
Atherosclerosis and the Bidirectional Relationship between Cancer and Cardiovascular Disease: From Bench to Bedside—Part 1
 ,
,  , , and
, , and 
Abstract
1. Introduction
2. The Changing Paradigm of Atherosclerosis and Related Cardiovascular Disease

{kind=link}
| Study | Anti-Inflammatory Agent | Target | Population | Effect on Inflammation Biomarker | Clinical Effects | References |
|---|---|---|---|---|---|---|
| CANTOS (main study) subgroup analysis of CANTOS | 3 doses of canakinumab (50 mg, 150 mg, and 300 mg) subcutaneously (s.c.) every 3 months vs. placebo | IL-1β | 10,061 patients with previous myocardial infarction and hsCRP ≥ 2 mg/L Subgroup of 338 patients with clonal haematopoiesis and variants in TET2 more common than DNMT3A | Reduction in hsCRP (for all the doses) | The dose of 150 mg s.c. every 3 months was associated with a significant reduction in recurrent CV events Patients with CHIP due to somatic variants in TET2 had reduced risk for MACE | Ref. [11] NEJM 2017;377:1119 Ref. [100] JAMA Cardiol 2022;7:521 |
| CIRT | Low-dose methotrexate (15–20 mg weekly) | No specific target | 4786 patients with known atherosclerosis and either DM orMS | No reduction in IL-1β or IL-6 | No reduction in CV event rates | Ref. [87] NEJM 2019;380:752 |
| RESCUE | Ziltivekimab (7·5 mg, 15 mg, or 30 mg every 4 weeks up to 24 weeks) | IL-6 | 264 patients with high CV risk> (age ≥ 18 years, moderate to severe CKD, hsCRP ≥ 2 mg/L) | Reduction in biomarkers of inflammation (hsCRP) and thrombosis (e.g., fibrinogen) | Reduction in biomarkers of inflammation (hsCRP) and thrombosis (e.g., fibrinogen) | Ref. [12] Lancet 2021;397:2060 |
| COLCOT | Low-dose colchicine (0.5 mg daily) | Inhibition of tubulin polymerization and alteration in leukocyte responsiveness | Patients with recent myocardial infarction: 2366 patients assigned to colchicine and 2379 to placebo | Significant reduction in ischemic CV events | Ref. [13] NEJM 2019;381:2497 | |
| Lo-Do-Co2 | Low-dose colchicine (0.5 mg daily) | Inhibition of tubulin polymerization and alteration in leukocyte responsiveness | Patients with chronic coronary artery disease in stable condition: 2762 patients assigned to colchicine and 2760 to placebo. | 31% lower relative risk of CV death, spontaneous myocardial infarction, ischemic stroke, or coronary revascularization in patients treated with colchicine compared to placebo | Ref. [14] NEJM 2020;383:1838 |
3. Atherosclerosis and Cancer: The Unexpected Link
4. Atherosclerosis and Cardio-Oncology: The Bidirectional Relationship Has Highlighted the Novel Issues That Need to Be Addressed
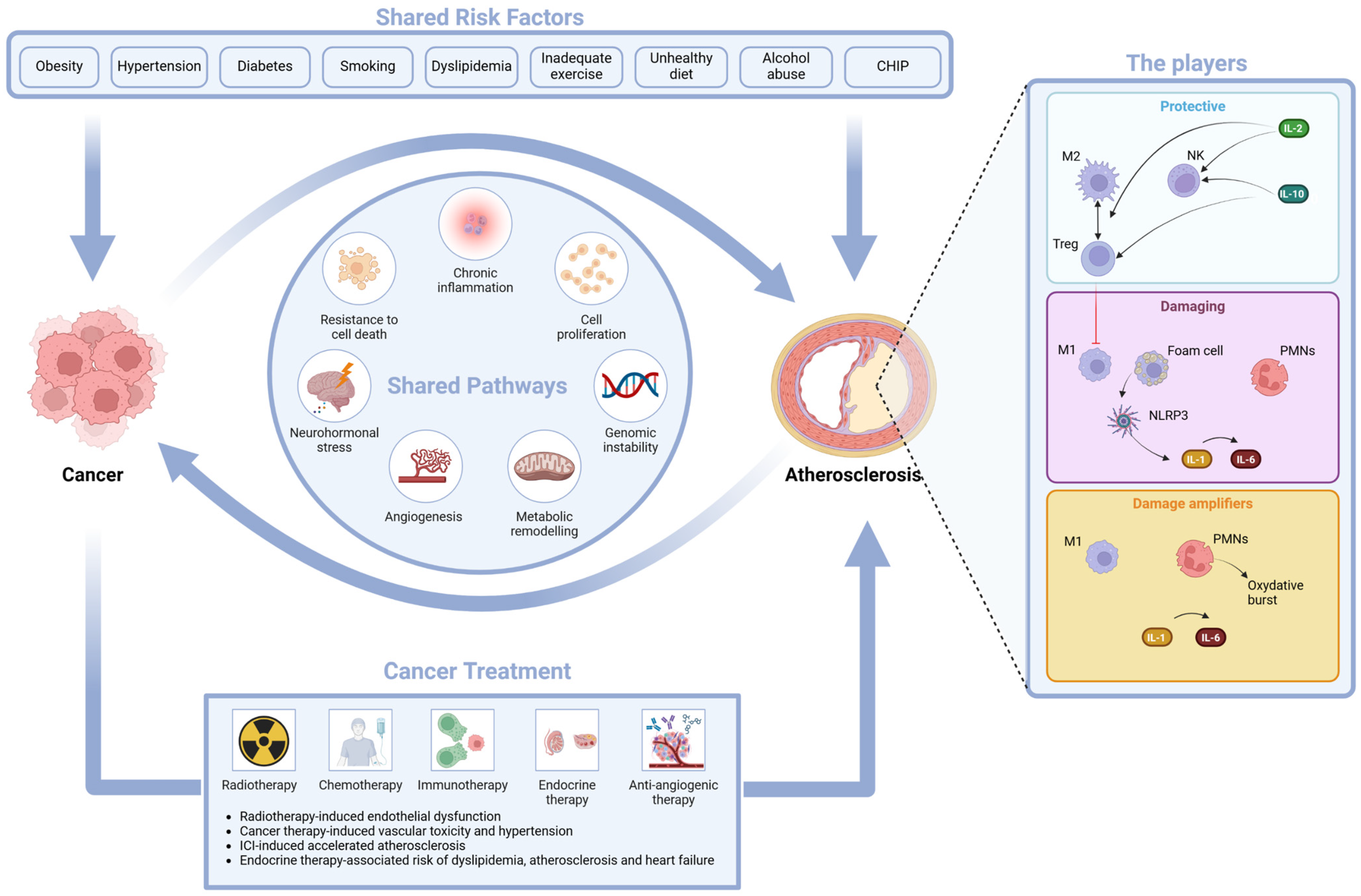
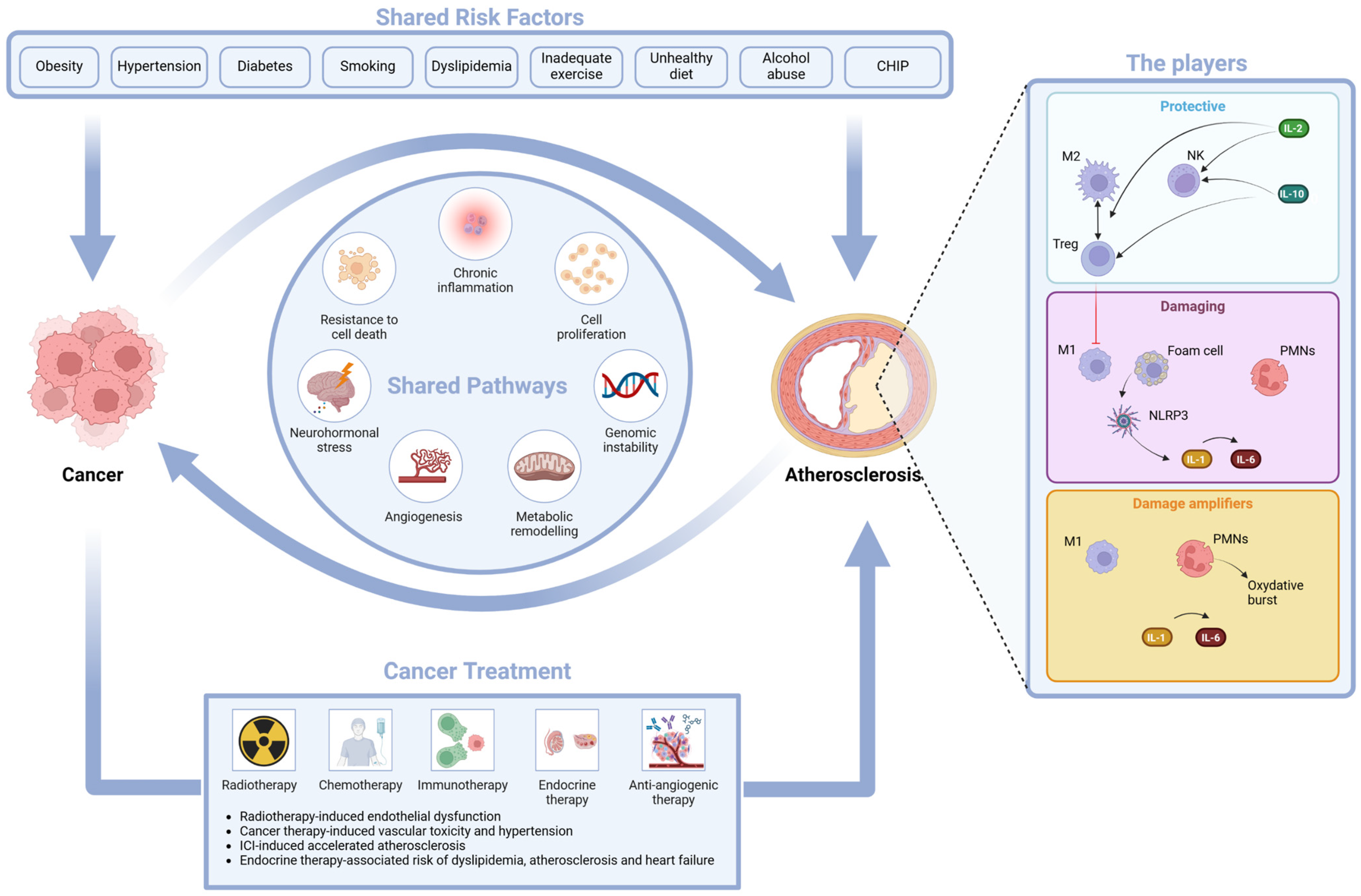
4.1. The Shared Risk Factors
4.2. The Shared Pathways
- -
- Inflammation, an epiphenomenon of immune dysregulation and cell senescence, is associated with increased levels of hsCRP and IL-6 and suppression of tumorigenicity 2 (ST2). These biomarkers are linked to tissue invasion and metastasis associated with cancer but also with tissue damage that underlies the atherosclerotic process [116,117].
