3820–3831. [Google Scholar] [CrossRef]
- Shigenobu, S.; Watanabe, H.; Hattori, M.; Sakaki, Y.; Ishikawa, H. Genome sequence of the endocellular bacterial symbiont of Aphids buchnera sp. APS. Nature 2000, 407, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Charles, H.; Balmand, S.; Lamelas, A.; Cottret, L.; Pérez-Brocal, V.; Burdin, B.; Latorre, A.; Febvay, G.; Colella, S.; Calevro, F.; et al. A genomic reappraisal of symbiotic function in the aphid/Buchnera symbiosis: Reduced transporter sets and variable membrane organisations. PLoS ONE 2011, 6, e29096. [Google Scholar] [CrossRef] [PubMed]
- Mergaert, P.; Kikuchi, Y.; Shigenobu, S.; Nowack, E.C.M. Metabolic integration of bacterial endosymbionts through antimicrobial peptides. Trends Microbiol. 2017, 25, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Mergaert, P. Role of antimicrobial peptides in controlling symbiotic bacterial populations. Nat. Prod. Rep. 2018, 35, 336–356. [Google Scholar] [CrossRef] [PubMed]
- Login, F.H.; Balmand, S.; Vallier, A.; Vincent-Monégat, C.; Vigneron, A.; Weiss-Gayet, M.; Rochat, D.; Heddi, A. Antimicrobial peptides keep insect endosymbionts under control. Science 2011, 334, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Login, F.H.; Heddi, A. Insect immune system maintains long-term resident bacteria through a local response. J. Insect Physiol. 2013, 59, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Masson, F.; Zaidman-Rémy, A.; Heddi, A. Antimicrobial peptides and cell processes tracking endosymbiont dynamics. Philos. Trans. R. Soc. B 2016, 371, 20150298. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.J.; Muñoz-Benavent, M.; García-Ferris, C.; Latorre, A. Blattella germanica displays a large arsenal of antimicrobial peptide genes. Sci. Rep. 2020, 10, 21058. [Google Scholar] [CrossRef]
- Zuber, L.; Domínguez-Santos, R.; García-Ferris, C.; Silva, F.J. Identification of the gene repertoire of the IMD pathway and expression of antimicrobial peptide genes in several tissues and hemolymph of the cockroach Blattella germanica. Int. J. Mol. Sci. 2022, 23, 8444. [Google Scholar] [CrossRef]
- Maire, J.; Parisot, N.; Galvao Ferrarini, M.; Vallier, A.; Gillet, B.; Hughes, S.; Balmand, S.; Vincent-Monégat, C.; Zaidman-Rémy, A.; Heddi, A. Spatial and morphological reorganization of endosymbiosis during metamorphosis accommodates adult metabolic requirements in a weevil. Proc. Natl. Acad. Sci. USA 2020, 117, 19347–19358. [Google Scholar] [CrossRef]
- Wang, Q.; Ren, M.; Liu, X.; Xia, H.; Chen, K. Peptidoglycan recognition proteins in insect immunity. Mol. Immunol. 2019, 106, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Shigenobu, S.; Stern, D.L. Aphids evolved novel secreted proteins for symbiosis with bacterial endosymbiont. Proc. R. Soc. B 2013, 280, 20121952. [Google Scholar] [CrossRef] [PubMed]
