
Understanding the Tumor Microenvironment in Melanoma Patients with In-Transit Metastases and Its Impacts on Immune Checkpoint Immunotherapy Responses
Abstract
1. Overview of Melanoma In-Transit Metastases (ITM)
1.1. Melanoma Biology and Staging
1.2. ITM of Melanoma
1.3. Treatments for Melanoma and ITM
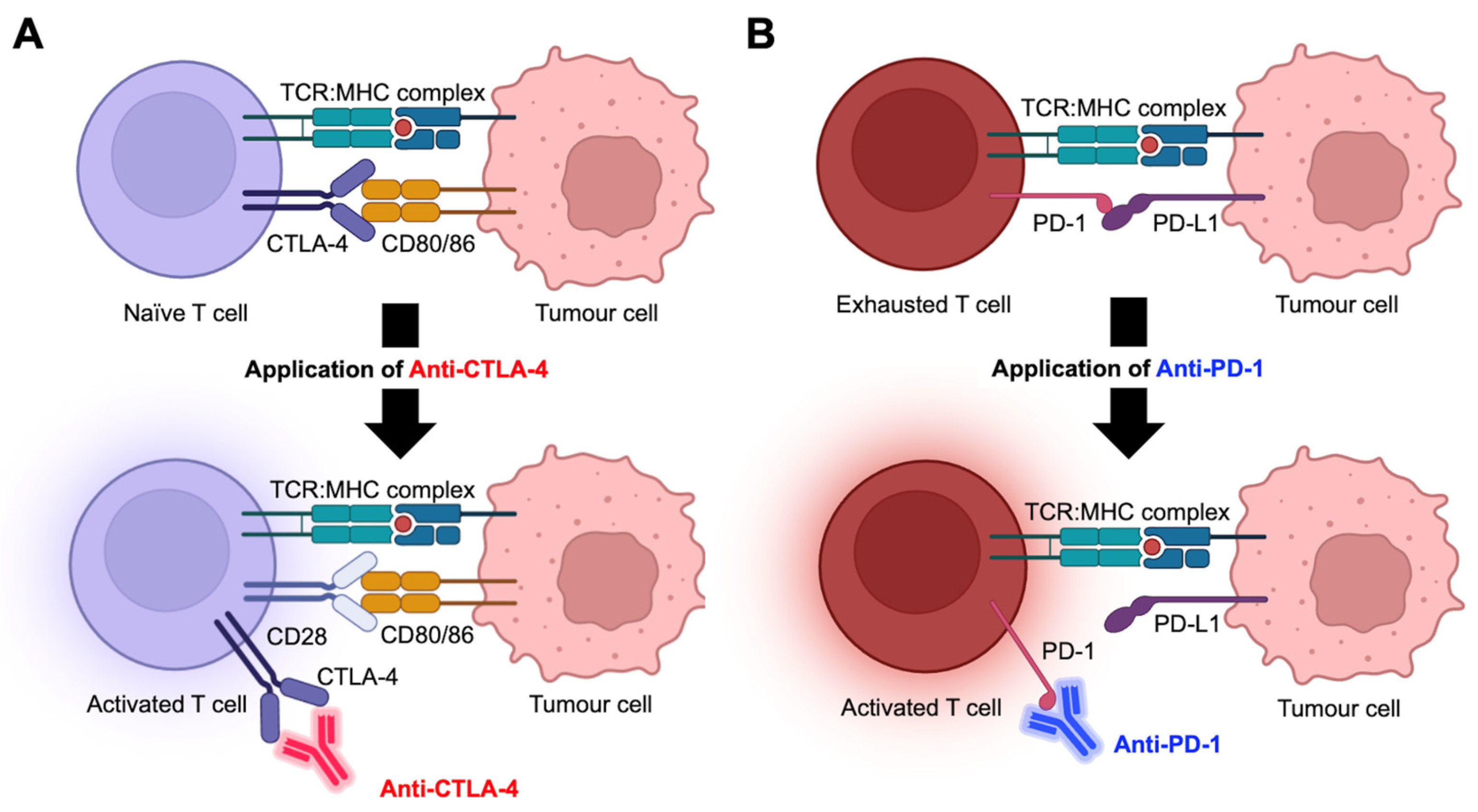
2. Immunotherapy: Immune Checkpoint Inhibitors (ICIs)
2.1. Types of ICIs
2.2. Biological Factors Associated with Response to Immunotherapy
2.3. Biological Factors Associated with Resistance to Immunotherapy
2.4. Application of Immunotherapy in Melanoma ITM
3. Tumor Microenvironment of Advanced Melanoma
3.1. Immune Characterization of Melanoma Microenvironment
3.1.1. Adaptive Immunity
3.1.2. Innate Immunity
3.2. Cell–Cell Communications in Melanoma Microenvironment
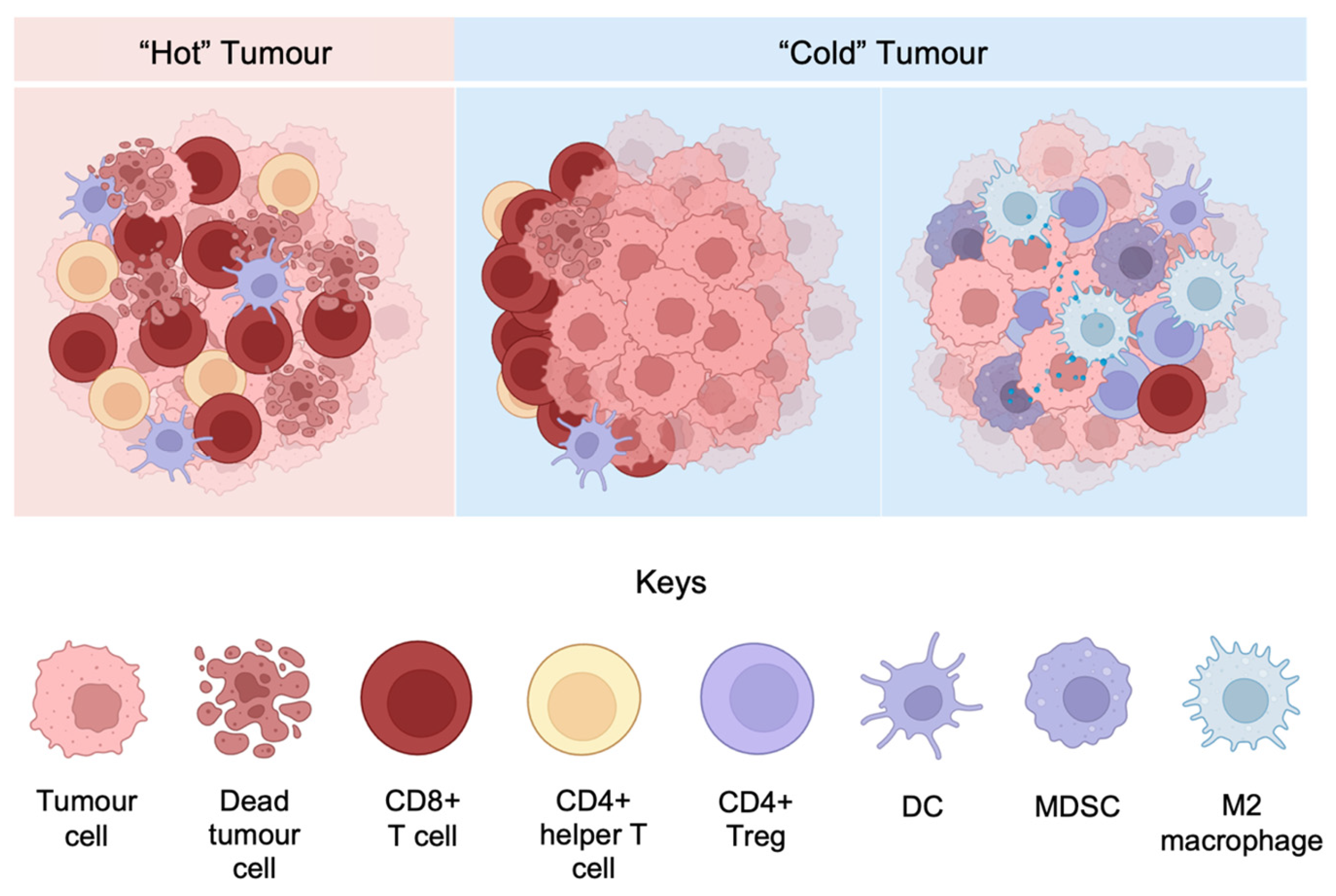
3.3. Inflamed and Non-Inflamed Phenotypes of Melanoma Microenvironment
3.4. Melanoma Microenvironment in the Spatial Context
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arnold, M.; Singh, D.; Laversanne, M.; Vignat, J.; Vaccarella, S.; Meheus, F.; Cust, A.E.; de Vries, E.; Whiteman, D.C.; Bray, F. Global Burden of Cutaneous Melanoma in 2020 and Projections to 2040. JAMA Dermatol. 2022, 158, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef]
- Crocetti, E.; Mallone, S.; Robsahm, T.E.; Gavin, A.; Agius, D.; Ardanaz, E.; Lopez, M.C.; Innos, K.; Minicozzi, P.; Borgognoni, L.; et al. Survival of patients with skin melanoma in Europe increases further: Results of the EUROCARE-5 study. Eur. J. Cancer 2015, 51, 2179–2190. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghi, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy. Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef]
- Becker, S.W. Black lesions of the skin. Calif. Med. 1958, 88, 228–233. [Google Scholar]
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef] [PubMed]
- Elder, D.E.; Barnhill, R.L.; Bastian, B.C.; Cook, M.G.; de la Fouchardiere, A.; Gerami, P.; Lazar, A.J.; Massi, D.; Mihm, M.C.J.; Nagore, E. Melanocytic tumour classification and the pathway concept of melanoma pathogenesis. World Health Organ. Classif. Tumours 2018, 11, 66–71. [Google Scholar]
- Long, G.V.; Swetter, S.M.; Menzies, A.M.; Gershenwald, J.E.; Scolyer, R.A. Cutaneous melanoma. Lancet 2023, 402, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Keung, E.Z.; Gershenwald, J.E. The eighth edition American Joint Committee on Cancer (AJCC) melanoma staging system: Implications for melanoma treatment and care. Expert Rev. Anticancer Ther. 2018, 18, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Faries, M.B.; Thompson, J.F.; Cochran, A.J.; Andtbacka, R.H.; Mozzillo, N.; Zager, J.S.; Jahkola, T.; Bowles, T.L.; Testori, A.; Beitsch, P.D.; et al. Completion Dissection or Observation for Sentinel-Node Metastasis in Melanoma. N. Engl. J. Med. 2017, 376, 2211–2222. [Google Scholar] [CrossRef] [PubMed]
- Read, R.L.; Haydu, L.; Saw, R.P.; Quinn, M.J.; Shannon, K.; Spillane, A.J.; Stretch, J.R.; Scolyer, R.A.; Thompson, J.F. In-transit melanoma metastases: Incidence, prognosis, and the role of lymphadenectomy. Ann. Surg. Oncol. 2015, 22, 475–481. [Google Scholar] [CrossRef]
- Sandru, A.; Voinea, S.; Panaitescu, E.; Blidaru, A. Survival rates of patients with metastatic malignant melanoma. J. Med. Life 2014, 7, 572–576. [Google Scholar] [PubMed]