- -
- -
- Resistance to cell death is linked to cellular stress response and apoptosis; its biomarker, growth differentiation factor (GDF-15), has a prognostic role in cancer mortality and in CVD (myocardial infarction, thromboembolic stroke, heart failure, and stroke) [120,121]; cardiac Troponin T (cTnT), a well-known marker of myocardial cell death, is also a useful marker in light chain amyloidosis [122,123].
- -
- Neurohormonal stress leads to increased levels of cardiovascular neurohormones such as N-terminal pro-B-type natriuretic peptide, mid-regional pro-atrial natriuretic peptide and other neurohormones, and diuretic hormones. These cardiovascular neurohormones have a relevant role in patients with acute or chronic heart failure (HF), but they might also play a role in cancer, since they may be produced by some malignant cells in the vascular bed of tumors [124,125].
- -
- Angiogenesis, with a role in EC survival and in tumorigenesis, invasion, and metastasis, may be measured by angiogenic biomarkers such as soluble fms-like tyrosine kinase 1, a variant of Flt-1 known also as vascular endothelial growth factor (VEGF) receptor, and placental growth factor (PlGF). VEGF biology has a relevant impact on tumorigenesis and on normal cardiovascular function [126,127].
- -
- Genomic instability, such as CHIP, has an impact on both CVD and cancer. CHIP [128] is a risk factor for CVD [95], but it may be caused by atherosclerosis due to the continuous stimulation of stem cell proliferation [129]. This reverse CHIP effect has uncovered an unexpected link between oncogenesis and atherogenesis [38]. The biological explanation of the impact of a potentially precancerous lesion on CVD involves an interaction between clonally derived monocytes and macrophages and the vascular endothelium that leads to vascular inflammation and accelerated atherogenesis [95,128].
4.3. The Atherogenic Effect of Some Oncologic Treatments
- -
- Radiotherapy-induced endothelial dysfunction. Radiation-associated damage induces the secretion of proinflammatory cytokines; increases the release of reactive oxygen species (ROS); causes a dysregulation of glycolysis, lipid metabolic pathways, and angiogenesis; and may have a negative impact on telomere function and immunity homeostasis. The final effect is EC death (either acutely via apoptosis or chronically via EC senescence) and a disrupted EC environment [130]. Clinical phenotypes of radiation-associated vascular damage are accelerated CAD, cerebral events due to carotid artery disease, calcification of the ascending aorta and aortic arch, and lesions of other vascular segments in the radiation field [131]. The recent BACCARAT study evaluated the association between cardiac exposure and the risk of developing calcified and non-calcified atheromatous plaques within 2 years of RT. As both calcified and non-calcified plaques were found, it may be hypothesized that cardiac radiation exposure accelerates the process of atherosclerosis in already existing plaques with an increase in their calcium content (calcified plaques) and starts new non-calcified plaques [132].
- -
- Cancer therapy-induced vasculotoxicity is associated with traditional chemotherapies (alkylating agents, microtubule inhibitors, and antimetabolites), with targeted therapies such as VEGF inhibitors, with breakpoint cluster region–Abelson oncogene locus tyrosine kinase inhibitors, and with multiple myeloma drugs. The majority of these drugs induce hypertension that may eventually drive atherosclerosis. They may also produce CV injury due to damage-associated molecular patterns (DAMPs) that sustain inflammation. There are many clinical phenotypes of vasculotoxicity, including CAD, stroke, systemic and pulmonary hypertension, vasospasm, and thrombosis [133,134].
- -
- Accelerated atherosclerosis is induced by immune checkpoint inhibitor (ICI) treatment. Oncologic studies of ICI-induced cardiotoxicity have indeed shed light on the complex relationship between the immune system, inflammation, and atherosclerosis. Preclinical studies have shown that the targets of ICIs [PD-1 (programmed cell death protein 1), PD-L1 (programmed death ligand 1), CTLA-4 (cytotoxic T-lymphocyte–associated protein4)] are proteins with a negative regulatory role in atherosclerosis [135]. Blockage of the checkpoints may predictably lead to accelerated atherosclerosis through enhanced T cell responses, limited Treg function, and infiltration of the vascular endothelium [136,137,138,139]. Preclinical studies have also shown that short-term ICI treatment promotes DAMPs and proinflammatory cytokine production [140]. In a clinical setting, a seminal study of 2842 patients and 2842 controls matched by age, a history of cardiovascular events and cancer type showed a 3-fold increase in atherosclerotic cardiovascular events (myocardial infarction, coronary revascularization, and ischemic stroke) after starting ICI treatment. Moreover, in a case-crossover analysis performed by the same authors and comparing an at-risk period (defined as the 2-year period after ICIs) and a control period (defined as the 2-year period before ICIs), atherosclerotic cardiovascular events significantly increased from 1.37 to 6.55 per 100 person-years at 2 years; in a subgroup of 40 patients, a 3-fold-higher rate of aortic plaque progression was also documented [141]. In a more recent retrospective cohort study on 1458 patients diagnosed with stage III or IV non-small-cell lung cancer (NSCLC) treated with (487 patients) and without (971 patients) ICI therapy and followed-up for a median time of 23.1 months, ICI therapy was associated with a 3.6-fold increase in the total risk of ASCVD events before propensity score matching [142].
- -
- Hormone therapy-associated risk of dyslipidemia, atherosclerosis, and heart failure. This effect has been proven in BC women treated with aromatase inhibitors that increase the risk of atherosclerosis, HF, and dyslipidemia [143] and in prostate cancer patients treated with androgen deprivation therapy (ADT) in which the increased risk of CV events is linked to indirect modifications of CVRFs. More specifically, Gonadotropin-releasing hormone (GnRH) agonists increase LDL-cholesterol and triglyceride levels, visceral fat, and insulin resistance and decrease lean body mass and glucose tolerance, leading to accelerated atherosclerosis and coronary artery disease (CAD) events, HF, and arrhythmias [144,145]. In preclinical studies, orchiectomy and GnRH agonists, but not GnRH antagonists, induced long- or short-term follicular stimulating hormone elevation that, acting synergistically with TNF-α, induced an amplified endothelial inflammation through elevation of vascular cell adhesion protein-1 expression, thus accelerating atherosclerosis [146].
5. How Do We Measure and Quantify Atherosclerosis?
5.1. Risk Scores and Mendelian Randomization
5.2. Biomarkers
5.3. Imaging Techniques
- (i)
- Coronary artery calcium (CAC) has become a useful tool to detect and quantify calcified subclinical atherosclerotic burden. The most widely used method to quantify CAC is the Agatston method, which uses the product of the total calcium area and a quantized peak calcium density weighting factor defined by the calcification attenuation in Hounsfied units on non-contrast computed tomography [159]; in addition, CAC may be identified on scans scheduled for other reasons [160]. In the ITALUNG trial, CAC was assessed in baseline, low-dose computed tomography performed on 1364 participants aged 59–69 years and with a smoking history ≥ 20 pack-years in a lung cancer screening program with a follow-up of 22 years. CAC score was graded as absent, mild, moderate, and severe. In the study, moderate or severe CAC was significantly associated with CV mortality after adjustment for traditional CVRFs [161]. CAC progression may also be a marker of accelerated atherosclerosis, as shown in a recent study regarding ICI therapy in cancer [142]. CAC score is unreliable with statin or PCSK9 inhibitor treatment [162,163].
- (ii)
- Computed Tomography Angiography. The “actionable lump” concept underscores the importance of early detection and proactive monitoring of sublinical atherosclerosis when preventive interventions, such as conversion to a healthy lifestyle and early treatment of CVRFs, may limit the progression of the atherosclerotic process [164].