- Uchi, N.; Fukudome, M.; Nozaki, N.; Suzuki, M.; Osuki, K.I.; Shigenobu, S.; Uchiumi, T. Antimicrobial activities of cysteine-rich peptides specific to bacteriocytes of the pea aphid Acyrthosiphon pisum. Microbes Environ. 2019, 34, 155. [Google Scholar] [CrossRef] [PubMed]
- Loth, K.; Parisot, N.; Paquet, F.; Terrasson, H.; Sivignon, C.; Rahioui, I.; Ribeiro Lopes, M.; Gaget, K.; Duport, G.; Delmas, A.F.; et al. Aphid BCR4 structure and activity uncover a new defensin peptide superfamily. Int. J. Mol. Sci. 2022, 23, 12480. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Santos, R.; Pérez-Cobas, A.E.; Cuti, P.; Pérez-Brocal, V.; García-Ferris, C.; Moya, A.; Latorre, A.; Gil, R. Interkingdom gut microbiome and resistome of the cockroach Blattella germanica. mSystems 2021, 6, e01213-20. [Google Scholar] [CrossRef] [PubMed]
- Rosas, T.; García-Ferris, C.; Domínguez-Santos, R.; Llop, P.; Latorre, A.; Moya, A. Rifampicin treatment of Blattella germanica evidences a fecal transmission route of their gut microbiota. FEMS Microbiol. Ecol. 2018, 94, fiy002. [Google Scholar] [CrossRef]
- Muñoz-Benavent, M.; Latorre, A.; Alemany-Cosme, E.; Marín-Miret, J.; Domínguez-Santos, R.; Silva, F.J.; Gil, R.; García-Ferris, C. Gut microbiota cannot compensate the impact of (quasi) aposymbiosis in Blattella germanica. Biology 2021, 10, 1013. [Google Scholar] [CrossRef]
- Cazzaniga, M.; Domínguez-Santos, R.; Marín-Miret, J.; Gil, R.; Latorre, A.; García-Ferris, C. Exploring gut microbial dynamics and symbiotic interaction in Blattella germanica using rifampicin. Biology 2023, 12, 955. [Google Scholar] [CrossRef]
- Valovage, W.D.; Brooks, M.A. Uric acid quantities in the fat body of normal and aposymbiotic German cockroaches, Blattella germanica. Ann. Entomol. Soc. Am. 1979, 72, 687–689. [Google Scholar] [CrossRef]
- Lehnert, E.M.; Mouchka, M.E.; Burriesci, M.S.; Gallo, N.D.; Schwarz, J.A.; Pringle, J.R. Extensive differences in gene expression between symbiotic and aposymbiotic cnidarians. G3 Genes Genomes Genet. 2014, 4, 277–295. [Google Scholar] [CrossRef]
- Bing, X.; Attardo, G.M.; Vigneron, A.; Aksoy, E.; Scolari, F.; Malacrida, A.; Weiss, B.L.; Aksoy, S. Unravelling the relationship between the tsetse fly and its obligate symbiont Wigglesworthia: Transcriptomic and metabolomic landscapes reveal highly integrated physiological networks. Proc. R. Soc. B 2017, 284, 20170360. [Google Scholar] [CrossRef]
- Hickin, M.L.; Kakumanu, M.L.; Schal, C. Effects of Wolbachia elimination and B-vitamin supplementation on bed bug development and reproduction. Sci. Rep. 2022, 12, 10270. [Google Scholar] [CrossRef]
- Sterkel, M.; Ons, S.; Oliveira, P.L. DOPA decarboxylase is essential for cuticle tanning in Rhodnius prolixus (Hemiptera: Reduviidae), affecting ecdysis, survival and reproduction. Insect Biochem. Mol. Biol. 2019, 108, 24–31. [Google Scholar] [CrossRef]
- Downer, R.G.H. Fat body and metabolism. In The American Cockroach; Bell, W.J., Adiyodi, K.G., Eds.; Chapman and Hall: London, UK, 1981; pp. 151–174. [Google Scholar] [CrossRef]