- Weide, B.; Faller, C.; Buttner, P.; Pflugfelder, A.; Leiter, U.; Eigentler, T.K.; Bauer, J.; Forschner, A.; Meier, F.; Garbe, C. Prognostic factors of melanoma patients with satellite or in-transit metastasis at the time of stage III diagnosis. PLoS ONE 2013, 8, e63137. [Google Scholar] [CrossRef] [PubMed]
- Karakousis, C.P.; Choe, K.J.; Holyoke, E.D. Biologic behavior and treatment of intransit metastasis of melanoma. Surg. Gynecol. Obstet. 1980, 150, 29–32. [Google Scholar] [PubMed]
- Holmberg, C.J.; Ny, L.; Hieken, T.J.; Block, M.S.; Carr, M.J.; Sondak, V.K.; Ortenwall, C.; Katsarelias, D.; Dimitriou, F.; Menzies, A.M.; et al. The efficacy of immune checkpoint blockade for melanoma in-transit with or without nodal metastases—A multicenter cohort study. Eur. J. Cancer 2022, 169, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Heenan, P.J.; Ghaznawie, M. The pathogenesis of local recurrence of melanoma at the primary excision site. Br. J. Plast. Surg. 1999, 52, 209–213. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, W.H. Melanoma: Margins for error—Another view. ANZ J. Surg. 2002, 72, 304–306. [Google Scholar] [CrossRef] [PubMed]
- Wilmott, J.; Haydu, L.; Bagot, M.; Zhang, Y.; Jakrot, V.; McCarthy, S.; Lugassy, C.; Thompson, J.; Scolyer, R.; Barnhill, R. Angiotropism is an independent predictor of microscopic satellites in primary cutaneous melanoma. Histopathology 2012, 61, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Nan Tie, E.; Lai-Kwon, J.; Rtshiladze, M.A.; Na, L.; Bozzi, J.; Read, T.; Atkinson, V.; Au-Yeung, G.; Long, G.V.; McArthur, G.A.; et al. Efficacy of immune checkpoint inhibitors for in-transit melanoma. J. Immunother. Cancer 2020, 8, e000440. [Google Scholar] [CrossRef] [PubMed]
- Read, T.A.; Smith, A.; Thomas, J.; David, M.; Foote, M.; Wagels, M.; Barbour, A.; Smithers, B.M. Intralesional PV-10 for the treatment of in-transit melanoma metastases-Results of a prospective, non-randomized, single center study. J. Surg. Oncol. 2018, 117, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Zaremba, A.; Philip, M.; Hassel, J.C.; Glutsch, V.; Fiocco, Z.; Loquai, C.; Rafei-Shamsabadi, D.; Gutzmer, R.; Utikal, J.; Haferkamp, S.; et al. Clinical characteristics and therapy response in unresectable melanoma patients stage IIIB-IIID with in-transit and satellite metastases. Eur. J. Cancer 2021, 152, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, T.M.; Ross, M.I.; Johnson, M.M.; Schacherer, C.W.; McClain, D.M.; Mansfield, P.F.; Lee, J.E.; Cormier, J.N.; Gershenwald, J.E. Predictors and natural history of in-transit melanoma after sentinel lymphadenectomy. Ann. Surg. Oncol. 2005, 12, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Joyce, D.; Skitzki, J.J. Surgical Management of Primary Cutaneous Melanoma. Surg. Clin. N. Am. 2020, 100, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.K.; Lourdault, K.; Ostad, T.; Stern, S.; Essner, R. Is therapeutic lymph node dissection of value for lymph node recurrence in melanoma? Am. J. Surg. 2024, 228, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Rozeman, E.A.; Menzies, A.M.; van Akkooi, A.C.J.; Adhikari, C.; Bierman, C.; van de Wiel, B.A.; Scolyer, R.A.; Krijgsman, O.; Sikorska, K.; Eriksson, H.; et al. Identification of the optimal combination dosing schedule of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma (OpACIN-neo): A multicentre, phase 2, randomised, controlled trial. Lancet Oncol. 