6. What Is New on the Horizon for Early Atherosclerosis Imaging?
6.1. Magnetic Resonance Imaging (MRI)
6.2. Computed Tomography Angiography (CTA)
7. Limitations and Unresolved Questions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABI | ankle–brachial index |
| ADT | androgen deprivation therapy |
| AHA | American Heart Association |
| AMI | acute myocardial infarction |
| ASCVD | atherosclerotic cardiovascular disease |
| BAC | breast arterial calcification |
| BC | breast cancer |
| CAC | coronary artery calcium |
| CAD | coronary artery disease |
| CHIP | clonal hematopoiesis of indeterminate potential |
| CT | computed tomography |
| CCTA | coronary computed tomography angiography |
| CTA | computed tomography angiography |
| cTnT | cardiac troponin T |
| CV | cardiovascular |
| CVD | cardiovascular disease |
| CVRFs | cardiovascular risk factors |
| DAMPs | danger-associated molecular patterns |
| EAT | epicardial adipose tissue |
| EC | endothelial cell |
| FAI | Fat Attenuation Index |
| GDF-15 | growth differentiation factor-15 |
| GnRH | gonadotropin releasing hormone |
| GWAS | genome wide association studies |
| HDL | high-density lipoprotein |
| HF | heart failure |
| HR | hazard ratio |
| hs-CRP | high-sensitivity C-reactive protein |
| GOF | gain of function |
| ICI | immune checkpoint inhibitor |
| IL | interleukin |
| LDL-C | low-density lipoprotein |
| LOF | loss of function |
| Lp(a) | lipoprotein(a) |
| MACE | major adverse CV event |
| MCP-1 | monocyte chemoattractant protein-1 |
| M1 | M1 macrophage |
| M2 | M2 macrophage |
| MRI | magnetic resonance imaging |
| NAFLD | non-alcoholic fatty liver disease |
| NET | neutrophil extracellular trap |
| NK | natural killer |
| NLRP3 | nucleotide oligomerization domain-containing, leucine-rich repeat-containing, and pyrin domain-containing protein3 |
| NSCLC | non-small-cell lung cancer |
| PESA | Progression of Early Subclinical Atherosclerosis |
| PET | positron emission tomography |
| PlGF | placental growth factor |
| PMNs | polymorphonuclear neutrophils |
| PVAT | perivascular adipose tissue |
| RCT | randomized controlled trial |
| ROS | reactive oxygen species |
| ST2 | suppression of tumorigenicity 2 |
| Th2 | type 2 helper T cells |
| TG | triglycerides |
| T reg | regulatory T cells |
| TNF-α | tumor necrosis factor-α |
| VEGF | vascular endothelial growth factor |
References
- World Health Organization. WHO Department of Data and Analytics. WHO Methods and Data Sources for Global Burden of Disease Estimates, 2000–2019; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Global Cardiovascular Risk Consortium; Magnussen, C.; Ojeda, F.M.; Leong, D.P.; Alegre-Diaz, J.; Amouyel, P.; Aviles-Santa, L.; De Bacquer, D.; Ballantyne, C.M.; Bernabé-Ortiz, A.; et al. Global Effect of Modifiable Risk Factors on Cardiovascular Disease and Mortality. N. Engl. J. Med. 2023, 389, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Townsend, N.; Kazakiewicz, D.; Lucy Wright, F.; Timmis, A.; Huculeci, R.; Torbica, A.; Gale, C.P.; Achenbach, S.; Weidinger, F.; Vardas, P. Epidemiology of cardiovascular disease in Europe. Nat. Rev. Cardiol. 2022, 19, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Cancer Risk Factors Collaborators. The global burden of cancer attributable to risk factors, 2010–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 563–591. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.S.; Paniagua, S.M.; Liu, E.; Jovani, M.; Li, S.X.; Takvorian, K.; Suthahar, N.; Cheng, S.; Splansky, G.L.; Januzzi, J.L., Jr.; et al. Cardiovascular Risk Factors are Associated with Future Cancer. JACC Cardio Oncol. 2021, 3, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Dzaye, O.; Berning, P.; Dardari, Z.A.; Mortensen, M.B.; Marshall, C.H.; Nasir, K.; Budoff, M.J.; Blumenthal, R.S.; Whelton, S.P.; Blaha, M.J. Coronary artery calcium is associated with increased risk for lung and colorectal cancer in men and women: The Multi-Ethnic Study of Atherosclerosis (MESA). Eur. Heart J. Cardiovasc. Imaging 2022, 23, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Kobold, S. Inflammation: A common contributor to cancer, aging, and cardiovascular diseases-expanding the concept of cardiooncology. Cardiovasc. Res. 2019, 115, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Bhatt, D.L.; Pradhan, A.D.; Glynn, R.J.; MacFadyen, J.G.; Nissen, S.E.; PROMINENT, REDUCE-IT, and STRENGTH Investigators. Inflammation and cholesterol as predictors of cardiovascular events among patients receiving statin therapy: A collaborative analysis of three randomised trials. Lancet 2023, 401, 1293–1301. [Google Scholar] [CrossRef]
- Ridker, P.M.; Lei, L.; Louie, M.J.; Haddad, T.; Nicholls, S.J.; Lincoff, A.M.; Libby, P.; Nissen, S.E.; CLEAR Outcomes Investigators. Inflammation and Cholesterol as Predictors of Cardiovascular Events among 13 970 Contemporary High-Risk Patients with Statin Intolerance. Circulation 2024, 149, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; Devalaraja, M.; Baeres, F.M.M.; Engelmann, M.D.M.; Hovingh, G.K.; Ivkovic, M.; Lo, L.; Kling, D.; Pergola, P.; Raj, D.; et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 2060–2069. [Google Scholar] [CrossRef]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.F.; Ireland, M.A.; Lenderink, T.; et al. LoDoCo2 Trial Investigators. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis—From experimental insights to the clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J. CANTOS Trial Group. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Kuo, M.C.; Chang, S.J.; Hsieh, M.C. Colchicine Significantly Reduces Incident Cancer in Gout Male Patients: A 12-Year Cohort Study. Medicine 2015, 94, e1570. [Google Scholar] [CrossRef]
- Yang, T.; Hao, L.; Yang, X.; Luo, C.; Wang, G.; Lin Cai, C.; Qi, S.; Li, Z. Prognostic value of derived neutrophil-to-lymphocyte ratio (dNLR) in patients with non-small cell lung cancer receiving immune checkpoint inhibitors: A meta-analysis. BMJ Open 2021, 11, e049123. [Google Scholar] [CrossRef]
- Han, C.L.; Meng, G.X.; Ding, Z.N.; Dong, Z.R.; Chen, Z.Q.; Hong, J.G.; Yan, L.J.; Liu, H.; Tian, B.W.; Yang, L.S.; et al. The Predictive Potential of the Baseline C-Reactive Protein Levels for the Efficiency of Immune Checkpoint Inhibitors in Cancer Patients: A Systematic Review and Meta-Analysis. Front. Immunol. 2022, 13, 827788. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.M.; Hong, Y.; Labarthe, D.; Mozaffarian, D.; Appel, L.J.; Van Horn, L.; Greenlund, K.; Daniels, S.; Nichol, G.; Tomaselli, G.F.; et al. American Heart Association Strategic Planning Task Force and Statistics Committee. Defining and setting national goals for cardiovascular health promotion and disease reduction: The American Heart Association’s strategic Impact Goal through 2020 and beyond. Circulation 2010, 121, 586–613. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.M.; Allen, N.B.; Anderson, C.A.M.; Black, T.; Brewer, L.C.; Foraker, R.E.; Grandner, M.A.; Lavretsky, H.; Perak, A.M.; Sharma, G.; et al. American Heart Association. Life’s Essential 8: Updating and Enhancing the American Heart Association’s Construct of Cardiovascular Health: A Presidential Advisory From the American Heart Association. Circulation 2022, 14, e18–e43. [Google Scholar] [CrossRef] [PubMed]
- Boehm, J.K.; Soo, J.; Chen, Y.; Zevon, E.S.; Hernandez, R.; Lloyd-Jones, D.; Kubzansky, L.D. Psychological well-being’s link with cardiovascular health in older adults. Am. J. Prev. Med. 2017, 53, 791–798. [Google Scholar] [CrossRef]
- Ogunmoroti, O.; Osibogun, O.; Spatz, E.S.; Okunrintemi, V.; Mathews, L.; Ndumele, C.E.; Michos, E.D. A systematic review of the bidirectional relationship between depressive symptoms and cardiovascular health. Prev. Med. 2022, 154, 106891. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Wang, Y.X.; Jiang, C.L. Inflammation: The Common Pathway of Stress-Related Diseases. Front. Hum. Neurosci. 2017, 11, 316. [Google Scholar] [CrossRef]
- Münzel, T.; Sørensen, M.; Hahad, O.; Nieuwenhuijsen, M.; Daiber, A. The contribution of the exposome to the burden of cardiovascular disease. Nat. Rev. Cardiol. 2023, 20, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Montone, R.A.; Camilli, M.; Calvieri, C.; Magnani, G.; Bonanni, A.; Bhatt, D.L.; Rajagopalan, S.; Crea, F.; Niccoli, G. Exposome in ischaemic heart disease: Beyond traditional risk factors. Eur. Heart J. 2024, 45, 419–438. [Google Scholar] [CrossRef]