- Harrison, M.C.; Jongepier, E.; Robertson, H.M.; Arning, N.; Bitard-Feildel, T.; Chao, H.; Childers, C.P.; Dinh, H.; Doddapaneni, H.; Dugan, S.; et al. Hemimetabolous genomes reveal molecular basis of termite eusociality. Nat. Ecol. Evol. 2018, 2, 557–566. [Google Scholar] [CrossRef]
- Xia, X.; You, M.; Rao, X.J.; Yu, X.Q. Insect C-type lectins in innate immunity. Dev. Comp. Immunol. 2018, 83, 70–79. [Google Scholar] [CrossRef]
- Noda, T.; Okude, G.; Meng, X.-Y.; Koga, R.; Moriyama, M.; Fukatsu, T. Bacteriocytes and Blattabacterium endosymbionts of the German cockroach Blattella germanica, the forest cockroach Blattella nipponica, and other cockroach species. Zool. Sci. 2020, 37, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Everaert, C.; Luypaert, M.; Maag, J.L.V.; Cheng, Q.X.; Marcel, E.; Hellemans, J.; Mestdagh, P. Benchmarking of RNA-sequencing analysis workflows using whole-transcriptome RT-qPCR expression data. Sci. Rep. 2017, 7, 1559. [Google Scholar] [CrossRef] [PubMed]
- Coenye, T. Do results obtained with RNA-sequencing require independent verification? Biofilm 2021, 3, 100043c. [Google Scholar] [CrossRef]
- Page, T.M.; Lawley, J.W. The next generation is here: A review of transcriptomic approaches in marine ecology. Front. Mar. Sci. 2022, 9, 757921. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Cui, X. Design and validation issues in RNA-seq experiments. Brief. Bioinform. 2011, 12, 280–287. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Seyednasrollah, F.; Laiho, A.; Elo, L.L. Comparison of software packages for detecting differential expression in RNA-seq studies. Brief. Bioinform. 2015, 16, 59–70. [Google Scholar] [CrossRef]
- Ayayee, P.A.; Larsen, T.; Sabree, Z. Symbiotic essential amino acids provisioning in the American cockroach, Periplaneta americana (Linnaeus) under various dietary conditions. PeerJ 2016, 4, e2046. [Google Scholar] [CrossRef]
- Chen, P.; Li, L.; Wang, J.; Li, H.; Li, Y.; Lv, Y.; Lu, C. BmPAH catalyzes the initial melanin biosynthetic step in Bombyx mori. PLoS ONE 2013, 8, e71984. [Google Scholar] [CrossRef]
- Simonet, P.; Gaget, K.; Parisot, N.; Duport, G.; Rey, M.; Febvay, G.; Charles, H.; Callaerts, P.; Colella, S.; Calevro, F. Disruption of phenylalanine hydroxylase reduces adult lifespan and fecundity, and impairs embryonic development in parthenogenetic pea aphids. Sci. Rep. 2016, 6, 34321. [Google Scholar] [CrossRef]
- Guo, H.; Lona, G.J.; Liu, X.Z.; Ma, Y.F.; Zhang, M.Q.; Gong, L.L.; Dewer, Y.; Hull, J.J.; Wang, M.M.; Wang, Q.; et al. Functional characterization of tyrosine melanin genes in the white-backed planthopper and utilization of a spray-based nanoparticle-wrapped dsRNA technique for pest control. Int. J. Biol. Macromol. 2023, 230, 123123. [Google Scholar] [CrossRef]