2019, 20, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Versluis, J.M.; Menzies, A.M.; Sikorska, K.; Rozeman, E.A.; Saw, R.P.M.; van Houdt, W.J.; Eriksson, H.; Klop, W.M.C.; Ch’ng, S.; van Thienen, J.V.; et al. Survival update of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma in the OpACIN and OpACIN-neo trials. Ann. Oncol. 2023, 34, 420–430. [Google Scholar] [CrossRef]
- Patel, S.P.; Othus, M.; Chen, Y.; Wright, G.P., Jr.; Yost, K.J.; Hyngstrom, J.R.; Hu-Lieskovan, S.; Lao, C.D.; Fecher, L.A.; Truong, T.G.; et al. Neoadjuvant-Adjuvant or Adjuvant-Only Pembrolizumab in Advanced Melanoma. N. Engl. J. Med. 2023, 388, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Nan Tie, E.; Henderson, M.A.; Gyorki, D.E. Management of in-transit melanoma metastases: A review. ANZ J. Surg. 2019, 89, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Read, T.; Lonne, M.; Sparks, D.S.; David, M.; Wagels, M.; Schaider, H.; Soyer, H.P.; Smithers, B.M. A systematic review and meta-analysis of locoregional treatments for in-transit melanoma. J. Surg. Oncol. 2019, 119, 887–896. [Google Scholar] [CrossRef]
- Thompson, J.F.; Agarwala, S.S.; Smithers, B.M.; Ross, M.I.; Scoggins, C.R.; Coventry, B.J.; Neuhaus, S.J.; Minor, D.R.; Singer, J.M.; Wachter, E.A. Phase 2 Study of Intralesional PV-10 in Refractory Metastatic Melanoma. Ann. Surg. Oncol. 2015, 22, 2135–2142. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O‘Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Del Vecchio, M.; Mandala, M.; Gogas, H.; Arance, A.M.; Dalle, S.; Cowey, C.L.; Schenker, M.; Grob, J.J.; Chiarion-Sileni, V.; et al. Adjuvant nivolumab versus ipilimumab in resected stage IIIB-C and stage IV melanoma (CheckMate 238): 4-year results from a multicentre, double-blind, randomised, controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Hodi, F.S.; Thompson, J.A.; McDermott, D.F.; Hwu, W.J.; Lawrence, D.P.; Dawson, N.A.; Wong, D.J.; Bhatia, S.; James, M.; et al. Pembrolizumab Plus Pegylated Interferon alfa-2b or Ipilimumab for Advanced Melanoma or Renal Cell Carcinoma: Dose-Finding Results from the Phase Ib KEYNOTE-029 Study. Clin. Cancer Res. 2018, 24, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Callahan, M.K.; Kluger, H.; Postow, M.A.; Segal, N.H.; Lesokhin, A.; Atkins, M.B.; Kirkwood, J.M.; Krishnan, S.; Bhore, R.; Horak, C.; et al. Nivolumab Plus Ipilimumab in Patients With Advanced Melanoma: Updated Survival, Response, and Safety Data in a Phase I Dose-Escalation Study. J. Clin. Oncol. 2018, 36, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-Year Overall Survival for Patients With Advanced Non-Small-Cell Lung Cancer Treated With Pembrolizumab: Results From the Phase I KEYNOTE-001 Study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, C.; Meyer, N.; Mortier, L.; Marquez-Rodas, I.; Robert, C.; Rutkowski, P.; Menzies, A.M.; Eigentler, T.; Ascierto, P.A.; Smylie, M.; et al. Evaluation of Two Dosing Regimens for Nivolumab in Combination With Ipilimumab in Patients With Advanced Melanoma: Results From the Phase IIIb/IV CheckMate 511 Trial. J. Clin. Oncol. 2019, 37, 867–875. [Google Scholar] [CrossRef]