- Beulens, J.W.J.; Pinho, M.G.M.; Abreu, T.C.; den Braver, N.R.; Lam, T.M.; Huss, A.; Vlaanderen, J.; Sonnenschein, T.; Siddiqui, N.Z.; Yuan, Z.; et al. Environmental risk factors of type 2 diabetes-an exposome approach. Diabetologia 2022, 65, 263–274. [Google Scholar] [CrossRef]
- Wild, C.P. Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1847–1850. [Google Scholar] [CrossRef]
- Rappaport, S.M.; Smith, M.T. Epidemiology. Environment and disease risks. Science 2010, 330, 460–461. [Google Scholar] [CrossRef] [PubMed]
- Vaduganathan, M.; Mensah, G.A.; Turco, J.V.; Fuster, V.; Roth, G.A. The Global Burden of Cardiovascular Diseases and Risk: A Compass for Future Health. J. Am. Coll. Cardiol. 2022, 80, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; Prattichizzo, F.; Sardu, C.; Fulgenzi, G.; Graciotti, L.; Spadoni, T.; D’Onofrio, N.; Scisciola, L.; La Grotta, R.; Frigé, C.; et al. Microplastics and Nanoplastics in Atheromas and Cardiovascular Events. N. Engl. J. Med. 2024, 390, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Yoon, H.K.; Kim, D.Y.; Jeong, T.S.; Park, Y.S. An Emerging Role of Micro- and Nanoplastics in Vascular Diseases. Life 2024, 14, 255. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, A. Environmental Determinants of Cardiovascular Disease. Circ. Res. 2017, 121, 162–180. [Google Scholar] [CrossRef]
- Sheikh, A.M.; Yano, S.; Tabassum, S.; Nagai, A. The Role of the Vascular System in Degenerative Diseases: Mechanisms and Implications. Int. J. Mol. Sci. 2024, 25, 2169. [Google Scholar] [CrossRef]
- Bagby, S.P.; Martin, D.; Chung, S.T.; Rajapakse, N. From the Outside In: Biological Mechanisms Linking Social and Environmental Exposures to Chronic Disease and to Health Disparities. Am. J. Public Health 2019, 109, S56–S63. [Google Scholar] [CrossRef]
- Janssen, H.; Koekkoek, L.L.; Swirski, F.K. Effects of lifestyle factors on leukocytes in cardiovascular health and disease. Nat. Rev. Cardiol. 2023. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. ESC National Cardiac Societies; ESC Scientific Document Group. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Bundy, J.D.; Mills, K.T.; Bazzano, L.A.; Kelly, T.N.; Zhang, Y.; Chen, J.; He, J. Secular Trends in Cardiovascular Health in US Adults (from NHANES 2007 to 2018). Am. J. Cardiol. 2021, 159, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Elías-López, D.; Vargas-Vázquez, A.; Mehta, R.; Cruz Bautista, I.; Del Razo Olvera, F.; Gómez-Velasco, D.; Almeda Valdes, P.; Aguilar-Salinas, C.A. Metabolic Syndrome Study Group. Natural course of metabolically healthy phenotype and risk of developing Cardiometabolic diseases: A three years follow-up study. BMC Endocr. Disord. 2021, 21, 85. [Google Scholar] [CrossRef]
- Lhoste, V.P.F.; Zhou, B.; Mishra, A.; Bennett, J.E.; Filippi, S.; Asaria, P.; Gregg, E.W.; Danaei, G.; Ezzati, M. Cardiometabolic and renal phenotypes and transitions in the United States population. Nat. Cardiovasc. Res. 2023, 3, 46–59. [Google Scholar] [CrossRef]
- He, J.; Zhu, Z.; Bundy, J.D.; Dorans, K.S.; Chen, J.; Hamm, L.L. Trends in Cardiovascular Risk Factors in US Adults by Race and Ethnicity and Socioeconomic Status, 1999-2018. JAMA 2021, 326, 1286–1298. [Google Scholar] [CrossRef]
- Kouvari, M.; Panagiotakos, D.B.; Yannakoulia, M.; Georgousopoulou, E.; Critselis, E.; Chrysohoou, C.; Tousoulis, D.; Pitsavos, C.; ATTICA Study Investigators. Transition from metabolically benign to metabolically unhealthy obesity and 10-year cardiovascular disease incidence: The ATTICA cohort study. Metabolism 2019, 93, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.X.; Chaudhary, N.; Akinyemiju, T. Metabolic Syndrome Prevalence by Race/Ethnicity and Sex in the United States, National Health and Nutrition Examination Survey, 1988–2012. Prev. Chronic Dis. 2017, 14, E24. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Friera, L.; Peñalvo, J.L.; Fernández-Ortiz, A.; Ibañez, B.; López-Melgar, B.; Laclaustra, M.; Oliva, B.; Mocoroa, A.; Mendiguren, J.; Martínez de Vega, V.; et al. Prevalence, Vascular Distribution, and Multiterritorial Extent of Subclinical Atherosclerosis in a Middle-Aged Cohort: The PESA (Progression of Early Subclinical Atherosclerosis) Study. Circulation 2015, 131, 2104–2113. [Google Scholar] [CrossRef]
- Raposeiras-Roubin, S.; Rosselló, X.; Oliva, B.; Fernández-Friera, L.; Mendiguren, J.M.; Andrés, V.; Bueno, H.; Sanz, J.; Martínez de Vega, V.; Abu-Assi, E.; et al. Triglycerides and Residual Atherosclerotic Risk. J. Am. Coll. Cardiol. 2021, 77, 3031–3041. [Google Scholar] [CrossRef]
- Wadström, B.N.; Pedersen, K.M.; Wulff, A.B.; Nordestgaard, B.G. Elevated remnant cholesterol, plasma triglycerides, and cardiovascular and non-cardiovascular mortality. Eur. Heart J. 2023, 44, 1432–1445. [Google Scholar] [CrossRef]
- Park, H.B.; Arsanjani, R.; Hong, S.J.; Yi, J.J.; Yi, S.W. Impact of hypertriglyceridaemia on cardiovascular mortality according to low-density lipoprotein cholesterol in a 15.6-million population. Eur. J. Prev. Cardiol. 2024, 31, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Sterling, K.; Wang, Z.; Zhang, Y.; Song, W. The role of inflammasomes in human diseases and their potential as therapeutic targets. Signal Transduct. Target. Ther. 2024, 9, 10. [Google Scholar] [CrossRef]
- Tall, A.R.; Bornfeldt, K.E. Inflammasomes and Atherosclerosis: A Mixed Picture. Circ. Res. 2023, 132, 1505–1520. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015, 116, 1231–1244. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.; Nguyen-Tran, H.H.; Trojanowska, M. Active roles of dysfunctional vascular endothelium in fibrosis and cancer. J. Biomed. Sci. 2019, 26, 86. [Google Scholar] [CrossRef] [PubMed]
- Engelen, S.E.; Robinson, A.J.B.; Zurke, Y.X.; Monaco, C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: How to proceed? Nat. Rev. Cardiol. 2022, 19, 522–542. [Google Scholar] [CrossRef]
- Crainiciuc, G.; Palomino-Segura, M.; Molina-Moreno, M.; Sicilia, J.; Aragones, D.G.; Li, J.L.Y.; Madurga, R.; Adrover, J.M.; Aroca-Crevillén, A.; Martin-Salamanca, S. Behavioural immune landscapes of inflammation. Nature 2022, 601, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Quintin, J.; van der Meer, J.W. Trained immunity: A memory for innate host defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef]
- Netea, M.G.; Dominguez-Andres, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 365–388. [Google Scholar] [CrossRef]
- Domínguez-Andrés, J.; Dos Santos, J.C.; Bekkering, S.; Mulder, W.J.M.; van der Meer, J.W.M.; Riksen, N.P.; Joosten, L.A.B.; Netea, M.G. Trained immunity: Adaptation within innate immune mechanisms. Physiol. Rev. 2023, 103, 313–346. [Google Scholar] [CrossRef]
- Anitschkow, N.; Chalatow, S. On experimental cholesterin steatosis and its significance in the origin of some pathological processes (1913). Arteriosclerosis 1983, 3, 178–182. [Google Scholar]
- Ross, R. Atherosclerosis-an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Gao, Y.; Galis, Z.S. Exploring the Role of Endothelial Cell Resilience in Cardiovascular Health and Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 179–185. [Google Scholar] [CrossRef]
- Hayes, K.L. Pericytes in Type 2 Diabetes. In Pericyte Biology in Disease; Advances in Experimental Medicine and Biology; Birbrair, A., Ed.; Springer International Publishing: Cham, Switzerland, 2019; Volume 1147, pp. 265–278. ISBN 978-3-030-16907-7. [Google Scholar]
- Dabravolski, S.A.; Markin, A.M.; Andreeva, E.R.; Eremin, I.I.; Orekhov, A.N.; Melnichenko, A.A. Molecular Mechanisms Underlying Pathological and Therapeutic Roles of Pericytes in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 11663. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; D’Souza, S.S.; Moskvin, O.V.; Toh, H.; Wang, B.; Zhang, J.; Swanson, S.; Guo, L.-W.; Thomson, J.A.; Slukvin, I.I. Specification and Diversification of Pericytes and Smooth Muscle Cells from Mesenchymoangioblasts. Cell Rep. 2017, 19, 1902–1916. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Sun, Z.; Zang, G.; Zhang, L.; Hou, L.; Shao, C.; Wang, Z. Epidemiological Research Advances in Vascular Calcification in Diabetes. J. Diabetes Res. 2021, 2021, 4461311. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Cui, Z.Y.; Huang, X.F.; Zhang, D.D.; Guo, R.J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef]