- Martínez-Ramírez, A.C.; Ferré, J.; Silva, F.J. Catecholamines in Drosophila melanogaster: DOPA and dopamine accumulation during development. Insect Biochem. Mol. Biol. 1992, 22, 491–494. [Google Scholar] [CrossRef]
- Gaskell, E.A.; Smith, J.E.; Pinney, J.W.; Westhead, D.R.; McConkey, G.A. A unique dual activity amino acid hydroxylase in Toxoplasma gondii. PLoS ONE 2009, 4, e4801. [Google Scholar] [CrossRef]
- Ren, X.; Guo, R.; Akami, M.; Niu, C. Nitrogen acquisition strategies mediated by insect symbionts: A review of their mechanisms, methodologies, and case studies. Insects 2022, 13, 84. [Google Scholar] [CrossRef]
- Lee, I.R.; Yang, L.; Sebetso, G.; Allen, R.; Doan, T.H.; Blundell, R.; Lui, E.Y.; Morrow, C.A.; Fraser, J.A. Characterization of the complete uric acid degradation pathway in the fungal pathogen Cryptococcus neoformans. PLoS ONE 2013, 8, e64292. [Google Scholar] [CrossRef]
- Ramazzina, I.; Folli, C.; Secchi, A.; Berni, R.; Percudani, R. Completing the uric acid degradation pathway through phylogenetic comparison of whole genomes. Nat. Chem. Biol. 2006, 2, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Price, D.R.G.; Feng, H.; Baker, J.D.; Bavan, S.; Luetje, C.W.; Wilson, A.C.C. Aphid amino acid transporter regulates glutamine supply to intracellular bacterial symbionts. Proc. Natl. Acad. Sci. USA 2014, 111, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.P.; Anderson, C.M.H.; Thwaites, D.T.; Luetje, C.W.; Wilson, A.C.C. Co-option of a conserved host glutamine transporter facilitates aphid/Buchnera metabolic integration. Proc. Natl. Acad. Sci. USA 2023, 120, e2308448120. [Google Scholar] [CrossRef]
- Feng, H.; Edwards, N.; Anderson, C.M.H.; Althaus, M.; Duncan, R.P.; Hsu, Y.C.; Luetje, C.W.; Price, D.R.G.; Wilson, A.C.C.; Thwaites, D.T. Trading amino acids at the aphid–Buchnera symbiotic interface. Proc. Natl. Acad. Sci. USA 2019, 116, 16003–16011. [Google Scholar] [CrossRef] [PubMed]
- Husnik, F.; Hypsa, V.; Darby, A. Insect-symbiont gene expression in the midgut bacteriocytes of a blood-sucking parasite. Genome Biol. Evol. 2020, 12, 429–442. [Google Scholar] [CrossRef]
- Stączek, S.; Cytryńska, M.; Zdybicka-Barabas, A. Unraveling the role of antimicrobial peptides in insects. Int. J. Mol. Sci. 2023, 24, 5753. [Google Scholar] [CrossRef]
- Ratzka, C.; Gross, R.; Feldhaar, H. Gene expression analysis of the endosymbiont-bearing midgut tissue during ontogeny of the carpenter ant Camponotus floridanus. J. Insect Physiol. 2013, 59, 611–623. [Google Scholar] [CrossRef]
- Maire, J.; Vincent-Monégat, C.; Balmand, S.; Vallier, A.; Hervé, M.; Masson, F.; Parisot, N.; Vigneron, A.; Anselme, C.; Perrin, J.; et al. Weevil pgrp-lb prevents endosymbiont TCT dissemination and chronic host systemic immune activation. Proc. Natl. Acad. Sci. USA 2019, 116, 5623–5632. [Google Scholar] [CrossRef]