- Perets, R.; Bar, J.; Rasco, D.W.; Ahn, M.J.; Yoh, K.; Kim, D.W.; Nagrial, A.; Satouchi, M.; Lee, D.H.; Spigel, D.R.; et al. Safety and efficacy of quavonlimab, a novel anti-CTLA-4 antibody (MK-1308), in combination with pembrolizumab in first-line advanced non-small-cell lung cancer. Ann. Oncol. 2021, 32, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Rupp, T.; Genest, L.; Babin, D.; Legrand, C.; Hunault, M.; Froget, G.; Castagne, V. Anti-CTLA-4 and anti-PD-1 immunotherapies repress tumor progression in preclinical breast and colon model with independent regulatory T cells response. Transl. Oncol. 2022, 20, 101405. [Google Scholar] [CrossRef] [PubMed]
- Willsmore, Z.N.; Coumbe, B.G.T.; Crescioli, S.; Reci, S.; Gupta, A.; Harris, R.J.; Chenoweth, A.; Chauhan, J.; Bax, H.J.; McCraw, A.; et al. Combined anti-PD-1 and anti-CTLA-4 checkpoint blockade: Treatment of melanoma and immune mechanisms of action. Eur. J. Immunol. 2021, 51, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255.e6. [Google Scholar] [CrossRef] [PubMed]
- Alqurashi, Y.E. Lymphocyte-activation gene 3 (LAG-3) as a promising immune checkpoint in cancer immunotherapy: From biology to the clinic. Pathol. Res. Pract. 2024, 254, 155124. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutierrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Ganguli, N.; Kumari, P.; Dash, S.; Samanta, D. Molecular and structural basis of TIGIT: Nectin-4 interaction, a recently discovered pathway crucial for cancer immunotherapy. Biochem. Biophys. Res. Commun. 2023, 677, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Tian, Y.; Peng, J.; Chen, C.; Peng, X.; Zhao, F.; Li, Z.; Li, M.; Zhao, F.; Sheng, X.; et al. Identification of a small-molecule Tim-3 inhibitor to potentiate T cell-mediated antitumor immunotherapy in preclinical mouse models. Sci. Transl. Med. 2023, 15, eadg6752. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A.; Sahasranaman, S.; Budha, N. Targeting TIGIT for Immunotherapy of Cancer: Update on Clinical Development. Biomedicines 2021, 9, 1277. [Google Scholar] [CrossRef]
- Zhao, L.; Cheng, S.; Fan, L.; Zhang, B.; Xu, S. TIM-3: An update on immunotherapy. Int. Immunopharmacol. 2021, 99, 107933. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Fast, E.; Liu, C.L.; Muftuoglu, Y.; Sworder, B.J.; Diehn, M.; Levy, R.; et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol. 2019, 37, 1332–1343. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.M.; Cheon, I.S.; Wu, Y.; Li, C.; Wang, Z.; Gao, X.; Chen, Y.; Takahashi, Y.; Fu, Y.X.; Dent, A.L.; et al. Tissue-resident CD4(+) T helper cells assist the development of protective respiratory B and CD8(+) T cell memory responses. Sci. Immunol. 2021, 6, eabb6852. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Fu, L.Q.; Du, W.L.; Cai, M.H.; Yao, J.Y.; Zhao, Y.Y.; Mou, X.Z. The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell Immunol. 2020, 353, 104119. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; Loo, K.; Pauli, M.L.; Sanchez-Rodriguez, R.; Sandoval, P.M.; Taravati, K.; Tsai, K.; Nosrati, A.; Nardo, L.; Alvarado, M.D.; et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J. Clin. Investig. 