- Alencar, G.F.; Owsiany, K.M.; Karnewar, S.; Sukhavasi, K.; Mocci, G.; Nguyen, A.T.; Williams, C.M.; Shamsuzzaman, S.; Mokry, M.; Henderson, C.A.; et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 2020, 142, 2045–2059. [Google Scholar] [CrossRef]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef]
- Clement, M.; Raffort, J.; Lareyre, F.; Tsiantoulas, D.; Newland, S.; Lu, Y.; Masters, L.; Harrison, J.; Saveljeva, S.; Ma, M.K.L.; et al. Impaired Autophagy in CD11b+ Dendritic Cells Expands CD4+ Regulatory T Cells and Limits Atherosclerosis in Mice. Circ. Res. 2019, 125, 1019–1034. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Lichtman, A.H. Monocyte- macrophages and T cells in atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef]
- Bekkering, S.; Blok, B.A.; Joosten, L.A.; Riksen, N.P.; van Crevel, R.; Netea, M.G. In vitro experimental model of trained innate immunity in human primary monocytes. Clin. Vaccine Immunol. 2016, 23, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.J. Macrophages in atherosclerosis regression. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef]
- Fredman, G.; Tabas, I. Boosting Inflammation Resolution in Atherosclerosis: The Next Frontier for Therapy. Am. J. Pathol. 2017, 187, 1211–1221. [Google Scholar] [CrossRef]
- Owsiany, K.M.; Alencar, G.F.; Owens, G.K. Revealing the origins of foam cells in atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 836–838. [Google Scholar] [CrossRef]
- Yurdagul, A., Jr.; Doran, A.C.; Cai, B.; Fredman, G.; Tabas, I.A. Mechanisms and Consequences of Defective Efferocytosis in Atherosclerosis. Front. Cardiovasc. Med. 2018, 4, 86. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Ketelhuth, D.F.J.; Hansson, G.K. Adaptive response of T and B cells in atherosclerosis. Circ. Res. 2016, 118, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Salomon, B.L.; Potteaux, S.; Robertson, A.K.; Gourdy, P.; Zoll, J.; Merval, R.; Esposito, B.; Cohen, J.L.; Fisson, S.; et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 2006, 12, 178–180. [Google Scholar] [CrossRef]
- Sage, A.P.; Tsiantoulas, D.; Binder, C.J.; Mallat, Z. The role of B cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 180–196. [Google Scholar] [CrossRef]
- Nus, M.; Sage, A.P.; Lu, Y.; Masters, L.; Lam, B.Y.H.; Newland, S.; Weller, S.; Tsiantoulas, D.; Raffort, J.; Marcus, D.; et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat. Med. 2017, 23, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.; Newland, S.A.; Jiang, W.; Giakomidi, D.; Zhao, X.; Clement, M.; Masters, L.; Corovic, A.; Zhang, X.; Drago, F.; et al. Marginal zone B cells produce ‘natural’ atheroprotective IgM antibodies in a T cell-dependent manner. Cardiovasc. Res. 2024, 120, 318–328. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Wantha, S.; Simsekyilmaz, S.; Döring, Y.; Megens, R.T.; Mause, S.F.; Drechsler, M.; Smeets, R.; Weinandy, S.; Schreiber, F.; et al. Neutrophil-derived cathelicidin protects from neointimal hyperplasia. Sci. Transl. Med. 2011, 3, 103ra98. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. CIRT Investigators. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef]
- Ridker, P.M. Anticytokine Agents: Targeting Interleukin Signaling Pathways for the Treatment of Atherothrombosis. Circ. Res. 2019, 124, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Wang, M.; Pirruccello, J.P.; Ellinor, P.T.; Ng, K.; Kathiresan, S.; Khera, A.V. Lp(a) (Lipoprotein[a]) Concentrations and Incident Atherosclerotic Cardiovascular Disease: New Insights From a Large National Biobank. Arterioscler. Thromb. Vasc. Biol. 2021, 4, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Koschinsky, M.L.; Maher, L.; Mehta, A.; Orringer, C.E.; Santos, R.D.; Shapiro, M.D.; Saseen, J.J. Global think tank on the clinical considerations and management of lipoprotein(a): The top questions and answers regarding what clinicians need to know. Prog. Cardiovasc. Dis. 2022, 73, 32–40. [Google Scholar] [CrossRef]
- Candelli, M.; Franza, L.; Cianci, R.; Pignataro, G.; Merra, G.; Piccioni, A.; Ojetti, V.; Gasbarrini, A.; Franceschi, F. The Interplay between Helicobacter pylori and Gut Microbiota in Non-Gastrointestinal Disorders: A Special Focus on Atherosclerosis. Int. J. Mol. Sci. 2023, 24, 17520. [Google Scholar] [CrossRef]
- Luqman, A.; Hassan, A.; Ullah, M.; Naseem, S.; Ullah, M.; Zhang, L.; Din, A.U.; Ullah, K.; Ahmad, W.; Wang, G. Role of the intestinal microbiome and its therapeutic intervention in cardiovascular disorder. Front. Immunol. 2024, 15, 1321395. [Google Scholar] [CrossRef]
- Afshar, M.; Thanassoulis, G. Lipoprotein(a): New insights from modern genomics. Curr. Opin. Lipidol. 2017, 28, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, B.J.; Pelletier, W.; Kaiser, Y.; Perrot, N.; Couture, C.; Khaw, K.T.; Wareham, N.J.; Bossé, Y.; Pibarot, P.; Stroes, E.S.G.; et al. Association of Long-term Exposure to Elevated Lipoprotein(a) Levels with Parental Life Span, Chronic Disease-Free Survival, and Mortality Risk: A Mendelian Randomization Analysis. JAMA Netw. Open. 2020, 3, e200129. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Marnell, C.S.; Bick, A.; Natarajan, P. Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J. Mol. Cell. Cardiol. 2021, 161, 98–105. [Google Scholar] [CrossRef]
- Pasupuleti, S.K.; Ramdas, B.; Burns, S.S.; Palam, L.R.; Kanumuri, R.; Kumar, R.; Pandhiri, T.R.; Dave, U.P.; Yellapu, N.K.; Zhou, X.; et al. Obesity-induced inflammation exacerbates clonal hematopoiesis. J. Clin. Investig. 2023, 133, e163968. [Google Scholar] [CrossRef]
- Fuster, J.J. Clonal hematopoiesis and cardiovascular disease in cancer patients and survivors. Thromb. Res. 2022, 213 (Suppl. S1), S107–S112. [Google Scholar] [CrossRef]
- Libby, P. Interleukin-1 β as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef]
- Svensson, E.C.; Madar, A.; Campbell, C.D.; He, Y.; Sultan, M.; Healey, M.L.; Xu, H.; D’Aco, K.; Fernandez, A.; Wache-Mainier, C.; et al. TET2-Driven Clonal Hematopoiesis and Response to Canakinumab: An Exploratory Analysis of the CANTOS Randomized Clinical Trial. JAMA Cardiol. 2022, 7, 521–528. [Google Scholar] [CrossRef]
- McAninch, E.A.; Fonseca, T.L.; Poggioli, R.; Panos, A.L.; Salerno, T.A.; Deng, Y.; Li, Y.; Bianco, A.C.; Iacobellis, G. Epicardial adipose tissue has a unique transcriptome modified in severe coronary artery disease. Obesity 2015, 23, 1267–1278. [Google Scholar] [CrossRef]
- Iacobellis, G. Epicardial adipose tissue in contemporary cardiology. Nat. Rev. Cardiol. 2022, 19, 593–606. [Google Scholar] [CrossRef]
- Matter, M.A.; Paneni, F.; Libby, P.; Frantz, S.; Stähli, B.E.; Templin, C.; Mengozzi, A.; Wang, Y.J.; Kündig, T.M.; Räber, L. Inflammation in acute myocardial infarction: The good, the bad and the ugly. Eur. Heart J. 2024, 45, 89–103. [Google Scholar] [CrossRef]
- van Dam, R.M.; Li, T.; Spiegelman, D.; Franco, O.H.; Hu, F.B. Combined impact of lifestyle factors on mortality: Prospective cohort study in US women. BMJ 2008, 337, a1440. [Google Scholar] [CrossRef]
- Rasmussen-Torvik, L.J.; Shay, C.M.; Abramson, J.G.; Friedrich, C.A.; Nettleton, J.A.; Prizment, A.E.; Folsom, A.R. Ideal cardiovascular health is inversely associated with incident cancer: The Atherosclerosis Risk In Communities study. Circulation 2013, 127, 1270–1275. [Google Scholar] [CrossRef]
- van Sloten, T.T.; Climie, R.E.D.; Deraz, O.; Périer, M.C.; Valentin, E.; Fayosse, A.; Sabia, S.; Weiderpass, E.; Jouven, X.; Goldberg, M.; et al. Is the number of ideal cardiovascular health metrics in midlife associated with lower risk of cancer? Evidence from 3 European prospective cohorts. CMAJ Open 2023, 11, E774–E781. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, H.; Huang, T.; Huang, N.; Liang, H. Importance of ideal cardiovascular health metrics in the risk of colorectal cancer among people aged 50 years or older: A UK Biobank cohort study. BMJ Open 2022, 12, e059642. [Google Scholar] [CrossRef] [PubMed]