- Yao, Z.; Cai, Z.; Ma, Q.; Bai, S.; Wang, Y.; Zhang, P.; Guo, Q.; Gu, J.; Lemaitre, B.; Zhang, H. Compartmentalized PGRP expression along the dipteran Bactrocera dorsalis gut forms a zone of protection for symbiotic bacteria. Cell Rep. 2022, 41, 111523. [Google Scholar] [CrossRef] [PubMed]
- Park, K.E.; Jang, S.H.; Lee, J.S.; Lee, A.; Kikuchi, Y.; Seo, Y.S.; Lee, B.L. The roles of antimicrobial peptide, rip-thanatin, in the midgut of Riptortus pedestris. Dev. Comp. Immunol. 2018, 78, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Zaidman-Rémy, A.; Hervé, M.; Poidevin, M.; Pili-Floury, S.; Kim, M.-S.; Blanot, D.; Oh, B.-H.; Ueda, R.; Mengin-Lecreulx, D.; Lemaitre, B. The Drosophila amidase PGRP-LB modulates the immune response to bacterial infection. Immunity 2006, 24, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Mellroth, P.; Karlsson, J.; Steiner, H. A scavenger function for a Drosophila peptidoglycan recognition protein. J. Biol. Chem. 2003, 278, 7059–7064. [Google Scholar] [CrossRef] [PubMed]
- Kordaczuk, J.; Sułek, M.; Wojda, I. General overview on the role of Peptidoglycan Recognition Proteins in insect immunity. Acta Biochim. Pol. 2020, 67, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Orlans, J.; Vincent-Monegat, C.; Rahioui, I.; Sivignon, C.; Butryn, A.; Soulère, L.; Zaidman-Remy, A.; Orville, A.M.; Heddi, A.; Aller, P.; et al. PGRP-LB: An inside view into the mechanism of the amidase reaction. Int. J. Mol. Sci. 2021, 22, 4957. [Google Scholar] [CrossRef] [PubMed]
- Masson, F.; Moné, Y.; Vigneron, A.; Vallier, A.; Parisot, N.; Vincent-Monégat, C.; Balmand, S.; Carpentier, M.C.; Zaidman-Rémy, A.; Heddi, A. Weevil endosymbiont dynamics is associated with a clamping of immunity. BMC Genom. 2015, 16, 819. [Google Scholar] [CrossRef]
- Nakabachi, A.; Shigenobu, S.; Sakazume, N.; Shiraki, T.; Hayashizaki, Y.; Carninci, P.; Ishikawa, H.; Kudo, T.; Fukatsu, T. Transcriptome analysis of the aphid bacteriocyte, the symbiotic host cell that harbors an endocellular mutualistic bacterium, Buchnera. Proc. Natl. Acad. Sci. USA 2005, 102, 5477–5482. [Google Scholar] [CrossRef] [PubMed]
- Feldhaar, H.; Gross, R. Insects as hosts for mutualistic bacteria. Int. J. Med. Microbiol. 2009, 299, 1–8. [Google Scholar] [CrossRef]
- Futahashi, R.; Tanaka, K.; Tanahashi, M.; Nikoh, N.; Kikuchi, Y.; Lee, B.L.; Fukatsu, T. Gene expression in gut symbiotic organ of stinkbug affected by extracellular bacterial symbiont. PLoS ONE 2013, 8, e64557. [Google Scholar] [CrossRef]
- Vigneron, A.; Charif, D.; Vincent-Monégat, C.; Vallier, A.; Gavory, F.; Wincker, P.; Heddi, A. Host gene response to endosymbiont and pathogen in the cereal weevil Sitophilus oryzae. BMC Microbiol. 2012, 12, S14. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Bai, Y.; Zheng, X.; Zheng, Y. Coral-algal endosymbiosis characterized using RNAi and single-cell RNA-seq. Nat. Microbiol. 2023, 8, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Jimbo, M.; Yamashita, H.; Koike, K.; Sakai, R.; Kamiya, H. Effects of lectin in the scleractinian coral Ctenactis echinata on symbiotic zooxanthellae. Fish. Sci. 2010, 76, 355–363. [Google Scholar] [CrossRef]