2016, 126, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Dongye, S.; Liu, X.; Xu, X.; Wang, L.; Jin, C.Q.; Yao, M.; Gong, Z.; Jiang, D.; Zhang, K.; et al. Immunotherapy of targeting MDSCs in tumor microenvironment. Front. Immunol. 2022, 13, 990463. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Li, Y.; Tan, J.; Xu, L.; Li, Y. Targeting LAG-3, TIM-3, and TIGIT for cancer immunotherapy. J. Hematol. Oncol. 2023, 16, 101. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Balatoni, T.; Mohos, A.; Papp, E.; Sebestyen, T.; Liszkay, G.; Olah, J.; Varga, A.; Lengyel, Z.; Emri, G.; Gaudi, I.; et al. Tumor-infiltrating immune cells as potential biomarkers predicting response to treatment and survival in patients with metastatic melanoma receiving ipilimumab therapy. Cancer Immunol. Immunother. 2018, 67, 141–151. [Google Scholar] [CrossRef]
- Hamid, O.; Molinero, L.; Bolen, C.R.; Sosman, J.A.; Munoz-Couselo, E.; Kluger, H.M.; McDermott, D.F.; Powderly, J.D.; Sarkar, I.; Ballinger, M.; et al. Safety, Clinical Activity, and Biological Correlates of Response in Patients with Metastatic Melanoma: Results from a Phase I Trial of Atezolizumab. Clin. Cancer Res. 2019, 25, 6061–6072. [Google Scholar] [CrossRef] [PubMed]
- Uryvaev, A.; Passhak, M.; Hershkovits, D.; Sabo, E.; Bar-Sela, G. The role of tumor-infiltrating lymphocytes (TILs) as a predictive biomarker of response to anti-PD1 therapy in patients with metastatic non-small cell lung cancer or metastatic melanoma. Med. Oncol. 2018, 35, 25. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.F.; Wei, W.; Smithy, J.W.; Acs, B.; Toki, M.I.; Blenman, K.R.M.; Zelterman, D.; Kluger, H.M.; Rimm, D.L. Multiplex Quantitative Analysis of Tumor-Infiltrating Lymphocytes and Immunotherapy Outcome in Metastatic Melanoma. Clin. Cancer Res. 2019, 25, 2442–2449. [Google Scholar] [CrossRef] [PubMed]
- Zander, R.; Schauder, D.; Xin, G.; Nguyen, C.; Wu, X.; Zajac, A.; Cui, W. CD4(+) T Cell Help Is Required for the Formation of a Cytolytic CD8(+) T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 2019, 51, 1028–1042.e4. [Google Scholar] [CrossRef]
- Sledzinska, A.; Vila de Mucha, M.; Bergerhoff, K.; Hotblack, A.; Demane, D.F.; Ghorani, E.; Akarca, A.U.; Marzolini, M.A.V.; Solomon, I.; Vargas, F.A.; et al. Regulatory T Cells Restrain Interleukin-2- and Blimp-1-Dependent Acquisition of Cytotoxic Function by CD4(+) T Cells. Immunity 2020, 52, 151–166.e6. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013.e20. [Google Scholar] [CrossRef] [PubMed]
- Buttner, R.; Longshore, J.W.; Lopez-Rios, F.; Merkelbach-Bruse, S.; Normanno, N.; Rouleau, E.; Penault-Llorca, F. Implementing TMB measurement in clinical practice: Considerations on assay requirements. ESMO Open 2019, 4, e000442. [Google Scholar] [CrossRef] [PubMed]
- Fietz, S.; Zarbl, R.; Niebel, D.; Posch, C.; Brossart, P.; Gielen, G.H.; Strieth, S.; Pietsch, T.; Kristiansen, G.; Bootz, F.; et al. CTLA4 promoter methylation predicts response and progression-free survival in stage IV melanoma treated with anti-CTLA-4 immunotherapy (ipilimumab). Cancer Immunol. Immunother. 2021, 70, 1781–1788. [Google Scholar] [CrossRef]
- Goltz, D.; Gevensleben, H.; Vogt, T.J.; Dietrich, J.; Golletz, C.; Bootz, F.; Kristiansen, G.; Landsberg, J.; Dietrich, D. CTLA4 methylation predicts response to anti-PD-1 and anti-CTLA-4 immunotherapy in melanoma patients. JCI Insight 2018, 3, e96793. [Google Scholar] [CrossRef] [PubMed]