- de Boer, R.A.; Meijers, W.C.; van der Meer, P.; van Veldhuisen, D.J. Cancer and heart disease: Associations and relations. Eur. J. Heart Fail. 2019, 21, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Hasin, T.; Iakobishvili, Z.; Weisz, G. Associated Risk of Malignancy in Patients with Cardiovascular Disease: Evidence and Possible Mechanism. Am. J. Med. 2017, 130, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Hasin, T.; Gerber, Y.; Weston, S.; Jjang, R.; Killian, J.M.; Manemann, S.M.; Cerhan, J.R.; Roger, V.R. Heart Failure After Myocardial Infarction Is Associated with Increased Risk of Cancer. J. Am. Coll. Cardiol. 2016, 68, 265–271. [Google Scholar] [CrossRef]
- Koelwyn, G.J.; Aboumsallem, J.P.; Moore, K.J.; de Boer, R.A. Reverse cardio-oncology: Exploring the effects of cardiovascular disease on cancer pathogenesis. J. Mol. Cell. Cardiol. 2022, 163, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Koene, R.J.; Prizment, A.E.; Blaes, A.; Konety, S.H. Shared risk factors in cardiovascular disease and cancer. Circulation 2016, 133, 1104–1114. [Google Scholar] [CrossRef]
- Hibler, E.A.; Lloyd-Jones, D.M. Addressing the “Common Soil” of Risk Factors for Cardiovascular Disease and Cancer. JACC CardioOncol. 2021, 3, 59–61. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Narayan, V.; Thompson, E.W.; Demissei, B.; Ho, J.E.; Januzzi, J.L., Jr.; Ky, B. Mechanistic Biomarkers Informative of Both Cancer and Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 2726–2737. [Google Scholar] [CrossRef]
- Larsen, K.M.; Minaya, M.K.; Vaish, V.; Pena, M.M.O. The role of IL-33/ST2 pathway in tumorigenesis. Int. J. Mol. Sci. 2018, 19, 2676. [Google Scholar] [CrossRef]
- Aimo, A.; Januzzi, J.L., Jr.; Bayes-Genis, A.; Vergaro, G.; Sciarrone, P.; Passino, C.; Emdin, M. Clinical and Prognostic Significance of sST2 in Heart Failure: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 2193–2203. [Google Scholar] [CrossRef]
- Wang, L.; Guo, X. Molecular regulation of galectin-3 expression and therapeutic implication in cancer progression. Biomed. Pharmacother. 2016, 78, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Sun, Y.; Zhao, Y.; Zhang, W.; Yang, Z.; Gao, Y.; Cai, H.; Li, Y.; Wang, Q.; Bian, B.; et al. Prognostic value of plasma galectin-3 levels in patients with coronary heart disease and chronic heart failure. Int. Heart J. 2015, 56, 314–318. [Google Scholar] [CrossRef]
- Emmerson, P.J.; Duffin, K.L.; Chintharlapalli, S.; Wu, X. GDF15 and growth control. Front. Physiol. 2018, 9, 1712. [Google Scholar] [CrossRef]
- Wollert, K.C.; Kempf, T.; Wallentin, L. Growth differentiation factor 15 as a biomarker in cardiovascular disease. Clin. Chem. 2017, 63, 140–151. [Google Scholar] [CrossRef]
- Cardinale, D.; Sandri, M.T.; Colombo, A.; Colombo, N.; Boeri, M.; Lamantia, G.; Civelli, M.; Peccatori, F.; Martinelli, G.; Fiorentini, C.; et al. Prognostic value of troponin I in cardiac risk stratification of cancer patients undergoing high-dose chemotherapy. Circulation 2004, 109, 2749–2754. [Google Scholar] [CrossRef]
- Kumar, S.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Colby, C.; Laumann, K.; Zeldenrust, S.R.; Leung, N.; Dingli, D.; et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J. Clin. Oncol. 2012, 30, 989–995. [Google Scholar] [CrossRef]
- Pavo, N.; Raderer, M.; Hülsmann, M.; Neuhold, S.; Adlbrecht, C.; Strunk, G.; Goliasch, G.; Gisslinger, H.; Steger, G.G.; Hejna, M.; et al. Cardiovascular biomarkers in patients with cancer and their association with all-cause mortality. Heart 2015, 101, 1874–1880. [Google Scholar] [CrossRef] [PubMed]
- Widiapradja, A.; Chunduri, P.; Levick, S.P. The role of neuropeptides in adverse myocardial remodeling and heart failure. Cell. Mol. Life Sci. 2017, 74, 2019–2038. [Google Scholar] [CrossRef] [PubMed]
- Ky, B.; French, B.; Ruparel, K.; Sweitzer, N.K.; Fang, J.C.; Levy, W.C.; Sawyer, D.B.; Cappola, T.P. The vascular marker soluble fms-like tyrosine kinase 1 is associated with disease severity and adverse outcomes in chronic heart failure. J. Am. Coll. Cardiol. 2011, 58, 386–394. [Google Scholar] [CrossRef]
- Yang, F.; Jin, C.; Jiang, Y.J.; Li, J.; Di, Y.; Fu, D.L. Potential role of soluble VEGFR-1 in antiangiogenesis therapy for cancer. Expert Rev. Anticancer Ther. 2011, 11, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020, 586, 763–768, Erratum in Nature 2021, 591, E27. [Google Scholar] [CrossRef] [PubMed]
- Heyde, A.; Rohde, D.; McAlpine, C.S.; Zhang, S.; Hoyer, F.F.; Gerold, J.M.; Cheek, D.; Iwamoto, Y.; Schloss, M.J.; Vandoorne, K.; et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell 2021, 184, 1348–1361.e22. [Google Scholar] [CrossRef] [PubMed]
- Venkatesulu, B.P.; Mahadevan, L.S.; Aliru, M.L.; Yang, X.; Bodd, M.H.; Singh, P.K.; Yusuf, S.W.; Abe, J.I.; Krishnan, S. Radiation-Induced Endothelial Vascular Injury: A Review of Possible Mechanisms. JACC Basic Transl. Sci. 2018, 3, 563–572. [Google Scholar] [CrossRef]
- Desai, M.Y.; Windecker, S.; Lancellotti, P.; Bax, J.J.; Griffin, B.P.; Cahlon, O.; Johnston, D.R. Prevention, Diagnosis, and Management of Radiation-Associated Cardiac Disease: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2019, 74, 905–927. [Google Scholar] [CrossRef]
- Honaryar, M.K.; Allodji, R.; Jimenez, G.; Lapeyre, M.; Panh, L.; Camilleri, J.; Broggio, D.; Ferrières, J.; De Vathaire, F.; Jacob, S. Early Development of Atherosclerotic Plaques in the Coronary Arteries after Radiotherapy for Breast Cancer (BACCARAT Study). J. Cardiovasc. Dev. Dis. 2023, 10, 299. [Google Scholar] [CrossRef]
- Herrmann, J. Vascular toxic effects of cancer therapies. Nat. Rev. Cardiol. 2020, 17, 503–522. [Google Scholar] [CrossRef]
- Terwoord, J.D.; Beyer, A.M.; Gutterman, D.D. Endothelial dysfunction as a complication of anti-cancer therapy. Pharmacol. Ther. 2022, 237, 108116. [Google Scholar] [CrossRef]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5, eaay1863. [Google Scholar] [CrossRef]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef]
- Bu, D.X.; Tarrio, M.; Maganto-Garcia, E.; Stavrakis, G.; Tajima, G.; Lederer, J.; Jarolim, P.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1100–1107. [Google Scholar] [CrossRef]
- Lee, J.; Zhuang, Y.; Wei, X.; Shang, F.; Wang, J.; Zhang, Y.; Liu, X.; Yang, Y.; Liu, L.; Zheng, Q. Contributions of PD-1/PD-L1 pathway to interactions of myeloid DCs with T cells in atherosclerosis. J. Mol. Cell. Cardiol. 2009, 46, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.M.; Sasaki, N.; Yamashita, T.; Emoto, T.; Kasahara, K.; Mizoguchi, T.; Hayashi, T.; Yodoi, K.; Kitano, N.; Saito, T.; et al. Overexpression of Cytotoxic T-Lymphocyte-Associated Antigen-4 Prevents Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1141–1151. [Google Scholar] [CrossRef]
- Quagliariello, V.; Passariello, M.; Di Mauro, A.; Cipullo, C.; Paccone, A.; Barbieri, A.; Palma, G.; Luciano, A.; Buccolo, S.; Bisceglia, I.; et al. Immune checkpoint inhibitor therapy increases systemic SDF-1, cardiac DAMPs, Fibronectin-EDA, S100/Calgranulin, galectine-3, and NLRP3-MyD88-chemokine pathways. Front. Cardiovasc. Med. 2022, 9, 930797. [Google Scholar] [CrossRef]
- Drobni, Z.D.; Alvi, R.M.; Taron, J.; Zafar, A.; Murphy, S.P.; Rambarat, P.K.; Mosarla, R.C.; Lee, C.; Zlotoff, D.A.; Raghu, V.K.; et al. Association between immune checkpoint inhibitors with cardiovascular events and atherosclerotic plaque. Circulation 2020, 142, 2299–2311. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Guo, Y.; Li, Y.; Wang, J.; Zhou, G.; Chen, Y.H.; Nie, T.; Yang, M.; Luo, K.; Zheng, C.; et al. Immune checkpoint inhibitors in cancer: The increased risk of atherosclerotic cardiovascular disease events and progression of coronary artery calcium. BMC Med. 2024, 22, 44. [Google Scholar] [CrossRef] [PubMed]
- Matthews, A.; Stanway, S.; Farmer, R.E.; Strongman, H.; Thomas, S.; Lyon, A.R. Cardiovascular disease in female breast cancer survivors: Systematic review. BMJ 2018, 363, k3845. [Google Scholar] [CrossRef]
- Bhatia, N.; Santos, M.; Jones, L.W.; Beckman, J.A.; Penson, D.F.; Morgans, A.K.; Moslehi, J. Cardiovascular effects of androgen deprivation therapy for the treatment of prostate cancer: ABCDE steps to reduce cardiovascular disease in patients with prostate cancer. Circulation 2016, 133, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.R.; Duncan, M.S.; Morgans, A.K.; Brown, J.D.; Meijers, W.C.; Freiberg, M.S.; Salem, J.E.; Beckman, J.A.; Moslehi, J.J. Cardiovascular effects of androgen deprivation therapy in prostate cancer: Contemporary meta-analyses. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e55–e64. [Google Scholar] [CrossRef]