- Dinh, C.; Farinholt, T.; Hirose, S.; Zhuchenko, O.; Kuspa, A. Lectins modulate the microbiota of social amoebae. Science 2018, 361, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Xiao, X.; Liu, Y.; Zhang, R.; Liu, J.; Liu, Q.; Wang, P.; Cheng, G. Mosquito C-type lectins maintain gut microbiome homeostasis. Nat. Microbiol. 2016, 1, 16023. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Florea, L. Rcorrector: Efficient and accurate error correction for Illumina RNA-seq reads. GigaScience 2015, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Saier, M.H.; Reddy, V.S.; Moreno-Hagelsieb, G.; Hendargo, K.J.; Zhang, Y.; Iddamsetty, V.; Lam, K.J.K.; Tian, N.; Russum, S.; Wang, J.; et al. The Transporter Classification Database (TCDB): 2021 update. Nucleic Acids Res. 2021, 49, D461–D467. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Transcript | Over | Under |
|---|---|---|
| Coding transcripts with annotation in eggnog | 160 | 227 |
| Coding transcripts with a BLASTP hit in NCBI but without eggNOG annotation | 27 | 55 |
| Coding transcripts without either annotation in eggNOG or hit in NCBI | 59 | 60 |
| Non-coding transcripts | 113 | 105 |
| Transcripts from Blattabacterium genome | 0 | 13 |
| TOTAL | 359 | 460 |
| Transcript 1 | Fold Change 2 | TPM q-Apo | TPM Control | Annotation from UniProt or Modified | ENA Accession Equivalency | TransDecoder Length (aa) | TransDecoder Information |
|---|---|---|---|---|---|---|---|
| DN206_c21_g1_i1 | 6.5 | 750.5 | 91.2 | Phenylalanine 4-monooxygenase | PSN42902.1 | 453 | complete |
| DN25_c0_g1_i4 | 4.2 | 42.7 | 7.7 | Aminotran_1_2 domain-containing protein | PSN48874.1 | 361 | complete |
| DN37368_c0_g1_i1 | 15.1 | 166.3 | 9.3 | Aromatic-L-amino acid decarboxylase | PSN57389.1 | 480 | complete |
| DN5435_c0_g1_i1 | 4 | 69.9 | 11.2 | y(+)L-type amino acid transporter 2 | PSN41882.1 | 495 | complete |
| DN85219_c0_g1_i1 | 18.3 | 10 | 0.4 | Cuticular protein RR2 type | PSN41655.1 | 154 | complete |
| DN8606_c0_g1_i2 | 7.7 | 14 | 1.3 | Endochitinase | PSN50302.1 | 431 | complete |
| DN15_c0_g1_i2 | 32.9 | 2452.8 | 54.3 | Storage protein/ hexamerin | PSN36665.1 | 626 | 3prime_partial |
| DN15_c0_g1_i4 | 22.7 | 5041.9 | 159.6 | Storage protein/ hexamerin | PSN36665.1 | 676 | complete |
| DN15_c0_g1_i6 | 5.8 | 15,488.8 | 1797.4 | Storage protein/ hexamerin | Absent | 676 | complete |
| Transcript 1 | Domain | Fold Change | TPM Mean q-Apo | TPM Mean Control | DeepLoc Localizations | TMSs No. | TCDB Family Homology | Putative Functions |
|---|---|---|---|---|---|---|---|---|
| DN12960_c0_g1_i2 | PF07690 | 20.9 | 0.8 | 11.5 | Cell membrane | 12 | Anion:Cation Symporter Family (TC# 2.A.1.14) | Glutamate or phosphate transporter |
| DN1327_c1_g1_i8 | PF00083 | 12.9 | 3.4 | 35.7 | Cell membrane | 12 | Sugar Porter Family (TC# 2.A.1.1) | Trehalose transporter |