- Vilain, R.E.; Menzies, A.M.; Wilmott, J.S.; Kakavand, H.; Madore, J.; Guminski, A.; Liniker, E.; Kong, B.Y.; Cooper, A.J.; Howle, J.R.; et al. Dynamic Changes in PD-L1 Expression and Immune Infiltrates Early During Treatment Predict Response to PD-1 Blockade in Melanoma. Clin. Cancer Res. 2017, 23, 5024–5033. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Oida, T.; Zhang, X.; Goto, M.; Hachimura, S.; Totsuka, M.; Kaminogawa, S.; Weiner, H.L. CD4+CD25− T cells that express latency-associated peptide on the surface suppress CD4+CD45RBhigh-induced colitis by a TGF-beta-dependent mechanism. J. Immunol. 2003, 170, 2516–2522. [Google Scholar] [CrossRef] [PubMed]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Zappasodi, R. A decade of checkpoint blockade immunotherapy in melanoma: Understanding the molecular basis for immune sensitivity and resistance. Nat. Immunol. 2022, 23, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, C.; Sevko, A.; Jiang, H.; Lichtenberger, R.; Reith, M.; Tarnanidis, K.; Holland-Letz, T.; Umansky, L.; Beckhove, P.; Sucker, A.; et al. Myeloid Cells and Related Chronic Inflammatory Factors as Novel Predictive Markers in Melanoma Treatment with Ipilimumab. Clin. Cancer Res. 2015, 21, 5453–5459. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Zha, H.; Wang, X.; Zhu, Y.; Chen, D.; Han, X.; Yang, F.; Gao, J.; Hu, C.; Shu, C.; Feng, Y.; et al. Intracellular Activation of Complement C3 Leads to PD-L1 Antibody Treatment Resistance by Modulating Tumor-Associated Macrophages. Cancer Immunol. Res. 2019, 7, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Huang, S.; Huang, C.; Fay, N.S.; Wang, Y.; Putrevu, S.; Wright, K.; Zaman, M.S.; Cai, W.; Huang, B.; et al. Fc-competent multispecific PDL-1/TIGIT/LAG-3 antibodies potentiate superior anti-tumor T cell response. Sci. Rep. 2023, 13, 9865. [Google Scholar] [CrossRef]
- Machiraju, D.; Wiecken, M.; Lang, N.; Hulsmeyer, I.; Roth, J.; Schank, T.E.; Eurich, R.; Halama, N.; Enk, A.; Hassel, J.C. Soluble immune checkpoints and T-cell subsets in blood as biomarkers for resistance to immunotherapy in melanoma patients. Oncoimmunology 2021, 10, 1926762. [Google Scholar] [CrossRef]
- Gide, T.N.; Paver, E.C.; Yaseen, Z.; Maher, N.; Adegoke, N.; Menzies, A.M.; Pires da Silva, I.; Wilmott, J.S.; Long, G.V.; Scolyer, R.A. Lag-3 expression and clinical outcomes in metastatic melanoma patients treated with combination anti-lag-3 + anti-PD-1-based immunotherapies. Oncoimmunology 2023, 12, 2261248. [Google Scholar] [CrossRef] [PubMed]
- Storey, L.; Abdul-Latif, M.; Kreft, S.; Barrett, E.; Pickering, L.M.; Rohaan, M.W.; Ahmed, S.; Eigentler, T.K.; Hassel, J.C.; Haferkamp, S.; et al. Checkpoint inhibitor treatment in patients with isolated in-transit melanoma metastases. J. Clin. Oncol. 2020, 38, 10070. [Google Scholar] [CrossRef]
- Speiser, D.E.; Chijioke, O.; Schaeuble, K.; Munz, C. CD4(+) T cells in cancer. Nat. Cancer 2023, 4, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-gamma in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Erdag, G.; Schaefer, J.T.; Smolkin, M.E.; Deacon, D.H.; Shea, S.M.; Dengel, L.T.; Patterson, J.W.; Slingluff, C.L., Jr. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012, 72, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, R.; Zhang, G.; Fukunaga-Kalabis, M.; Perego, M.; Krepler, C.; Xu, X.; Wagner, C.; Hristova, D.; Zhang, J.; Tian, T.; et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat. Commun. 2017, 8, 607. [Google Scholar] [CrossRef] [PubMed]