- Wang, Q.; Han, J.; Liang, Z.; Geng, X.; Du, Y.; Zhou, J.; Yao, W.; Xu, T. FSH Is Responsible for Androgen Deprivation Therapy-Associated Atherosclerosis in Mice by Exaggerating Endothelial Inflammation and Monocyte Adhesion. Arterioscler. Thromb. Vasc. Biol. 2024. ahead of print. [Google Scholar] [CrossRef]
- Dawber, T.R.; Meadors, G.F.; Moore, F.E., Jr. Epidemiological approaches to heart disease: The Framingham Study. Am. J. Public Health Nations Health 1951, 41, 279–281. [Google Scholar] [CrossRef]
- Dawber, T.T.; Moore, F.E.; Mann, G.V. Coronary heart disease in the Framingham study. Am. J. Public Health Nations Health 1957, 47, 4–24. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; Castelli, W.P.; Gordon, T. Cholesterol in the prediction of atherosclerotic disease. New perspectives based on the Framingham study. Ann. Intern. Med. 1979, 90, 85–91. [Google Scholar] [CrossRef]
- Mahmood, S.S.; Levy, D.; Vasan, R.S.; Wang, T.J. The Framingham Heart Study and the Epidemiology of Cardiovascular Diseases: A Historical Perspective. Lancet 2014, 383, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, R.B., Sr.; Vasan, R.S.; Pencina, M.J.; Wolf, P.A.; Cobain, M.; Massaro, J.M.; Kannel, W.B. General cardiovascular risk profile for use in primary care: The Framingham Heart Study. Circulation 2008, 117, 743–753. [Google Scholar] [CrossRef]
- SCORE2 working group and ESC Cardiovascular risk collaboration. SCORE2 risk prediction algorithms: New models to estimate 10-year risk of cardiovascular disease in Europe. Eur. Heart J. 2021, 42, 2439–2454. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Assael, F.; Ribaudo, M.C.; Zappaterreno, A.; Alessi, G.; Di Mario, U.; Leonetti, F. Epicardial fat from echocardiography: A new method for visceral adipose tissue prediction. Obes. Res. 2003, 11, 304–310. [Google Scholar] [CrossRef]
- Iacobellis, G.; Ribaudo, M.C.; Assael, F.; Vecci, E.; Tiberti, C.; Zappaterreno, A.; Di Mario, U.; Leonetti, F. Echocardiographic epicardial adipose tissue is related to anthropometric and clinical parameters of metabolic syndrome: A new indicator of cardiovascular risk. J. Clin. Endocrinol. Metab. 2003, 88, 5163–5168. [Google Scholar] [CrossRef] [PubMed]
- Lesko, L.J.; Atkinson, A.J., Jr. Use of biomarkers and surrogate endpoints in drug development and regulatory decision making: Criteria, validation, strategies. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 347–366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Biomarkers of endothelial activation and dysfunction in cardiovascular diseases. Rev. Cardiovasc. Med. 2022, 23, 73. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Z.; Liu, Y.; Yuan, L. Exosomes in atherosclerosis: Performers, bystanders, biomarkers, and therapeutic targets. Theranostics 2021, 11, 3996–4010. [Google Scholar] [CrossRef]
- Lao, K.H.; Zeng, L.; Xu, Q. Endothelial and smooth muscle cell transformation in atherosclerosis. Curr. Opin. Lipidol. 2015, 26, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Agatston, A.S.; Janowitz, W.R.; Hildner, F.J.; Zusmer, N.R.; Viamonte, M.; Detrano, R. Quantification of coronary artery calcium using ultrafast computed tomography. J. Am. Coll. Cardiol. 1990, 15, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Razavi, A.C.; Agatston, A.S.; Shaw, L.J.; De Cecco, C.N.; van Assen, M.; Sperling, L.S.; Bittencourt, M.S.; Daubert, M.A.; Nasir, K.; Blumenthal, R.S.; et al. Evolving Role of Calcium Density in Coronary Artery Calcium Scoring and Atherosclerotic Cardiovascular Disease Risk. JACC Cardiovasc. Imaging 2022, 15, 1648–1662. [Google Scholar] [CrossRef]
- Mascalchi, M.; Puliti, D.; Romei, C.; Picozzi, G.; De Liperi, A.; Diciotti, S.; Bartolucci, M.; Grazzini, M.; Vannucchi, L.; Falaschi, F. Moderate-severe coronary calcification predicts long-term cardiovascular death in CT lung cancer screening: The ITALUNG trial. Eur. J. Radiol. 2021, 145, 110040. [Google Scholar] [CrossRef]
- Robinson, J.G.; Williams, K.J.; Gidding, S.; Borén, J.; Tabas, I.; Fisher, E.A.; Packard, C.; Pencina, M.; Fayad, Z.A.; Mani, V.; et al. Eradicating the Burden of Atherosclerotic Cardiovascular Disease by Lowering Apolipoprotein B Lipoproteins Earlier in Life. J. Am. Heart Assoc. 2018, 7, e009778. [Google Scholar] [CrossRef] [PubMed]
- Pérez de Isla, L.; Díaz-Díaz, J.L.; Romero, M.J.; Muñiz-Grijalvo, O.; Mediavilla, J.D.; Argüeso, R.; Sánchez Muñoz-Torrero, J.F.; Rubio, P.; Álvarez-Baños, P.; Ponte, P.; et al. Alirocumab and coronary atherosclerosis in asymptomatic patients with familial hypercholesterolemia:the ARCHITECT study. Circulation 2023, 147, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J. Eradicating Atherosclerotic Events by Targeting Early Subclinical Disease: It Is Time to Retire the Therapeutic Paradigm of Too Much, Too Late. Arterioscler. Thromb. Vasc. Biol. 2024, 44, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Selvam, P.V.; Grandhi, G.R.; Leucker, T.M.; Arbab-Zadeh, A.; Gulati, M.; Blumenthal, R.S.; Whelton, S.P. Recent advances in cardiovascular risk assessment: The added value of non-invasive anatomic imaging. J. Cardiovasc. Comput. Tomogr. 2024, 18, 113–119. [Google Scholar] [CrossRef]
- Ansari, S.; Pourafkari, L.; Kinninger, A.; Manubolu, V.; Budoff, M.J. Risk stratifying individuals with zero, minimal, and mild coronary artery calcium for cardiovascular disease by determining coronary plaque burden. J. Cardiovasc. Comput. Tomogr. 2024, 18, 137–141. [Google Scholar] [CrossRef]
- Grant, J.K.; Orringer, C.E. Coronary and Extra-coronary Subclinical Atherosclerosis to Guide Lipid-Lowering Therapy. Curr. Atheroscler. Rep. 2023, 25, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.K.; Bhatia, H.S.; Allen, T.S.; Grainger, T.; Pouncey, A.L.; Dichek, D.; Virmani, R.; Golledge, J.; Allison, M.A.; Powell, J.T. Assessment of Subclinical Atherosclerosis in Asymptomatic People In Vivo: Measurements Suitable for Biomarker and Mendelian Randomization Studies. Arterioscler. Thromb. Vasc. Biol. 2024, 44, 24–47. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, R.P.; Fuster, V.; Badimon, J.J.; Fisher, E.A.; Fayad, Z.A. MRI and characterization of atherosclerotic plaque: Emerging applications and molecular imaging. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1065–1074. [Google Scholar] [CrossRef]
- Wald, L.L.; McDaniel, P.C.; Witzel, T.; Stockmann, J.P.; Cooley, C.Z. Low-cost and portable MRI. J. Magn. Reson. Imaging 2020, 52, 686–696. [Google Scholar] [CrossRef]
- Senders, M.L.; Calcagno, C.; Tawakol, A.; Nahrendorf, M.; Mulder, W.J.M.; Fayad, Z.A. PET/MR imaging of inflammation in atherosclerosis. Nat. Biomed. Eng. 2023, 7, 202–220. [Google Scholar] [CrossRef] [PubMed]
- Gurgoglione, F.L.; Denegri, A.; Russo, M.; Calvieri, C.; Benatti, G.; Niccoli, G. Intracoronary Imaging of Coronary Atherosclerotic Plaque: From Assessment of Pathophysiological Mechanisms to Therapeutic Implication. Int. J. Mol. Sci. 2023, 24, 5155. [Google Scholar] [CrossRef] [PubMed]
- Channon, K.M.; Newby, D.E.; Nicol, E.D.; Deanfield, J. Cardiovascular computed tomography imaging for coronary artery disease risk: Plaque, flow and fat. Heart 2022, 108, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, A.S.; Sanna, F.; Sabharwal, N.; Thomas, S.; Oikonomou, E.K.; Herdman, L.; Margaritis, M.; Shirodaria, C.; Kampoli, A.M.; Akoumianakis, I.; et al. Detecting human coronary inflammation by imaging perivascular fat. Sci. Transl. Med. 2017, 9, eaal2658. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.K.; Antonopoulos, A.S.; Schottlander, D.; Marwan, M.; Mathers, C.; Tomlins, P.; Siddique, M.; Klüner, L.V.; Shirodaria, C.; Mavrogiannis, M.C.; et al. Standardized measurement of coronary inflammation using cardiovascular computed tomography: Integration in clinical care as a prognostic medical device. Cardiovasc. Res. 2021, 117, 2677–2690. [Google Scholar] [CrossRef] [PubMed]
| Players | Phenotypes | “Good” Behavior | “Bad” Behavior | “Ugly” Behavior |
|---|---|---|---|---|