| DN1382_c0_g1_i1 | PF02535 | 10.4 | 2.1 | 18.5 | Cell membrane| Lysosome/ Vacuole | 8 | Zinc (Zn2+)-Iron (Fe2+) Permease Family (TC# 2.A.5) | Zinc transporter |
| DN2082_c0_g1_i1 | PF03253 | 15.5 | 0.3 | 3 | Cell membrane| Lysosome/ Vacuole | 10 | Urea Transporter Family (TC# 1.A.28) | Urea transporter |
| DN22022_c0_g2_i1 | PF12832 | 19.6 | 0.7 | 9.5 | Cell membrane | 12 | Unidentified Major Facilitator-14 Family (TC# 2.A.1.65) | Unknown |
| DN2708_c1_g1_i2 | PF00083 | 8.7 | 4.2 | 29.5 | Cell membrane | 12 | Sugar Porter Family (TC# 2.A.1.1) | Trehalose transporter |
| DN3532_c0_g1_i1 | PF00023 | 11.5 | 2.3 | 18.5 | Cell membrane | 6 | Transient Receptor Potential Ca2+/Cation Channel Family (TC# 1.A.4) | Ca2+/cation channel |
| DN3824_c0_g1_i2 | PF01080 | 10.6 | 0.9 | 6.7 | Cell membrane| Endoplasmic reticulum|Golgi apparatus | 9 | Presenilin Endoplasmic Reticulum Ca2+ Leak Channel Family (TC# 1.A.54) | Ca2+ channel |
| DN4893_c0_g1_i1 | PF07690 | 5.9 | 4.5 | 17.3 | Cell membrane | 11 | Anion:Cation Symporter Family (TC# 2.A.1.14) | Glutamate or phosphate transporter |
| DN74365_c0_g1_i1 | PTHR34609 | 6.8 | 2.8 | 13.3 | Lysosome/ Vacuole | 4 | 4 TMS Multidrug Endosomal Transporter Family (TC# 2.A.74) | Unknown |
| DN8_c1_g1_i10 | PTHR34609 | 22.3 | 4.1 | 68.9 | Lysosome/ Vacuole | 4 | 4 TMS Multidrug Endosomal Transporter Family (TC# 2.A.74) | Unknown |
| DN82467_c0_g1_i1 | PF05978 | 7.2 | 19.7 | 96 | Cell membrane | 12 | N-Acetylglucosamine Transporter Family (TC# 2.A.1.58) | Potassium channel regulatory protein |
| DN9218_c0_g1_i4 | PF00005 | 7 | 3.9 | 18.5 | Cell membrane| Endoplasmic reticulum | 6 | Eye Pigment Precursor Transporter Family (TC# 3.A.1.204) | ABC transporter |
| Gene Name | Transcript 1 | Protein Length (aa) | Fold Change | TPM Mean q-Apo | TPM Mean Control | Localization 2 | Signal Peptide 2 |
|---|---|---|---|---|---|---|---|
| cactus | DN1475_c0_g1_i1 | 438 | 5 | 20.1 | 76.7 | Cytoplasm | |
| cactus | DN1475_c0_g1_i2 | 440 | 5.6 | 11.3 | 43.9 | Cytoplasm | |
| cactus | DN1475_c0_g1_i4 | 456 | 1319.5 | 0 | 31.3 | Cytoplasm | |
| pelle | DN733_c0_g1_i4 | 728 | 2.3 | 10.7 | 17.5 | Cytoplasm | |
| PGRP-LA-like | DN3159_c1_g1_i1 | 226 | 3.9 | 18.3 | 49.1 | Cell membrane | Yes |
| PGRP-LB_1 | DN3036_c0_g1_i2 | 182 | 5.5 | 8 | 30.1 | Cytoplasm| Extracellular | |
| 3 defensin_g9 | defensin_g9 | 71 | 7 | 36.9 | 162.2 | Extracellular | Yes |
| 3 defensin_g10 | defensin_g10 | 71 | 8.5 | 31.9 | 165.7 | Extracellular | Yes |
| 3 termicin_g4 | DN4880_c0_g1_i1 | 61 | 7.9 | 53.8 | 262.2 | Extracellular | Yes |
| PGRP-LB_1 | 4 DN3036_c0_g1_i1 | 237 | - | 1.3 | 0 | Extracellular | Yes |
| PGRP-LB_2 | 4 DN4891_c0_g1_i1 | 191 | - | 5.5 | 7 | Extracellular | Yes |
| Transcript 1 | Protein Length (aa) | Cys No. | Domain | SignalP | Fold Change | TPM Mean q-Apo | TPM Mean Control | DeepLoc Localizations | TMSs No. | AMP-Like Factor Candidate |