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 2019, 10, 4186. [Google Scholar] [CrossRef] [PubMed]
- Ferrari de Andrade, L.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F., Jr.; Harvey, C.J.; et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Amores-Iniesta, J.; Conde-Garrosa, R.; Khouili, S.C.; Melero, I.; Sancho, D. Effective cancer immunotherapy by natural mouse conventional type-1 dendritic cells bearing dead tumor antigen. J. Immunother. Cancer 2019, 7, 100. [Google Scholar] [CrossRef]
- Zhao, Z.; Chung, Y.H.; Steinmetz, N.F. Melanoma immunotherapy enabled by M2 macrophage targeted immunomodulatory cowpea mosaic virus. Mater. Adv. 2024, 5, 1473–1479. [Google Scholar] [CrossRef]
- Ozbay Kurt, F.G.; Lasser, S.; Arkhypov, I.; Utikal, J.; Umansky, V. Enhancing immunotherapy response in melanoma: Myeloid-derived suppressor cells as a therapeutic target. J. Clin. Investig. 2023, 133, e170762. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.K.; Frankel, A.E.; Feun, L.G.; Ekmekcioglu, S.; Kim, K.B. Arginine deprivation therapy for malignant melanoma. Clin. Pharmacol. 2013, 5, 11–19. [Google Scholar] [CrossRef]
- O‘Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Papaccio, F.; Kovacs, D.; Bellei, B.; Caputo, S.; Migliano, E.; Cota, C.; Picardo, M. Profiling Cancer-Associated Fibroblasts in Melanoma. Int. J. Mol. Sci. 2021, 22, 7255. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Guo, S.; Guan, X.; Kang, Y.; Liu, J.; Yang, X. Immunological Classification of Tumor Types and Advances in Precision Combination Immunotherapy. Front. Immunol. 2022, 13, 790113. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; He, Y.; He, W.; Wu, G.; Zhou, X.; Sheng, Q.; Zhong, W.; Lu, Y.; Ding, Y.; Lu, Q.; et al. Exhausted CD8+T Cells in the Tumor Immune Microenvironment: New Pathways to Therapy. Front. Immunol. 2020, 11, 622509. [Google Scholar] [CrossRef]
- Moro, A.; Gao, Z.; Wang, L.; Yu, A.; Hsiung, S.; Ban, Y.; Yan, A.; Sologon, C.M.; Chen, X.S.; Malek, T.R. Dynamic transcriptional activity and chromatin remodeling of regulatory T cells after varied duration of interleukin-2 receptor signaling. Nat. Immunol. 2022, 23, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Kirilovsky, A.; Angell, H.K.; Obenauf, A.C.; Tosolini, M.; Church, S.E.; Maby, P.; Vasaturo, A.; Angelova, M.; et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci. Transl. Med. 2016, 8, 327ra326. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Sajid, M.; Lv, R.; Liu, L.; Sun, C. A review of spatial profiling technologies for characterizing the tumor microenvironment in immuno-oncology. Front. Immunol. 2022, 13, 996721. [Google Scholar] [CrossRef] [PubMed]