| Endothelial cells (EC) secrete vasoactive substances (e.g., endothelin-1, nitric oxide, etc.) affecting vascular smooth muscle cells, platelets, and white blood cells. In atherosclerosis ECs are activated by trapped lipoproteins. Refs. [51,52,58,61] | Heterogeneous ECs to suit heterogeneous endothelium, capable of angiogenic and metabolic switch. EC may exhibit trained immunity. | Healthy ECs are excellent sensors of the hemodynamic forces of blood flow. ECs have a pivotal role in endothelial resilience, the ability to cope with many stressors or challenges (exposomes). | In the early stages of atherosclerosis, high levels of ox-LDL and remnants of tryglyceride-rich lipoproteins gain access to the subendothelial space, eliciting a danger signal that activates the NLRP3-inflammasome in innate immune cells and the inflammatory pathways, leading to endothelial dysfunction. | Inflammation begets inflammation, and this vicious circle leads to advanced stages of atherosclerosis. |
| Pericytes: perivascular cells derived from human pluripotent stem cells (HPSCs) and located around ECs. Refs. [62,63,64,65] | Due to their common origin from HPSCs, pericytes can differentiate into other cells of the mesenchymal lineage such as monocytes. | Support vascular stability by preventing matrix degradation; play a relevant role in differentiation, angiogenesis, regeneration, immunomodulation, and blood flow regulation. | Dysmetabolic-driven alteration of pericytes in diabetes contributes to plaque formation. | In advanced atherosclerosis, pericytes are involved in plaque neovascularization, inflammation, and vascular calcification processes. |
| Vascular smooth muscle cells exhibit a contractile phenotype in the healthy arterial wall. If stimulated by ECs through PDGF-BB and TNF-α, they can switch to a synthetic phenotype that increases the production of ECM, exosome, and proinflammatory cytokines. Refs. [66,67,68,69] | Various phenotypes with beneficial and detrimental role in atherogenesis. | VSMC stabilize fibrous cap in advanced atherosclerosis and produce ECM (fibroblast-like features). | Lipid-induced transformation in macrophage-like and foam cell-like phenotypes, exhibiting proinflammatory behavior and increasing vulnerability to plaque (macrophage-like features). | |
| Dendritic cells (DCs) bridge the innate and adaptive immune response involved in the scenario of evolving plaque. Refs. [66,70] | Preclinical studies: proatherogenic and anti-atherogenic function. | In Ldlr−/− mice fed with high-fat diet, autophagy disruption in DCs limits atherogenesis. | In humans, dendritic cell numbers are connected to vulnerability of atherosclerotic plaque. | |
| Monocytes in a homeostatic state populate blood, bone marrow, and spleen. Refs. [71,72] | Classical vs. non-classical monocytes. Monocytes may be rewired by metabolic stimuli (e.g., ox-LDL) to become a “trained” immune cell. | Non-classical monocytes are “on patrol” to maintain vascular endothelium. Recruited monocytes also have an impact on atherosclerosis regression. | Classical monocytes (CD14+ CD16− in humans, Ly6Chigh in mice) recruited to atherosclerotic plaque exhibit phenotypic heterogeneity, differentiating into dendritic cells and macrophages. | In preclinical studies in mice, splenic classical monocytes (Ly6Chigh) increase plaque and its instability. In humans and mice, monocytosis is associated with increased severity of atherosclerosis. |
| Macrophages Refs. [73,74,75,76,77] | M1 macrophages, M2 macrophages. Macrophages may be upgraded in “trained” immune cells. | M2 macrophages clear lipids and secrete anti-inflammatory factors (e.g., Il-10 and collagen). Recruited monocyte-derived macrophages remove apoptotic cells (efferocytosis), eliciting the secretion of anti-inflammatory cytokines and hampering the progression of atherosclerotic plaque. | M1 macrophages favor the accumulation of intracellular lipids and increase the secretion of proinflammatory factors (e.g., TNF-α IL-1 β and IL-6). Macrophages may appear as foamy cells. | M1: when reprogrammed macrophages lose their efferocytic ability, apoptotic cells undergo post-apoptotic necrosis, releasing proinflammatory mediators and providing a boost to the progression of plaque. |
| T cells have a role in all stages of atherosclerosis. CD4+T cells are prevalent in mouse atherosclerotic plaque and exhibit a proinflammatory atherogenic phenotype. Refs. [78,79,80] | Atheroprotective phenotype (T reg) and proatherogenic phenotype (T helper 1). | T reg can silence inflammation through the elaboration of the immunomodulatory cytokine transforming growth factor beta and by secreting IL-10 (preclinical studies). | Proinflammatory phenotype (T helper 1 cells): activated T cells have a direct role in the arterial wall or help B cells in antibody production. | |
| B cells are classified into B1 cells (subdivided into B1a and B1b cells), mainly produced in the fetal liver, and B2 cells (subdivided in T1 and T2 marginal zone progenitor). Refs. [81,82,83] | B1 cells are atheroprotective in mice. When challenged by a high-fat/high-cholesterol diet, marginal zone B cells switch to an atheroprotective programme mediated by Atf3, Nr4a1, and Pdl1. | B1 cells exhibit atheroprotective behavior in mice due to the production of IgM antibodies that block the uptake of oxLDL by macrophages in atherosclerotic lesions. | B2 cells exhibit proatherogenic behavior through antibody responses that stimulate adaptive immunity. | |
| Neutrophils have a role in all stages of atherosclerosis Refs. [84,85,86] | Proatherogenic phenotype. Reparative phenotype. | Reparative phenotype exhibited during thrombotic events when neutrophils promote endothelial repair and angiogenesis (arterial healing). | Neutrophils secrete ROS, increasing the permeability of ECs and inducing NLRP3 inflammasome. Neutrophils attract monocytes and can activate macrophages via extrusion of their NETs. | NET formation that stimulates the NLRP3 inflammasome and produces IL-1β (preclinical study). In this scenario, NLRP3 inflammasome requires a second hit to be fully activated. Defective efferocytosis that leads to accumulation of DAMPs. |
| Safe and easy to measure; |
| Reliable; |
| Sensitive and specific; |
| Capable of discriminating healthy patients from unhealthy patients; |
| Able to predict future cardiovascular events; |
| Should express early in the disease progression; |
| Can be applied to diagnosis, staging, and prognosis; |
| Cost efficient for follow-up; |
| Modifiable with treatment. |
| Experimental | |
|---|---|
| Biomarker | Atherosclerosis |
| IL-1 β | Anti IL-1 β-L:  late athero late atheroLOF IL-1 β: athero |
| IL-1 α | Anti IL-1 α: early athero LOF IL-1 α: athero |
| IL-6/IL-6 | LOF IL-6:  athero atherorIL-6: athero |
| IL-10 | GOF IL-10: athero |
| T reg | athero |
| Clinical | |
| Biomarker | Atherosclerosis |
| PMN | PMN late lesions and atherothrombosis |
| IL-1 β | Anti IL-1β-L: MACE |
| IL-6 | Anti IL-6 L: inflammation and thrombosis |
decrease; increase. Modified from Matter MA et al. [103].Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallucci, G.; Turazza, F.M.; Inno, A.; Canale, M.L.; Silvestris, N.; Farì, R.; Navazio, A.; Pinto, C.; Tarantini, L. Atherosclerosis and the Bidirectional Relationship between Cancer and Cardiovascular Disease: From Bench to Bedside—Part 1. Int. J. Mol. Sci. 2024, 25, 4232. https://doi.org/10.3390/ijms25084232
Gallucci G, Turazza FM, Inno A, Canale ML, Silvestris N, Farì R, Navazio A, Pinto C, Tarantini L. Atherosclerosis and the Bidirectional Relationship between Cancer and Cardiovascular Disease: From Bench to Bedside—Part 1. International Journal of Molecular Sciences. 2024; 25(8):4232. https://doi.org/10.3390/ijms25084232
Chicago/Turabian StyleGallucci, Giuseppina, Fabio Maria Turazza, Alessandro Inno, Maria Laura Canale, Nicola Silvestris, Roberto Farì, Alessandro Navazio, Carmine Pinto, and Luigi Tarantini. 2024. "Atherosclerosis and the Bidirectional Relationship between Cancer and Cardiovascular Disease: From Bench to Bedside—Part 1" International Journal of Molecular Sciences 25, no. 8: 4232. https://doi.org/10.3390/ijms25084232
APA StyleGallucci, G., Turazza, F. M., Inno, A., Canale, M. L., Silvestris, N., Farì, R., Navazio, A., Pinto, C., & Tarantini, L. (2024). Atherosclerosis and the Bidirectional Relationship between Cancer and Cardiovascular Disease: From Bench to Bedside—Part 1. International Journal of Molecular Sciences, 25(8), 4232. https://doi.org/10.3390/ijms25084232