|---|---|---|---|---|---|---|---|---|---|---|
| DN12569_c0_g1_i1 | 127 | 14 | PF00008 | Yes | 6.8 | 7.5 | 31.9 | Extracellular | No | |
| DN17653_c0_g1_i2 | 186 | 9 | PF00059 | Yes | 11 | 1 | 7.8 | Extracellular | Yes | |
| DN2464_c0_g1_i3 | 140 | 8 | PF00062 | Yes | 10.3 | 2.1 | 13.9 | Extracellular | Yes | |
| DN3965_c0_g1_i1 | 108 | 15 | Yes | 7.1 | 3.5 | 17.5 | Extracellular | Yes | ||
| DN43375_c0_g1_i1 | 106 | 8 | 14.3 | 1.7 | 16.2 | Mitochondrion | No | |||
| DN48391_c0_g1_i1 | 188 | 10 | PF00059 | Yes | 12.2 | 0.8 | 7.7 | Extracellular | Yes | |
| 2 DN4880_c0_g1_i1 termicin_g4 | 61 | 7 | PF11415 | Yes | 7.9 | 53.8 | 262.2 | Extracellular | No | |
| DN5401_c0_g2_i1 | 67 | 7 | 47.1 | 0.2 | 9.3 | Extracellular | 1 | Yes | ||
| DN54153_c0_g1_i1 | 102 | 6 | 22.2 | 0.7 | 10.1 | Cytoplasm| Nucleus | No | |||
| DN7034_c0_g1_i1 | 150 | 11 | PF17064 | Yes | 6.3 | 5.9 | 24.2 | Cell membrane | No | |
| DN74365_c0_g1_i1 | 195 | 6 | PTHR34609 | 6.8 | 2.8 | 13.3 | Lysosome/ Vacuole | 4 | No | |
| DN8_c1_g1_i10 | 174 | 10 | PTHR34609 | 22.3 | 4.1 | 68.9 | Lysosome/ Vacuole | 4 | No | |
| 2 defensin_g10 | 71 | 7 | PF01097 | Yes | 8.5 | 31.9 | 165.7 | Extracellular | No | |
| 2 defensin_g9 | 71 | 7 | PF01097 | Yes | 7 | 36.9 | 162.2 | Extracellular | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, F.J.; Domínguez-Santos, R.; Latorre, A.; García-Ferris, C. Comparative Transcriptomics of Fat Bodies between Symbiotic and Quasi-Aposymbiotic Adult Females of Blattella germanica with Emphasis on the Metabolic Integration with Its Endosymbiont Blattabacterium and Its Immune System. Int. J. Mol. Sci. 2024, 25, 4228. https://doi.org/10.3390/ijms25084228
Silva FJ, Domínguez-Santos R, Latorre A, García-Ferris C. Comparative Transcriptomics of Fat Bodies between Symbiotic and Quasi-Aposymbiotic Adult Females of Blattella germanica with Emphasis on the Metabolic Integration with Its Endosymbiont Blattabacterium and Its Immune System. International Journal of Molecular Sciences. 2024; 25(8):4228. https://doi.org/10.3390/ijms25084228
Chicago/Turabian StyleSilva, Francisco J., Rebeca Domínguez-Santos, Amparo Latorre, and Carlos García-Ferris. 2024. "Comparative Transcriptomics of Fat Bodies between Symbiotic and Quasi-Aposymbiotic Adult Females of Blattella germanica with Emphasis on the Metabolic Integration with Its Endosymbiont Blattabacterium and Its Immune System" International Journal of Molecular Sciences 25, no. 8: 4228. https://doi.org/10.3390/ijms25084228
APA StyleSilva, F. J., Domínguez-Santos, R., Latorre, A., & García-Ferris, C. (2024). Comparative Transcriptomics of Fat Bodies between Symbiotic and Quasi-Aposymbiotic Adult Females of Blattella germanica with Emphasis on the Metabolic Integration with Its Endosymbiont Blattabacterium and Its Immune System. International Journal of Molecular Sciences, 25(8), 4228. https://doi.org/10.3390/ijms25084228