- Brummel, K.; Eerkens, A.L.; de Bruyn, M.; Nijman, H.W. Tumour-infiltrating lymphocytes: From prognosis to treatment selection. Br. J. Cancer 2023, 128, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Sobottka, B.; Nowak, M.; Frei, A.L.; Haberecker, M.; Merki, S.; Tumor Profiler, c.; Levesque, M.P.; Dummer, R.; Moch, H.; Koelzer, V.H. Establishing standardized immune phenotyping of metastatic melanoma by digital pathology. Lab. Investig. 2021, 101, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Nirmal, A.J.; Maliga, Z.; Vallius, T.; Quattrochi, B.; Chen, A.A.; Jacobson, C.A.; Pelletier, R.J.; Yapp, C.; Arias-Camison, R.; Chen, Y.A.; et al. The Spatial Landscape of Progression and Immunoediting in Primary Melanoma at Single-Cell Resolution. Cancer Discov. 2022, 12, 1518–1541. [Google Scholar] [CrossRef] [PubMed]
- Sautes-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Factors | Descriptions | Association with Response/Resistance | References |
|---|---|---|---|
| Response | |||
| CD8+ T cells | Releasing IFN-γ, granzyme B, and perforin to eliminate tumor cells | High intra- or peri-tumoral proportion | [58] |
| CD4+ helper T cells | Assisting CD8+ T cells; secretion of IFN-γ and TNFα to directly target tumor cells | High proportion in tumor | [59] |
| CD20+ B cells | Forming tertiary lymphoid structure within the tumor | High proportion in tumor | [60] |
| M1 macrophages | Tumor-associated macrophage secreting IL-1 and TNFα that harm tumor cells | High proportion in tumor | [61] |
| CTLA-4 | Inhibiting CD4+ T cell differentiation and activation | High expression on naïve T cells | [62] |
| PD-1 | Interacting with PD-L1 to put CD8+ T cells into hyporesponsive exhausted state | High expression on CD8+ T cells | [62] |
| PD-L1 | Interacting with PD-1 to put CD8+ T cell into a hyporesponsive exhausted state | High expression on tumor cells | [63] |
| Resistance | |||
| CD4+ Tregs | Immunosuppressive T cells regulating immunity of cytotoxic T cells | High intra-tumoral proportion | [64] |
| MDSCs | Immune cells secreting immunosuppressive cytokines such as IL-10, IL-35, and TGF-β | High tumor-infiltrated proportion | [65] |
| M2 macrophages | Anti-inflammatory tumor-associated macrophage secreting immunosuppressive cytokines such as IL-10 | High tumor-infiltrated proportion | [61] |
| LAG-3 | Immune checkpoint molecules mediating tumor immune escaping | High expression on LAG-3+ TILs | [66] |
| TIM-3 | Immune checkpoint molecules | High expression on various immune cells | [67] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, J.; Quek, C. Understanding the Tumor Microenvironment in Melanoma Patients with In-Transit Metastases and Its Impacts on Immune Checkpoint Immunotherapy Responses. Int. J. Mol. Sci. 2024, 25, 4243. https://doi.org/10.3390/ijms25084243
Tian J, Quek C. Understanding the Tumor Microenvironment in Melanoma Patients with In-Transit Metastases and Its Impacts on Immune Checkpoint Immunotherapy Responses. International Journal of Molecular Sciences. 2024; 25(8):4243. https://doi.org/10.3390/ijms25084243
Chicago/Turabian StyleTian, Jiabao, and Camelia Quek. 2024. "Understanding the Tumor Microenvironment in Melanoma Patients with In-Transit Metastases and Its Impacts on Immune Checkpoint Immunotherapy Responses" International Journal of Molecular Sciences 25, no. 8: 4243. https://doi.org/10.3390/ijms25084243
APA StyleTian, J., & Quek, C. (2024). Understanding the Tumor Microenvironment in Melanoma Patients with In-Transit Metastases and Its Impacts on Immune Checkpoint Immunotherapy Responses. International Journal of Molecular Sciences, 25(8), 4243. https://doi.org/10.3390/ijms25084243